

Abstract
Phenol hydroxylase (PH) belongs to a family of bacterial multicomponent monooxygenases (BMMs) with carboxylate-bridged diiron active sites. Included are toluene/o-xylene (ToMO) and soluble methane (sMMO) monooxygenase. PH hydroxylates aromatic compounds, but unlike sMMO, it cannot oxidize alkanes despite having a similar dinuclear iron active site. Important for activity is formation of a complex between the hydroxylase and a regulatory protein component. To address how structural features of BMM hydroxylases and their component complexes may facilitate the catalytic mechanism and choice of substrate, we determined X-ray structures of native and SeMet forms of the PH hydroxylase (PHH) in complex with its regulatory protein (PHM) to 2.3 Å resolution. PHM binds in a canyon on one side of the (αβγ)2 PHH dimer, contacting α-subunit helices A, E, and F ∼12 Å above the diiron core. The structure of the dinuclear iron center in PHH resembles that of mixed-valent MMOH, suggesting an Fe(II)Fe(III) oxidation state. Helix E, which comprises part of the iron-coordinating four-helix bundle, has more π-helical character than analogous E helices in MMOH and ToMOH lacking a bound regulatory protein. Consequently, conserved active site Thr and Asn residues translocate to the protein surface, and an ∼6 Å pore opens through the four-helix bundle. Of likely functional significance is a specific hydrogen bond formed between this Asn residue and a conserved Ser side chain on PHM. The PHM protein covers a putative docking site on PHH for the PH reductase, which transfers electrons to the PHH diiron center prior to O2 activation, suggesting that the regulatory component may function to block undesired reduction of oxygenated intermediates during the catalytic cycle. A series of hydrophobic cavities through the PHH α-subunit, analogous to those in MMOH, may facilitate movement of the substrate to and/or product from the active site pocket. Comparisons between the ToMOH and PHH structures provide insights into their substrate regiospecificities.
Bacterial multicomponent monooxygenases (BMMs)1 are a diverse family of enzymes that hydroxylate and epoxidize an array of hydrocarbon substrates, including alkanes, alkenes, and aromatics in a regio- and enantioselective fashion (1-3). This chemistry takes place at a carboxylate-bridged diiron center similar to the ones found in the R2 subunit of type I ribonucleotide reductase (RNR-R2), stearoylacyl carrier protein (ACP) Δ9-desaturase, and ferritin (4, 5). Bacteria harboring different BMM family members, which include soluble methane monooxygenases (sMMOs), toluene monooxygenases (TMOs), phenol hydroxylase (PH), alkene monooxygenase (AMO), tetrahydrofuran monooxygenase (THFMO), and hyperthermophilic toluene monooxygenase (SsoMO), use small organic compounds as their sole source of carbon and energy. The substrate transformations performed by this family of enzymes are responsible, in part, for removing atmospheric methane, a greenhouse gas, and carcinogenic halogenated solvents like TCE from the environment (6, 7). Because of the broad range of substrates oxidized by these enzymes, they have generated much interest for bioremediation and synthetic industrial applications.
BMMs like sMMO and PH consist of a 200–255 kDa dimeric hydroxylase of the form (αβγ)2, a cofactorless 10–16 kDa regulatory protein that enhances catalytic turnover by 30–150-fold and a FAD- and [2Fe-2S]-containing 38–40 kDa reductase that supplies the hydroxylase with electrons by consuming NADH (1-3). TMOs and SsoMOs differ slightly since they utilize an additional, Rieske protein to assist with electron transfer between the reductase and hydroxylase components (2, 3). The active sites in the resting states of the methane (MMOH) and toluene/o-xylene (ToMOH) monooxygenase hydroxylases contain a diiron(III) center coordinated by four glutamate and two histidine ligands from a four-helix bundle composed of helices B, C, E, and F of the protein α-subunit (8, 9). Solvent-derived water and hydroxide ions complete the octahedral coordination spheres. Sequence, spectroscopic, and structural analyses suggest that all BMM hydroxylases have nearly identical dinuclear iron centers (1, 10-12). Because AMO, TMO, SsoMO, and PH are incapable of hydroxylating alkanes, whereas sMMO and THFMO are adept at this transformation, other features of the system must control substrate reactivity at their dioxygen-activated metal centers. For sMMO, methane hydroxylation is achieved exclusively by a di-μ-oxo diiron(IV) intermediate, Q, whereas other reactions can occur at a diiron(III) peroxo precursor of Q, Hperoxo (1, 13, 14). A diiron(IV) intermediate has not been detected in any other BMM hydroxylase, but recent work has identified oxygenated diiron(III) species capable of oxidizing aromatic substrates (15).2,3
Comparisons between the MMOH and ToMOH structures have provided insight into how the protein scaffold may tune the reactivity and substrate specificity of the diiron center by controlling the access of small molecules to the active site pocket. In particular, there is a large access channel in ToMOH linking the diiron center to the exterior environment that is absent in MMOH (8, 9). The regulatory proteins in these systems may also influence reactivity by inducing different structural changes at or near the diiron active site. In addition to enhancing the catalytic rate, the regulatory protein alters the substrate regiospecificity (16-18), influences the spectroscopic and redox properties of the diiron center (1), governs movement of the substrate and product to and from the active site pocket (5), and couples NADH consumption by the reductase with substrate hydroxylation at the diiron center (18, 19). Knowledge of the docking site on the hydroxylase and of structural changes at the diiron center imposed by the regulatory protein is therefore essential for improving our understanding of O2 and C–H bond activation in BMMs.
To address these questions and improve our structural knowledge about the BMM family members, we have determined in this study the X-ray crystal structure of a complex between the hydroxylase component of phenol hydroxylase, PHH, and its regulatory protein, PHM, from Pseudomonas sp. OX1 to 2.3 Å resolution. The information afforded provides novel insights into the functions of the regulatory protein and the tuning of the diiron active site chemistry in BMM enzyme systems.
MATERIALS AND METHODS
Purification of Phenol Hydroxylase
The hydroxylase (PHH), regulatory protein (PHM), and reductase (PHP) from Pseudomonas sp. OX1 were expressed simultaneously from the pJSX148 plasmid in Escherichia coli JM109 cells essentially as described by Cafaro et al. (20). The procedure was modified such that 100 μM Fe(NH4)2SO4·6H2O was added to each liter of medium at induction and every hour thereafter until the cells were harvested.
The frozen cells were resuspended in a 25 mM MOPS (pH 7.0), 10% glycerol, 50 mM NaCl buffer (buffer A) supplemented with 5 mM MgCl2, Pefabloc, 20 units of DNase, and PMSF and then lysed by sonication using 6 × 1 min pulses. Particulate debris was removed by centrifugation at 163000g for 45 min, after which the supernatant was filtered through a 0.2 μm membrane and loaded onto a DEAE column conditioned with buffer A. A salt gradient was run from 50 to 400 mM NaCl, and the hydroxylase-containing fractions were pooled and concentrated to <5 mL using an Amicon concentrator fitted with a YM100 membrane. The concentrated protein was loaded onto an S300 column preequilibrated with 25 mM MOPS (pH 8.0), 10% glycerol, and 300 mM NaCl (buffer B) and run in that same buffer. Fractions containing PHH were pooled, loaded onto a phenyl Sepharose column conditioned with 10 mM MOPS (pH 7.0), 10% glycerol, and 100 mM NaCl, and eluted by using a 100 to 0 mM NaCl gradient. The purified PHH, which contained noticeable amounts of PHM, was concentrated, frozen in liquid N2, and stored at −80 °C.
A selenomethionine (SeMet) derivative of PHH was generated by growing BL21(DE3) Gold E. coli cells transformed with the pJSX148 plasmid on LeMaster's medium supplemented with 90 mg/L Se-Met and ∼5 mg/L folate, vitamin B12, nicotinamide, pantothenate, thiamine, riboflavin, and p-aminobenzoic acid (21). The protein was expressed and purified as described above.
Activity and Iron Content
The enzyme activity, measured by using a catechol 2,3-dioxygenase (C2,3O) assay as described previously (20), was 470 ± 40 milliunits/mg. The C2,3O protein required for the assay was expressed and purified as described previously (20). The iron content of PHH was determined to be 3.6 Fe atoms/dimer by a colorimetric ferrozine assay (22).
Crystallization and Collection of X-ray Diffraction Data
Crystals were grown at 20 °C by the hanging drop vapor diffusion method. A protein solution containing 35 μM PHH in 10 mM MES (pH 7.1) and 10% glycerol was combined with an equal volume of crystallization buffer containing 100 mM Tris (pH 7.0), 150 mM Na2MoO4, 5% glycerol, and 17–20% PEG 8000 (w/w). This condition was initially identified by using the Hauptman-Woodward Institute high-throughput screen. The addition of varying amounts of PHM to the crystallization buffer did not improve the occupancy of the regulatory protein in the crystals (vide infra). Before data collection, PHH crystals were frozen in a cryo solution containing the precipitant and 20% glycerol. Data at the Se peak wavelength were collected on beamline 9-1 at SSRL, and the native data set was collected at APS on the NECAT 8BM beamline. The data were indexed and scaled with HKL2000 (23). The space group is P212121, and the unit cell dimensions are 87.8 Å × 146.3 Å × 190.0 Å. Further experimental details about data collection and refinement are given in Table 1.
Table 1.
Data Collection, Phasing, and Refinement Statistics
| Se SAD | native | |
|---|---|---|
| data collection | ||
| beamline | SSRL BL9-1 | NE-CAT (Sector 8) |
| wavelength (Å) | 0.979 | 0.979 |
| resolution (Å) | 30–2.7 | 30–2.3 |
| no. of unique observationsa | 109776 | 108695 |
| total no. of observationsa | 592029 | 773692 |
| completeness (%)a | 83.0 (49.8)b | 100 (100)b |
| redundancya | 5.4 (4.3)b | 7.1 (6.9)b |
| I/σa | 17.3 (5.0)b | 16.5 (4.9)b |
| Rsyma,c (%) | 5.9 (26.0)b | 8.9 (44.8)b |
| Se sites used for phasing | 71 of 77 | |
| figure of merit (after density modification) | 0.40 (0.75) | |
| refinement | ||
| Rwork (%)d | 20.2 | 19.8 |
| Rfree (%)e | 25.2 | 24.2 |
| no. of molecules in ASU | 1 | 1 |
| no. of protein non-hydrogen atoms | 16076 | 15307 |
| no. of non-protein atoms | 315 | 1042 |
| rmsd for bond lengths (Å) | 0.033 | 0.0065 |
| rmsd for bond angles (deg) | 2.55 | 1.28 |
| average B value (Å2) | 36.0 | 37.7 |
Values for the Se SAD data were calculated considering I+ and I− as separate reflections.
Values in parentheses are for the highestresolution shell.
, where Ii(hkl) is the ith measured diffraction intensity and 〈I(hkl)〉 is the mean of the intensity for the Miller index (hkl).
Rfree = Rwork for a test set of reflections (5%).
Structure Determination
Molecular replacement was attempted initially by using the most structurally conserved regions of the MMOH and ToMOH α- and β-subunits. None of the starting α, α2, β, β2, αβ, and α2β2 models used for molecular replacement afforded good starting phases, even though the PHH α- and β-subunits fold in a manner similar to the folding of these subunits in MMOH and ToMOH (see below).
Single-wavelength anomalous dispersion data for the selenomethionine derivative were then used to determine a set of starting phases. Attempts to determine the heavy atom substructure by using CNS (24), Solve (25), and SHELX (26) failed. SnB (27) was the only program able to locate 71 of 77 Se atoms in the asymmetric unit by using the peak wavelength. Native data were included in the phasing by refinement of both real and anomalous occupancies with MLPHARE from the CCP4 suite of programs (28). Initial phases were calculated with MLPHARE, improved, and extended to the limit of the native data by density modification with DM (28). Resolve (25) was used to autobuild ∼50% of PHH, after which the remainder of the molecule was manually built in XtalView (29) and O (30) and refined with REFMAC5 (28) and CNS (24). Because of structural and occupancy differences, models for both the native and SeMet data sets were generated. The relative occupancy of PHM was estimated by refining the PHM occupancies after its B-factors were fixed to reflect the average value of the entire PHH molecule. A Ramachandran plot calculation using PROCHECK (31) indicated that 99.8% of the residues occupied allowed regions.
RESULTS AND DISCUSSION
PHH Global Fold
The PH hydroxylase component has an (αβγ)2 homodimeric composition with a noncrystallo-graphic 2-fold symmetry axis similar to those in the ToMOH and MMOH structures (Figure 1A and Figure S1A–C of the Supporting Information) (8, 9). The folds of the PHH α- and β-subunits are nearly identical to those of the respective subunits in ToMOH and MMOH. The rms deviations between the α-carbon atoms are 1.5–1.6 Å for all possible α-to-α and β-to-β combinations. The largest topological differences between these evolutionarily related hydroxylase proteins occur for the γ-subunit, both in its fold and in its location. For PHH and ToMOH, the γ-subunits are positioned in the top left and right quadrants (11 and 1 o'clock, respectively) of the protein with similar ferredoxin-like folds despite having sequences that are only 13% identical (Figure S2) (9). Unlike the ToMOH γ-subunit, however, the PHH γ-subunit has an unstructured 22-amino acid N-terminus that forms extensive contacts with the α-subunit. For MMOH, the mostly α-helical γ-subunits localize to the far left- and right-hand sides of the protein (9 and 3 o'clock, respectively) and contact both the α- and β-subunits (Figure S1). Such structural differences are not surprising given the poor sequence conservation for this subunit among the different BMM family members. The function of this subunit, which is absent in some BMMs like alkene monooxygenases, is unclear (2, 3, 11).
Figure 1.
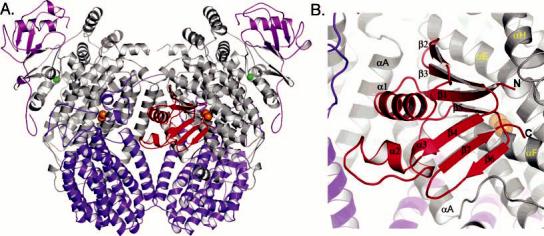
(A) Global structure of the PHH–PHM complex and (B) enlarged view of PHM in the PHH canyon. The PHH α-, β-, and γ-subunits are colored gray, purple, and pink, respectively, and PHM is colored red. Iron and zinc atoms are depicted as orange and green spheres, respectively. All figures were generated by using PyMOL (70).
PHM Fold and Binding Site on the Hydroxylase
The interface between the αβγ protomers of PHH forms a canyon, which we previously suggested would be the binding site for the BMM regulatory protein and reductase components on their hydroxylases (5, 8, 32, 33). On one face of the PHH protein, a single molecule of PHM binds in the canyon region across α-subunit helices A, E, F, and H, just 12 Å above the diiron center at its closest distance. On the opposite face of the hydroxylase, both a γ-subunit from a different, symmetry-related hydroxylase molecule in the unit cell and the N-terminus of the β-subunit occupy the space defined by the PHM binding site (Figure S3). The occupancy of the PHM component was estimated to be 40 and 50% in the native and selenomethionine (SeMet) structures, respectively. These different occupancies presumably reflect differing amounts of PHM that copurified with the hydroxylase component. We therefore generated models of the PHHPHM complex from both the native and SeMet data.
The PHM global fold comprises three α-helices flanked by two nearly perpendicular β-sheets (Figure 1B). The fold is nearly identical to those previously reported for the NMR and crystal structures of the methane monooxygenase (MMOB; PDB entries 1CKV and 2MOB) and toluene 4-monooxygenase (T4MOD; PDB entries 2BF5 and 1G10) regulatory proteins (34-37). The rms deviations between PHM and the T4MOD and MMOB proteins are between 1.5 and 1.6 Å for all backbone atoms (Figure S4). Comparisons between hydroxylase-bound PHM, T4MOD, and MMOB suggest there may be no significant structural rearrangements of the peptide backbone when these regulatory proteins form a complex with their respective hydroxylases.
Primarily in contact with the α-subunit of one protomer, PHM interacts with PHH helices A, E, F, and H via β-strands 1, 5, and 6, α-helix 3, the N-terminal region of α-helix 1, the loop between β-strands 2 and 3, and the peptide chain termini (Figures 1B and S5). The PHM binding site on the surface of the PHH α-subunit is formed by portions of the helices including residues 64–68 (helix A), 200–207 (helix E), and 226–245 (helix F), which collectively create a concave surface that complements the protrusive binding face of the regulatory protein. The PHH-PHM binding interaction is primarily hydrophobic, with some flanking polar and charged residues (Figure S1D). The N- and C-termini of the PHH regulatory protein are oriented away from the canyon region toward solvent and form some contacts with α-subunit helix H (Figure 1B).
Sequence alignments of the regulatory proteins from representative BMM family members indicate that very few of the amino acids are strictly conserved (Figure S5). Residues at the binding interface appear to be conserved only within a subfamily, suggesting why regulatory protein cross-reactivity with hydroxylases from different BMM subfamilies is rarely observed. Nevertheless, we generated electrostatic surface maps of what might be the homologous hydroxylase binding faces of T4MOD and MMOB from the information in Figure S5. As depicted in panels E and F of Figure S1, these protein surfaces, like that in PHM, have significant hydrophobic character, but with a somewhat greater number of charged residues. NMR line broadening, mutagenesis, cross-linking, and spin-labeling studies probing interactions of MMOB with MMOH are highly consistent with a similar surface being used by MMOB to bind its hydoxylase (Figure S5) (34, 38-40). Previously published spin-labeling experiments by our laboratory identified a different and probably incorrect MMOB binding face (41). A recent discussion of component interactions in the sMMO system is available (40).
The PHH–PHM crystal structure presented here thus serves as an excellent template for modeling interactions of the folded regulatory protein core with its cognate hydroxylase in related BMM systems. Although the unstructured Nand C-termini of some regulatory proteins (Figure S5), such as those on MMOB that affect steps in the catalytic cycle (42-45), cannot be placed on the hydroxylase surface with any degree of certainty, the specific interactions identified here (vide infra) should be quite valuable in guiding future mutagenesis, structural, and mechanistic studies of all the BMM enzymes.
Diiron Center
The diiron center of PHH is located in the α-subunit four-helix bundle 12 Å below the surface. Both protomers in the native and SeMet structures have nearly similar diiron active sites despite the binding of PHM to only one side of the hydroxylase. The iron atoms are coordinated by a bridging or semibridging glutamate, Glu-138, the δ-nitrogen atoms of His-141 and His-236, which lie distal to the active site pocket, and three additional glutamate residues, Glu-108, Glu-199, and Glu-233 (Figures 2 and S6). Glu-108 coordinates to Fe1 and hydrogen bonds to a terminal water molecule bound to the same iron atom. This structural motif is observed in all known BMM hydroxylase structures in their various oxidation and product-bound states (9, 33, 46-50). In the native structure and the SeMet α-subunit with bound PHM, Fe2 is coordinated by Glu-233 in a bidentate chelating mode, the geometry of which is most similar to that of the mixed-valent, Fe(II)Fe(III) form of MMOH (MMOHmv) (Figure 2C). In the other SeMet protomer without bound PHM, Glu-233 appears to coordinate to Fe2 by using only one oxygen atom, the other forming a hydrogen bond to the terminal water on Fe1. For most of the active sites, Glu-199 coordinates to Fe2 via just one oxygen atom while the other dangling oxygen atom is in hydrogen bond contact with nearby residues Tyr-115 and Gln-134 (Figures 2 and 3A). This ligand appears to adopt a bidentate chelating geometry in the SeMet α-subunit without bound PHM. The final iron ligand that we could identify is a water molecule bound exclusively to Fe1. Electron density corresponding to a bridging or semibridging water-derived ligand, like hydroxide or oxide, between the iron atoms was not observed. In the higher-resolution native structure, the Fe1–Fe2 distances are 3.6 and 3.3 Å for the PHM-bound and unbound protomers, respectively. The Fe1–Fe2 distances in the lower-resolution SeMet structure are 3.0–3.1 Å for both active sites. These shorter Fe–Fe distances may be a consequence of a fully bridging Glu-138 residue versus the semibridging one in the native structure. Overall, Fe1 has a five-coordinate, square-pyramidal geometry, whereas Fe2 adopts a more distorted structure.
Figure 2.
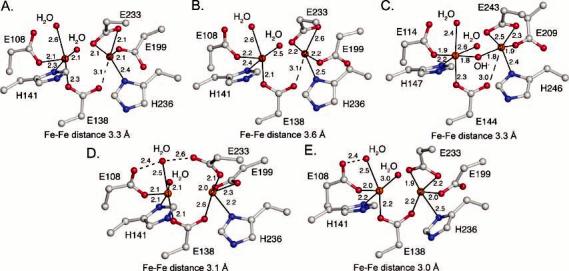
Structures of the diiron center. (A) Native PH diiron center without PHM bound. (B) Native PH diiron center with PHM bound. (C) Mixed-valent MMOH diiron center. (D) SeMet PH diiron center without PHM bound. (E) SeMet PH diiron center with PHM bound. Carbon, nitrogen, oxygen, and iron atoms are colored gray, blue, red, and orange, respectively. This color scheme will be used throughout unless otherwise noted.
Figure 3.
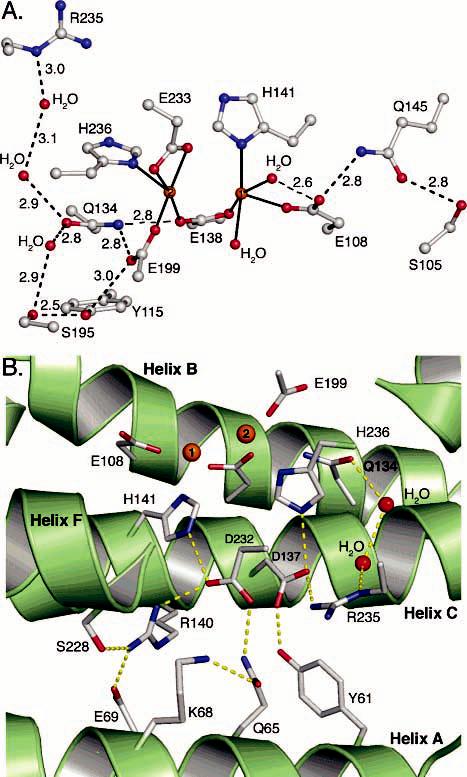
Hydrogen bonding networks in the second coordination sphere. (A) Hydrogen bonding networks flanking the diiron center. (B) Hydrogen bonding network behind the diiron center that extends to the surface of the hydroxylase canyon region where PHM binds.
The identity of the Fe2-coordinating histidine N-donor is notably different among the PHH, MMOH, and ToMOH dinuclear iron centers. In ToMOH, this histidine coordinates to iron by using its ε-nitrogen atom, whereas in MMOH and PHH, the histidine δ-nitrogen atom is employed. When the ToMOH structure was first determined, it was unclear whether this structural difference might influence the hydroxylation chemistry (9). Since PH and ToMO hydroxylate similar aromatic substrates at nearly equivalent rates (20), the structural difference is presumably of little or no consequence to this property of the dimetallic center.
At present, it is difficult to discern the true oxidation state of the different dinuclear iron centers in PHH enzymes. As purified, the enzyme has an optical band at 350 nm that is indicative of a (μ-oxo)diiron(III) species comprising ∼68–96% of the sample (ε350 = 4800–6000 M−1 cm−1 per dinuclear iron cluster) (12). PHH from Pseudomonas sp. strain CF600 is 81% identical to the P. stutzeri OX1 enzyme and forms a complex with its regulatory component involving the α-subunit as deduced by chemical cross-linking studies (51). Spectroscopic investigations of this enzyme indicate that the oxidized form contains both (μ-oxo)- and di-μ-(hydroxo)diiron(III) metal centers (12) and that the predominant, catalytically competent species is the oxo-bridged diiron(III) form. Preliminary EPR spectroscopic analysis of samples of P. stutzeri OX1 PHH in its resting and cryo-reduced states reveals that the purified protein contains a heterogeneous mixture of diiron(III) and mixed-valent Fe(II)Fe(III) species, the relative concentrations of which are at present unknown.4 If the PHH metal centers in these crystal structures were fully oxidized, one would expect to see either a short Fe–Fe distance of 3.0 Å and a dihydroxo-bridged dimetallic cluster (9, 48, 49) or a longer Fe–Fe distance of 3.2–3.6 Å and an oxo-bridged cluster (4, 5, 52). None of the PHH diiron centers, however, has an observable bridging oxide or hydroxide ligand. Given that the geometries of the active site metal clusters are most similar to that of the mixed-valent Fe(II)Fe(III) form of MMOH (49), one possibility is that photoreduction by synchrotron radiation occurred to produce the mixed-valent oxidation state. A similar phenomenon has been reported previously for structures of Δ9-desaturase and RNR-R2, although the oxidized forms of these proteins were reduced by two electrons (53-55). The structures of the PHH diiron centers presented here are not charge-balanced, however, and would require an additional negatively charged ligand, such as a bridging hydroxide ion, to achieve a neutral state similar to that observed in MMOHmv (Figure 2C). Although |2Fo| – |Fc| and |Fo| – |Fc| simulated-annealing omit electron density maps have not allowed us to identify such a ligand, the data quality at 2.3 and 2.7 Å resolution may not be sufficient to reveal a coordinating oxygen atom between the iron atoms (56).
A second possibility is that the metal centers have each been photoreduced by two electrons to the diiron(II) state, rendering the PHH active sites charge neutral. The structures do not, however, resemble the reduced forms of MMOH and ToMOH, which both have a 3.3–3.4 Å metal–metal distance and a glutamate residue that coordinates to Fe2 in a bidentate chelating fashion and simultaneously bridges the two iron atoms with one of its O atoms (48, 49, 57). Because the active sites on the PHM-bound and unbound sides of PHH are so similar, it is unclear whether PHM affects them both in a similar manner by an allosteric effect or not at all. The incomplete PHM occupancy may also be a factor. Further spectroscopic and structural investigations are required to improve our understanding of the PHH dinuclear iron center and to discern how the diiron site geometries are influenced by the oxidation state and the presence of bound PHM.
Second-Sphere Hydrogen Bonding Patterns
The second coordination sphere is critical to the formation and stability of the BMM diiron centers, providing ligand conformational constraints in the PHH, ToMOH, and MMOH structures (5). For PHH, the geometry of Glu-108, which coordinates to Fe1, is stabilized by a hydrogen bonding network involving Gln-145 and Ser-105 (Figure 3). A similar pattern exists in ToMOH involving Gln-141 and His-96 (9), but not in MMOH, which has a cysteine residue positioned 4.5 Å from the coordinating glutamate (8). Gln-145 is conserved among all PH and TMO family members, whereas Ser-105 and His-96 are not. Substituting the corresponding Gln with a Cys in toluene 4-monooxygenase (T4MO) resulted in a variant with a 10-fold diminished turnover rate that was still incapable of hydroxylating methane (58). A Gln-to-Val substitution in the same enzyme resulted in a 100-fold decrease in the rate of turnover compared to that of the wild-type protein. These results and the crystal structure presented here suggest that stabilization of the Fe1-coordinating terminal glutamate by hydrogen bonding may be critical for reactivity in the TMO and PH systems. The importance of such hydrogen bonding involving the analogous residue in MMOH, Glu-114, is less obvious because this residue has no hydrogen bonding partners other than an active site pocket water molecule that is not always detected in every crystal structure (49).
At the other end of the diiron center, there is extensive hydrogen bonding to conserved Gln-134, which may be essential for stabilizing the geometry by which the bridging Glu-138 and terminal Glu-199 residues coordinate to Fe2 (Figure 3). Glu-199 may also form a weak hydrogen bond with Tyr-115; this residue is a phenylalanine in homologous PH family members, however. The same general arrangement of amino acids is preserved in MMOH (9). ToMOH utilizes a water molecule (or hydronium ion) to mediate interactions between the Fe-coordinating glutamates and a second-sphere glutamate, Glu-111. In the PHH structure, hydrogen bonds between two ordered water molecules link Gln-134 with conserved Arg-235, an arrangement that differs from that in the other hydroxylase proteins (see below). It is clear through these comparisons that BMMs employ a variety of schemes to stabilize the coordination spheres around the two iron atoms, but it is unknown whether and how these different architectures might contribute to the dissimilar reactivity of the enzymes. Comparisons of the native and SeMet α-subunits with and without bound PHM do not reveal any significant alterations in these hydrogen bonding patterns.
A Conserved Mononuclear Zn2+ Site
In the C-terminal domain of the PHH α-subunit, 24 Å from the diiron center, there exists a four-cysteine site that coordinates a single metal ion with a tetrahedral geometry and an average metal–sulfur distance of 2.3 Å (Figures 1A and 4). Sequence homology indicates that the coordinating cysteines are conserved only among members of the PH and AMO families (Figure S7). UV–vis, EPR, and Mössbauer spectroscopic studies of recombinant PHH from P. stutzeri OX1 and native PHH from Pseudomonas sp. strain CF600 do not reveal the presence of a thiolate rich mononuclear iron center, or any other mononuclear paramagnetic metal center, in any of the protein preparations (12).3, 4 Anomalous difference electron density maps generated from data collected at the selenium edge reveal much stronger peaks at the mononuclear metal site than at the diiron center (Table 2), suggesting that the metal at this position is a stronger absorber of X-rays at 12 660 eV than iron, the K-edge of which is 7111 eV. The K-edge of zinc occurs at 9659 eV. Given the ligand content, geometry, and absence of a spectroscopic signal, Zn2+ is most likely bound to this position. In addition, it was observed previously that the toluene 2-monooxygenase hydroxylase, a PH family member, is purified with two zinc and four iron atoms (59). In light of this information, we conclude that zinc is the physiologically preferred metal ion bound to the four-cysteine site and suggest that it may be important for stabilizing the fold of the α-subunit C-terminal domain.
Figure 4.
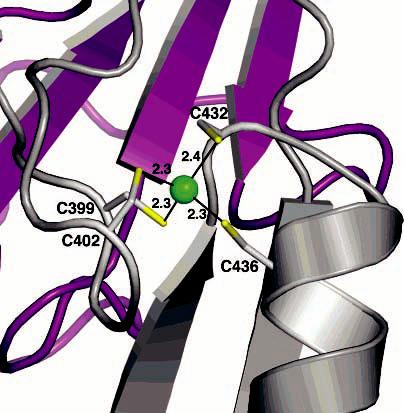
Zinc binding site in the C-terminal domain of the PHH α-subunit. Zinc is depicted as a green sphere, and sulfur atoms are colored yellow. The α-subunit is depicted as a gray ribbons and the γ-subunit as purple ribbons.
Table 2.
Heavy Atom Anomalous Peak Intensitiesa
| subunit | atom | peak height | subunit | atom | peak height |
|---|---|---|---|---|---|
| α1 | Fe1 | 11.3 | α2 | Fe1 | 11.4 |
| Fe2 | 9.62 | Fe2 | 8.9 | ||
| Zn | 20.2 | Zn | 22.9 |
Peak intensities were determined by using CCP4 (28).
Conserved Active Site Residues and Changes in Helix E
Situated just above the diiron center on helix E in PHH are two highly conserved residues found in every BMM, Thr-203 and Asn-204 (Thr-213 and Asn-214 in MMOH, respectively). Thr-203 is positioned at the interface between helices E and F above the Fe2-coordinating Glu-233 (Figure 5A), which undergoes oxidation state-dependent carboxylate shifts in MMOH and ToMOH (48, 49, 57). Asn-204 is located on the surface of the protein, 12 Å from the diiron site. Several ordered water molecules link these Thr and Asn residues to the diiron center by hydrogen bonds. In MMOH and ToMOH, the Thr residue forms the “roof” of the active site pocket. The Asn residue lies between helices E and F just above the iron-coordinating Glu that undergoes a carboxylate shift upon reduction of the diiron center (Figure 5B) (48, 49). The rotameric conformation of this Asn also changes with the active site oxidation state; it points inward toward the diiron center in the mixed-valent and reduced forms and away from it when the iron atoms are oxidized (Figure 5B) (48, 49). Mutagenesis studies on TMOs suggest that the Thr is not essential for catalysis under steady state conditions (60), whereas the Asn is important for turnover and protein component interactions.5
Figure 5.
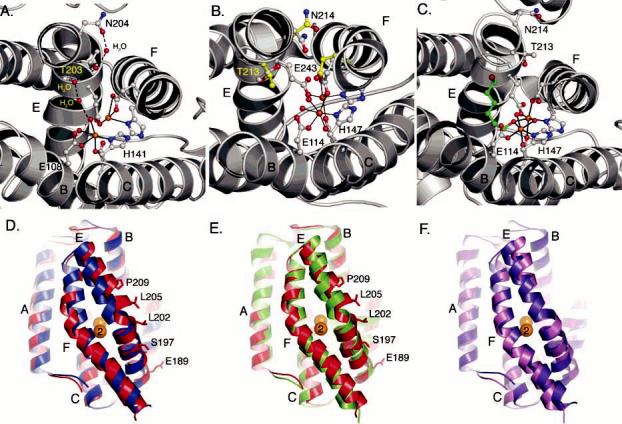
Structural differences in conserved helix E residues. (A) Positions of conserved Thr-203 and Asn-204 in PHH. (B) Redox-dependent structural changes in the positions of Thr-213, Asn-214, and Glu-243 in MMOH. Carbon atoms for the reduced and oxidized forms of MMOH are colored gray and yellow, respectively. (C) Positions of conserved Thr-213 and Asn-214 in the 6-bromohexan-1-ol-bound form (green) of MMOH in which helix E adopts a more π-helical character. (D) Superimposition of the PHH (red) and ToMOH (blue) four-helix bundles. Helices A–F are labeled accordingly. (E) Superimposition of the four-helix bundle from the 6-bromohexan-1-ol-bound form of MMOH (green) with the PHH (red) four-helix bundle. (F) Superimposition of the ToMOH (violet) and MMOH (pink) four-helix bundles.
The positions of this pair of Thr and Asn residues in both protomers from the native and SeMet PHH structures differ significantly from what has been observed for oxidized, reduced, and mixed-valent MMOH (8, 46, 48, 49) as well as for oxidized and Mn2+-substituted ToMOH (9, 57). The alternative positions that we observe here are the consequence of a different helix E peptide backbone configuration (Figure 5D). Residues 193–202 form a π-helix; residues 205–217 are α-helical, and Thr-203 and Asn-204 mark the transition from the π-to the α-helical forms. In MMOH and ToMOH, the backbone atoms of Thr-213 and Asn-214 assume an α-helical configuration (Figure 5B,F), whereas the residues preceding these two form a π-helix. The alternative configuration of helix E in PHH has been observed once before, in a 6-bromohexan-1-ol-bound structure of MMOH (Figure 5C,E) (33). In this particular MMOH crystal structure, residues 212–216 adopt a π-helical configuration to accommodate the bound product analogue by increasing the volume of the active site pocket. As a result, the orientation of the conserved Thr and Asn residues changes to adopt a configuration nearly identical to the one observed in PHH. We previously hypothesized that such a helical reconfiguration in MMOH may reflect one way by which the regulatory protein induces a change in the active site pocket to alter substrate regiospecificity and accommodate larger substrates. The PHH structure marks the second time that this alternative configuration of helix E has been observed and strongly supports our previous suggestion that the flexibility exhibited by this segment of the α-subunit may play a significant, and largely unexplored, role in the functions of BMMs. Because helix E in all of the PHH α-subunits adopts this configuration, we cannot conclude that PHM is directly responsible for this perturbation without further investigation. If both the conformational changes in helix E and the conserved Asn are relevant to the functions of BMMs, an additional challenge will be to understand when these conformational changes take place during the catalytic cycle.
Structural Changes in Helix F
The electron density for helix F residues 225–231 in the SeMet PHH α-subunit where PHM is bound at higher occupancy is different than that in the other α-subunits (Figure S8), suggesting that PHM may induce a slight reconfiguration, or disordering, of this helix when it is bound to the hydroxylase. The best interpretation of the electron density in this region, which may be a combination of the PHM-bound and unbound forms, is presented in Figure 6. In most α-subunits, F227 and Q230 comprise part of the canyon surface and typically point toward solvent. When the regulatory protein binds, PHM side chains Q10 and I71 occupy the former positions of Q230 and F227, respectively (Figure 6B), and presumably force helix F to reconfigure. The occupancy of PHM in the native structure is insufficient for observation of these differences or identification of the positions of the specific PHM side chains. The major consequences of the helix F rearrangement are the movement of Q230 toward the diiron center, such that it lies above Fe2-coordinating E233, and a shift in the position of the F207 side chain, such that it is closer to the terminal water ligand on Fe1. Residue Q230 is highly conserved among all of the different BMM subfamilies except that of sMMO, where it is a Glu in MMOH and butane monooxygenase (BMOH). It is unknown how mutations of this Gln would affect hydroxylation, electron transfer, or proton transfer chemistry. Residue F207 is conserved among the aromatic hydroxylating BMMs but is a Leu or Ile in the other BMM systems. The conformational changes in helix F do not alter any of the second-sphere hydrogen bonding networks, but they do decrease slightly the solvent accessibility of the diiron center.
Figure 6.

Structural changes in α-subunit helix F in the SeMet structure. (A) Comparison between PHM-altered (orange) and unaltered (gray) helix F in stereo. (B) PHH (gray) and PHM (red) binding interface at α-subunit helices E and F.
Possible Consequences of Regulatory Protein Binding on Transfer of Electrons to the Hydroxylase
Behind the diiron center is an extensive hydrogen bonding network extending from the active site to the surface of the hydroxylase canyon at α-subunit helix A (Figure 3B). Most of the residues contributing to this network are strictly conserved among the different BMM families (3), suggesting that they are critical to the function and folding of the protein. Several other diiron proteins, including bacterioferritin and rubrerythrin, have similar hydrogen bonding patterns that extend 10–14 Å to a redox active cofactor, such as a porphyrin or mononuclear iron–sulfur center (61, 62). In the R2 subunit of RNR, a tryptophan residue in an analogous hydrogen bonding network behind the diiron center facilitates generation of the catalytically important Fe(III)Fe(IV) X intermediate and tyrosyl radical (63). More recently, this pathway in Δ9-desaturase has been implicated in electron transfer (64). On the basis of these observations, we hypothesized that this hydrogen bonding network in BMMs may represent the electron transfer pathway from the reductase to the diiron center (5). Investigations of the electron transfer reactions between MMOH and its reductase, MMOR, indicate the [2Fe-2S] cluster of the latter and the MMOH diiron center may be 11–14 Å apart when this protein complex forms (65). Using this distance constraint, an analysis of the PHH surface structure places the reductase binding site very close to that of the PHM component. If our assumptions about the relative location of reductase binding site are correct, the PH regulatory and regulatory proteins must share a similar binding site on both faces of the hydroxylase surface.
This finding has important implications for how the regulatory protein influences the properties of the hydroxylase diiron center. One attractive scenario is that in which regulatory protein binding prevents the reductase from binding to the hydroxylase and donating extra electrons to the dioxygen-activated diiron center, quenching the peroxo or Q intermediates before they can achieve their hydroxylation chemistry. Such uncoupling, or premature reduction of the oxygenated metal cluster, would not only consume the reactive diiron species but also wastefully deplete the NADH supply of the cell. After completion of the hydroxylation reaction, the regulatory protein may temporarily dissociate from the hydroxylase so that the reductase can bind and reinitiate the catalytic cycle. This scenario would explain why, in the sMMO system, binding of MMOB to MMOH shifts the redox potentials to disfavor reduction of the diiron center, whereas reductase binding shifts the potentials so that reduction is favored (65-68). It is also possible that BMMs function via half-sites reactivity. During the catalytic cycle, the regulatory protein may bind on one face of the hydroxylase to facilitate catalysis while the reductase binds temporarily to the other side to reinitiate the cycle. Such a model may explain why excess regulatory protein often inhibits BMM hydroxylation chemistry, whereas substoichiometric amounts lead to uncoupling (18, 19). Pinning down the exact function of the regulatory protein has been a difficult task. Often, it has been termed a “coupling protein” because it couples hydroxylation with electron transfer and an “effector protein” because it effects substrate hydroxylation. The PHH–PHM structure demonstrates how it may carry out both functions.
Access of Substrate to the Diiron Center
The α-subunit scaffold is important for controlling the entrance and egress of small molecules to and from the diiron center. The pathway for substrate entrance, such as the hydrophobic cavities or channels that we previously identified in MMOH and ToMOH, may play an important role in determining the substrate specificity and reactivity of these enzymes (8, 9, 33, 69). Surface calculations of the PHH α-subunits using PyMOL (70) reveal the presence of three cavities leading from the active site pocket (cavity 1) to the protein surface near the γ-subunit (cavity 3) (Figure 7A). Cavities 1 and 3 have direct access to the exterior environment of the protein, whereas access to cavity 2 is gated by Leu-107 and Leu-272 at one end near the active site pocket and Leu-399 and Val-455 on the other near cavity 3. This putative route for the access of small molecules to the active site involves mostly hydrophobic residues. It traverses the α-subunit in a manner that is analogous to the pathway defined by the cavities in MMOH and ToMOH but different from the 40 Å channel in ToMOH (9, 33, 57, 69). Although the PHH cavities are large enough to accommodate aromatic substrates, in the absence of structures containing bound product or substrate, it is difficult to know whether these cavities are relevant to the function of PHH.
Figure 7.
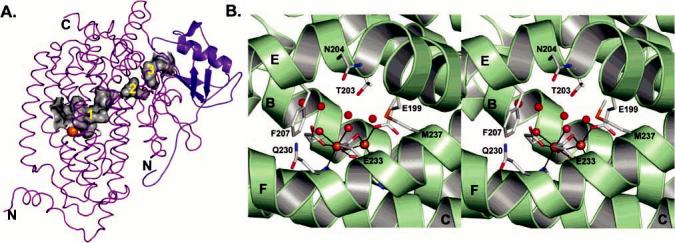
Cavities and pores in the PHH α-subunit. (A) α-subunit cavities (gray) that extend from the diiron center to the γ-subunit (blue). (B) Stereoview of the conserved residues and water molecules (red spheres) contributing to the pore through the α-subunit four-helix bundle.
Surface calculations of the PHH α-subunits without bound PHM reveal a large pore between helices E and F that offers the most direct route for access of small molecules to the active site pocket. This pore is ∼6 Å in diameter, lies above the diiron center, and is lined with the side chains of Thr-203, Asn-204, Phe-207, Glu-233, and Met-237 (Figure 7B), all of which are conserved among the different PH enzymes. The pore diameter may be large enough to accommodate phenol and alternative substrates like xylene, dimethylcatechol, and naphthalene, which have diameters between 5.0 and 7.0 Å. Several water molecules occupy the pore and delineate how a substrate may move through the four-helix bundle in the absence of PHM. The 6-bromohexanol-bound form of MMOH, which has a helix E configuration similar to that in PHH, does not have such an obvious mode of entry through helices E and F. The absence of an analogous pore in the MMOH structure may reflect the fact that the peptide backbones of helices E and F near the diiron center are closer together by approximately 2.0 Å, measuring from the C R atoms of Asn-214 and Gln-240 (Figure 5E). In some, but not all, crystal structures of reduced MMOH in which helix E has its typical configuration, a pore with a narrow 2.0 Å diameter forms as a result of Asn-214 (Asn-204 in PHH) shifting to point inward, toward the diiron center (33). This pore may become large enough to accommodate small substrates like methane, dioxygen, and protons in the form of H3O+.
Surface calculations of the PHH α-subunits with bound PHM reveal that direct access to the diiron center through helices E and F is restricted in this conformation of the complex (Figure 8). Binding of the regulatory protein also induces F207 and Q230 to reposition as described above, further limiting access to the diiron center. Previously, we suggested a similar barrier-like role for MMOB in the sMMO system because it significantly slowed the rate of diffusion of iron into and out of the active site during metal reconstitution studies (32). Investigations of Methylosinus trichosporium OB3b MMOB mutants in which residues 108–111 were changed to Ala revealed variable substrate access to the diiron center. On the basis of these results, MMOB was proposed to generate a size-selective pore on the hydroxylase surface that would serve as a substrate-specific “sieve” (44, 71, 72). We find no evidence for a PHM-generated substrate-specific pore in the PHH–PHM complex or in models of the MMOH–MMOB structure (not shown), suggesting that the MMOB mutations, which correspond to residues 70–73 in PHM (Figures 6B and S5), may simply decrease the diffusion barrier at the MMOH–MMOB interface to modify the access of the substrate and product to and from the diiron center. Kinetic and computational studies of the reactions of intermediate Q with a variety of substrates have revealed two mechanistic limits, depending upon whether access to the diiron center is rate-determining (73). These results suggest how the regulatory protein might be responsible for controlling the kinetics of these processes in the sMMO system.
Figure 8.
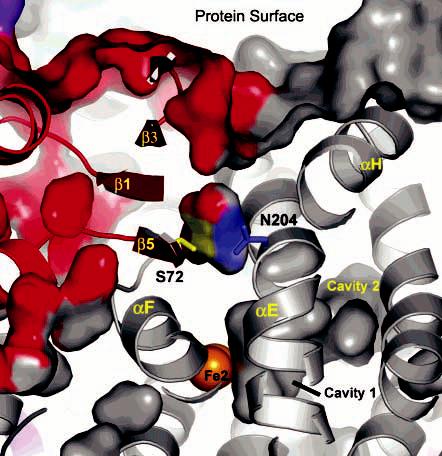
Access of the substrate to the diiron center at the PHH–PHM interface. PHH (gray) and PHM (red) surfaces and cavities are depicted. Interactions between conserved N204 (blue) and S72 (yellow) and how they may gate access to the metal center are also shown.
The PHM equivalent of S110 in M. trichosporium MMOB is S72. The side chain of this residue forms a hydrogen bond to conserved Asn-204 in both the native and SeMet crystal structures (Figures 6B and 8) and is highly conserved among the different BMM regulatory proteins. The link between these two residues has not yet been fully explored. Given that the PH enzymes act on substrates larger than MMOH and that only a deep crevice is observed on the surface of the PHH–PHM complex (Figure 8), the aromatic substrates may enter the PH hydroxylase by an alternative route, possibly via cavity 3. If a pore through the E and F helices of the α-subunit forms at specific stages of the catalytic cycle, such as when the diiron center is reduced and Asn-204 undergoes a rotameric shift, it may allow passage of O2, H2O, or H3O+ to or from the active site. Collectively, these findings reveal how the regulatory protein may control the access of small molecules and ions to the diiron center and possibly offers some type of protection of the dioxygen-activated metal center as it traverses the reaction cycle.
Active Site Pocket and Substrate Specificity
The arrangement of side chains in the active site pockets of BMMs is crucial for determining substrate specificity. Phenol hydroxylases, for the most part, are ortho-hydroxylating enzymes, whereas toluene monooxygenases hydroxylate in the para position (58, 74). PHH and ToMOH from P. stutzeri OX1, however, have relaxed regiospecificities and typically afford 70% o-cresol and 45% p-cresol, respectively (20, 75, 76). For alternative substrates, PH and ToMO perform highly regiospecific reactions. For example, PH and ToMO yield 96% 3-methylcatechol and 95% 4-methylcatechol, respectively, when m-cresol is used as the substrate (76, 77). The hydrophobic residues that line the active site pockets of PHH and ToMOH are mostly conserved among their specific subfamilies (Table 3) (3). A superimposition of the PHH and ToMOH active sites indicates that the side chains occupy nearly identical positions except for those residues on helix E because of its alternative conformation (Figure 9). Single-ring aromatic substrates can be most effectively docked manually into the active site by orienting their ring plane perpendicular to the Fe–Fe vector. A similar orientation for the aromatic ring has been suggested previously for ToMOH (77). On the basis of DFT calculations and structural investigations of MMOH, substrate hydroxylation presumably occurs at a bridging position between the two iron atoms (78). By docking the oxygen atom of a pentamethylphenol at such a position, we readily see that ortho hydroxylation is favored if a ring substituent points above or below the plane of the iron atoms and bridging oxygen (carbons 2 and 6 in Figure 9), whereas para and meta hydroxylation are favored only if the ring substituent is positioned at the back of the active site pocket. Para hydroxylation may be disfavored in PH enzymes because of steric interaction between Leu-107 and the substituent on the C4 atom (Figure 9). For TMOs, the residue analogous to Leu-107 is typically a glycine or a glutamate (Glu-103) in ToMOH that points away from the active site pocket. A smaller residue at this position would provide enough space to orient a substituent on C4 toward the back of the active site pocket to favor hydroxylation of the para carbon atom. Mutants of both T4MOH and ToMOH in which this residue was changed to leucine afforded 55–60% ortho hydroxylation (58, 77), suggesting that the space occupied by this residue is important for steering substrates. Conversely, mutagenesis studies on a related phenol hydroxylase family member demonstrated that changing the residue analogous to Leu-107 to an alanine produced an enzyme that yielded 50% o-cresol, compared to 98% for the wild-type form (74). Moreover, this variant was capable of hydroxylating naphthalene, anthracene, fluorene, and phenanthrene at increased rates. Thus, increasing the depth of the active site pocket promotes hydroxylation of larger substrates.
Table 3.
Residues Comprising the BMM Active Site Substrate Binding Pocketa
| PHH | I104 | L107 | A111 | Q145 | F179 | R183b | I194 | F198 | T203 | L206 | F207 |
| CF600 | V105 | L108 | A112 | Q146 | F180 | R184b | V195 | F199 | T204 | L207 | F208 |
| TOMH | V106 | L109 | A113 | E147 | F181 | A185b | V196 | F200 | T205 | L208 | F209 |
| ToMOH | I100 | E103 | A107 | Q141 | F176 | M180 | L192 | F196 | T201 | Q204 | F205 |
| T4MOH | I100 | G103 | A107 | Q141 | F176 | I180 | L192 | F196 | T201 | Q204 | F205 |
| AMOH | L91 | A94 | A98 | E133 | G167 | F172 | L184 | A188 | T193 | L196 | L197 |
| MMOH | L110 | G113 | A117 | C151 | F188 | F192 | L204 | G208 | T213 | L216 | I217 |
Abbreviations: PHH, Pseudomonas sp. OX1 PHH; CF600, Pseudomonas sp. strain CF600 PHH; TOMH, Burkholderia cepacia G4 toluene o-monooxygenase hydroxylase; ToMOH, Pseudomonas sp. OX1 ToMOH; T4MOH, Pseudomonas medonica KR1 T4MOH; AMOH, Rhodococcus corallinus B-276 alkene monooxygenase hydroxylase; MMOH, Methylococcus capsulatus (Bath).
A side chain that does not contribute to the active site pocket yet has a homologous BMM family member with a side chain that does.
Figure 9.
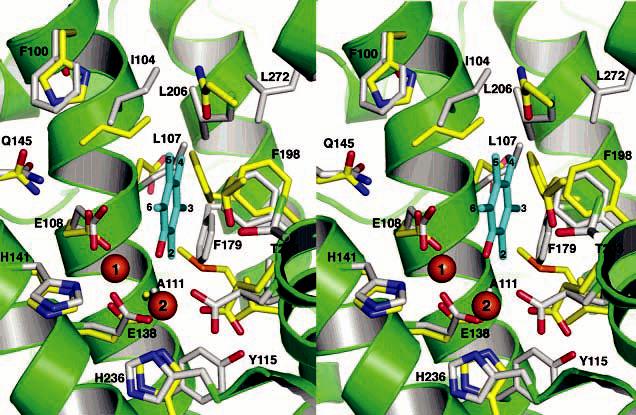
Stereoview of the superimposed PHH and ToMOH substrate binding pockets. The PHH and ToMOH carbon atoms are colored gray and yellow, respectively, and helices of PHH are depicted in green. Pentamethylphenol (cyan) is docked manually into the active site pocket. The aromatic carbon atoms of the docked aromatics as well as the PHH residues contributing to the active site are labeled. The identity of the ToMOH residues can be obtained from Table 3.
In essence, the PHH active site pocket is shallower than that of ToMOH. Because the regulatory proteins in these different BMM systems alter the product distributions by influencing the structure of the active site pocket (17, 18), possibly by changing the configuration of helix E, it is difficult to account fully for all of the residues and structural features that are responsible for orienting substrates in the absence of a crystal structure of the hydroxylase-regulatory protein complex at higher occupancy and in different oxidation states. Nevertheless, our structures of PHH and ToMOH, in addition to revealing the two different configurations of helix E, provide an improved understanding of the active site pockets and an initial structural basis from which one can proceed to rationally engineer these enzymes for industrial and bioremediation applications.
CONCLUSIONS AND PROSPECTS
The fundamental properties that are responsible for controlling the substrate reactivity of the BMM diiron centers lie within the protein scaffold of the hydroxylase subunit. The structure of the PHH–PHM complex represents a key step toward unraveling the principles that underlie the chemistry performed by the carboxylate-bridged diiron centers of these proteins. The structure indicates that PHM functions to block the entrance of the solvent and substrate to the diiron active site through the four-helix bundle, alter the configuration of helices E and F in a manner that could enhance the overall activity of the metal cluster, shape the active site cavity to control product distributions, and most likely affect electron transfer from the reductase by partially or wholly covering its binding site on the hydroxylase canyon walls. Sharing of the hydroxylase surface by the reductase and regulatory protein components would imply that a critical role for the regulatory proteins in all BMMs is to prevent reduction of the dioxygen-activated diiron center at inappropriate times during the catalytic cycle. The recombinant PH enzyme system is ideal for testing the various structural control features, such as hydrogen bonding networks, cavity gates, active site pores, and helix rearrangements, that we suggest may be functionally significant for substrate entrance, electron transfer, and component interactions. Moreover, the PH system will allow future testing of mechanistic theories related to the different reactivities of BMMs.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Viviana Izzo (Universitá di Napoli Federico II) for providing the plasmids and protein-expressing strains as well as Eugenio Notomista (Universitá di Napoli Federico II) and Leslie Muray (Massachusetts Institute of Technology) for useful discussions about substrate regiospecificity and electron transfer.
Footnotes
This research was supported by National Institute of General Medical Sciences Grant GM32134 (S.J.L.) and the Italian Ministry of University and Research PRIN/2004 (A.D.). Portions of this study were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
The coordinates and structure factors for the PHH–PHM complex have been deposited in the Protein Data Bank for the native and SeMet enzyme as entries 2INP and 2INN, respectively.
Abbreviations: ACP, acyl carrier protein; AMO, alkene monooxygenase; APS, Advanced Photon Source; BMM, bacterial multicomponent monooxygenase; C2,3O, catechol 2,3-dioxygenase; PH, phenol hydroxylase; PHH, phenol hydroxylase hydroxylase component; PHM, phenol hydroxylase regulatory protein; PHP, phenol hydroxylase reductase; MMOB, methane monooxygenase regulatory protein; MMOH, methane monooxygenase hydroxylase; MMOR, methane monooxygenase reductase; rmsd, root-mean-square deviation; sMMO, soluble methane monooxygenase; SsoMO, hyperthermophilic toluene monooxygenase; SSRL, Stanford Synchrotron Radiation Laboratory; T4MO, toluene 4-monooxygenase; T4MOD, toluene 4-monooxygenase regulatory protein; THFMO, tetrahydrofuran monooxygenase; TMO, toluene monooxygenase; TOM, toluene o-monooxygenase; ToMO, toluene/o-xylene monooxygenase; ToMOD, toluene/o-xylene monooxygenase regulatory protein; ToMOH, toluene/o-xylene monooxygenase hydroxylase.
V. Izzo, C. Tinberg, R. García-Serres, S. Naik, B. H. Huynh, and S. J. Lippard, unpublished results.
L. J. Murray, M. S. McCormick, R. García-Serres, S. Naik, B. H. Huynh, and S. J. Lippard, unpublished results.
V. Izzo, R. Davydov, M. H. Sazinsky, S. J. Lippard, and B. M. Hoffman, unpublished results.
E. Cadieux, L. J. Murray, M. S. McCormick, M. H. Sazinsky, and S. J. Lippard, unpublished results.
SUPPORTING INFORMATION AVAILABLE
BMM hydroxylase and regulatory protein structures and their electrostatic surfaces (Figures S1 and S2), packing of the β-subunit N-terminus and a γ-subunit from an adjacent molecule in the PHH canyon (Figure S3), comparison of known regulatory protein structures (Figure S4), sequence alignments of the different regulatory proteins (Figure S5), models of the PHH diiron center in electron density maps (Figure S6), comparison of the hydroxylase zinc-binding sequences (Figure S7), and electron density maps around helix F (Figure S8). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: A tale of two irons and three proteins. Angew. Chem., Int. Ed. 2001;40:2783–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. (and references cited therein) [DOI] [PubMed] [Google Scholar]
- 2.Notomista E, Lahm A, Di Donato A, Tramontano A. Evolution of bacterial and archaeal multicomponent monooxygenases. J. Mol. Evol. 2003;56:435–445. doi: 10.1007/s00239-002-2414-1. [DOI] [PubMed] [Google Scholar]
- 3.Leahy JG, Batchelor PJ, Morcomb SM. Evolution of the soluble diiron monooxygenases. FEMS Microbiol. Rev. 2003;27:449–479. doi: 10.1016/S0168-6445(03)00023-8. [DOI] [PubMed] [Google Scholar]
- 4.Kurtz DM., Jr. Structural similarity and functional diversity in diiron-oxo proteins. J. Biol. Inorg. Chem. 1997;2:159–167. [Google Scholar]
- 5.Sazinsky MH, Lippard SJ. Correlating structure with function in bacterial multicomponent monooxygenases and related diiron proteins. Acc. Chem. Res. 2006;39:558–566. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]
- 6.Fox BG, Borneman JG, Wackett LP, Lipscomb JD. Haloalkene oxidation by the soluble methane monooxygenase from Methylosinus trichosporium OB3b: Mechanistic and environmental implications. Biochemistry. 1990;29:6419–6427. doi: 10.1021/bi00479a013. [DOI] [PubMed] [Google Scholar]
- 7.Hanson RS, Hanson TE. Methanotrophic bacteria. Microbiol. Rev. 1996;60:439–471. doi: 10.1128/mr.60.2.439-471.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 9.Sazinsky MH, Bard J, Di Donato A, Lippard SJ. Structure of the toluene/o-xylene monooxygenase hydroxylase from Pseudomonas stutzeri OX1: Substrate channeling and active site tuning of multicomponent monooxygenases. J. Biol. Chem. 2004;279:30600–30610. doi: 10.1074/jbc.M400710200. [DOI] [PubMed] [Google Scholar]
- 10.Pikus JD, Studts JM, Achim C, Kauffmann KE, Münck E, Steffan RJ, McClay K, Fox BG. Recombinant toluene-4-monooxygenase: Catalytic and Mössbauer studies of the purified diiron and Rieske components of a four-protein complex. Biochemistry. 1996;35:9106–9119. doi: 10.1021/bi960456m. [DOI] [PubMed] [Google Scholar]
- 11.Gallagher SC, Cammack R, Dalton H. Alkene monooxygenase from Nocardia corallina B-276 is a member of the class of dinuclear iron proteins capable of stereospecific epoxygenation reactions. Eur. J. Biochem. 1997;247:635–641. doi: 10.1111/j.1432-1033.1997.00635.x. [DOI] [PubMed] [Google Scholar]
- 12.Cadieux E, Vrajmasu V, Achim C, Powlowski J, Münck E. Biochemical, Mössbauer, and EPR studies of the diiron cluster of phenol hydroxylase from Pseudomonas sp. strain CF 600. Biochemistry. 2002;41:10680–10691. doi: 10.1021/bi025901u. [DOI] [PubMed] [Google Scholar]
- 13.Valentine AM, Stahl SS, Lippard SJ. Mechanistic studies of the reaction of reduced methane monooxygenase hydroxylase with dioxygen and substrates. J. Am. Chem. Soc. 1999;121:3876–3887. [Google Scholar]
- 14.Beauvais LG, Lippard SJ. Reactions of the peroxo intermediate of soluble methane monooxygenase hydroxylase with ethers. J. Am. Chem. Soc. 2005;127:7370–7378. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- 15.Murray LJ, García-Serres R, Naik S, Huynh BH, Lippard SJ. Dioxygen activation at non-heme diiron centers: Characterization of intermediates in a mutant form of toluene/o-xylene monooxygenase hydroxylase. J. Am. Chem. Soc. 2006;128:7458–7459. doi: 10.1021/ja062762l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green J, Dalton H. Substrate specificity of soluble methane monooxygenase: Mechanistic implications. J. Biol. Chem. 1989;264:17698–17703. [PubMed] [Google Scholar]
- 17.Froland WA, Andersson KK, Lee SK, Liu Y, Lipscomb JD. Methane monooxygenase component B and reductase alter the regioselectivity of the hydroxylase component-catalyzed reactions. J. Biol. Chem. 1992;267:17588–17597. [PubMed] [Google Scholar]
- 18.Mitchell KH, Studts JM, Fox BG. Combined participation of hydroxylase active residues and effector protein binding in a para to ortho modulation of toluene 4-monooxygenase regiospecificity. Biochemistry. 2002;41:3176–3188. doi: 10.1021/bi012036p. [DOI] [PubMed] [Google Scholar]
- 19.Gassner GT, Lippard SJ. Component interactions in the soluble methane monooxygenase system from Methylococcus capsulatus (Bath) Biochemistry. 1999;38:12768–12785. doi: 10.1021/bi990841m. [DOI] [PubMed] [Google Scholar]
- 20.Cafaro V, Izzo V, Scognamiglio R, Notomista E, Capasso P, Casbarra A, Pucci P, Di Donato A. Phenol hydroxylase and toluene/o-xylene monooxygenase from Pseudomonas stutzeri OX1: Interplay between two enzymes. Appl. Environ. Microbiol. 2004;70:2211–2219. doi: 10.1128/AEM.70.4.2211-2219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LeMaster DM, Richards FM. 1H–15N heteronuclear NMR studies of Escherichia coli thioredoxin in samples isotopically labeled by residue type. Biochemistry. 1985;24:7263–7268. doi: 10.1021/bi00346a036. [DOI] [PubMed] [Google Scholar]
- 22.Gibbs CR. Characterization and application of ferrozine iron reagent as a ferrous iron indicator. Anal. Chem. 1976;48:1197–1201. [Google Scholar]
- 23.Otwinowski Z, Minor W. HKL Suite of programs. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 24.Brünger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 25.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheldrick GM, Uson I. Advances in direct methods for protein crystallography. Curr. Opin. Struct. Biol. 1999;9:643–648. doi: 10.1016/s0959-440x(99)00020-2. [DOI] [PubMed] [Google Scholar]
- 27.Weeks CM, Miller R. The design and implementation of SnB v2.0. J. Appl. Crystallogr. 1999;32:120–124. [Google Scholar]
- 28.Collaborative Computational Project Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. 1995;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 29.McRee DE. XtalView/Xfit: A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 30.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Moss DS, Thorton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 32.Sazinsky MH, Merkx M, Cadieux E, Tang S, Lippard SJ. Preparation and X-ray structures of metal-free, dicobalt and dimanganese forms of soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath) Biochemistry. 2004;43:16263–16276. doi: 10.1021/bi048140z. [DOI] [PubMed] [Google Scholar]
- 33.Sazinsky MH, Lippard SJ. Product bound structures of the soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): Protein motion in the α-subunit. J. Am. Chem. Soc. 2005;127:5814–5825. doi: 10.1021/ja044099b. [DOI] [PubMed] [Google Scholar]
- 34.Walters KJ, Gassner GT, Lippard SJ, Wagner G. Structure of the soluble methane monooxygenase regulatory protein B. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7877–7882. doi: 10.1073/pnas.96.14.7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang S-L, Wallar BJ, Lipscomb JD, Mayo KH. Solution structure of component B from methane monooxygenase derived through heteronuclear NMR and molecular modeling. Biochemistry. 1999;38:5799–5812. doi: 10.1021/bi982992f. [DOI] [PubMed] [Google Scholar]
- 36.Hemmi H, Studts JM, Chae YK, Song J, Markley JL, Fox BG. Solution structure of the toluene 4–monooxygenase effector protein (T4moD) Biochemistry. 2001;40:3512–3524. doi: 10.1021/bi0013703. [DOI] [PubMed] [Google Scholar]
- 37.Lountos GT, Mitchell KH, Studts JM, Fox BG, Orville AM. Crystal structures and functional studies of T4moD, the toluene 4–monooxygenase catalytic effector protein. Biochemistry. 2005;44:7131–7142. doi: 10.1021/bi047459g. [DOI] [PubMed] [Google Scholar]
- 38.Brazeau BJ, Wallar BJ, Lipscomb JD. Effector proteins from P450cam and methane monooxygenase: Lessons in tuning nature's powerful reagents. Biochem. Biophys. Res. Commun. 2003;312:143–148. doi: 10.1016/j.bbrc.2003.09.242. [DOI] [PubMed] [Google Scholar]
- 39.Chang S, Wallar BJ, Lipcomb JD, Mayo JD. Residues in Methylosinus trichosporium OB3b methane monooxyenase component B involved in molecular interactions with reduced- and oxidized-hydoxylase component: A role for the N-terminus. Biochemistry. 2001;40:9539–9551. doi: 10.1021/bi0103462. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Wallar BJ, Popescu CV, Renner DB, Thomas DD, Lipscomb JD. Methane monooxygenase hydroxylase and B component interactions. Biochemistry. 2006;45:2913–2926. doi: 10.1021/bi052256t. [DOI] [PubMed] [Google Scholar]
- 41.MacArthur R, Sazinsky MH, Kühne H, Whittington DA, Lippard SJ, Brudvig GW. Component B binding to the soluble methane monooxygenase hydroxylase by saturation recovery-EPR spectroscopy of spin-labeled MMOB. J. Am. Chem. Soc. 2002;124:13392–13393. doi: 10.1021/ja0279904. [DOI] [PubMed] [Google Scholar]
- 42.Lloyd JS, Bhambra A, Murrell JC, Dalton H. Inactivation of the regulatory protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath) by proteolysis can be overcome by a Gly to Gln modification. Eur. J. Biochem. 1997;248:72–79. doi: 10.1111/j.1432-1033.1997.t01-1-00072.x. [DOI] [PubMed] [Google Scholar]
- 43.Brandstetter H, Whittington DA, Lippard SJ, Frederick CA. Mutational and structural analyses of the regulatory protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath) Chem. Biol. 1999;6:441–449. doi: 10.1016/s1074-5521(99)80062-3. [DOI] [PubMed] [Google Scholar]
- 44.Wallar BJ, Lipscomb JD. Methane monooxygenase component B mutants alter the kinetic steps throughout the catalytic cycle. Biochemistry. 2001;40:2220–2233. doi: 10.1021/bi002298b. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Lipscomb JD. Role of the C-terminal region of the B component of Methylosinus trichosporium OB3b methane monooxygenase in the regulation of oxygen activation. Biochemistry. 2006;45:1459–1469. doi: 10.1021/bi051721j. [DOI] [PubMed] [Google Scholar]
- 46.Elango N, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Crystal structure of the hydroxylase component of methane monooxygenase from Methylosinus trichosporium OB3b. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenzweig AC, Lippard SJ. Determining the structure of a hydroxylase enzyme that catalyzes the conversion of methane to methanol in methanotrophic bacteria. Acc. Chem. Res. 1994;27:229–236. [Google Scholar]
- 48.Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Geometry of the soluble methane monooxygenase catalytic diiron center in two oxidation states. Chem. Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- 49.Whittington DA, Lippard SJ. Crystal structures of the soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath) demonstrating geometrical variability at the dinuclear iron active site. J. Am. Chem. Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- 50.Whittington DA, Sazinsky MH, Lippard SJ. X-ray crystal structure of alcohol products bound at the active site of soluble methane monooxygenase hydroxylase. J. Am. Chem. Soc. 2001;123:1794–1795. doi: 10.1021/ja0031725. [DOI] [PubMed] [Google Scholar]
- 51.Cadieux E, Powlowski J. Characterization of active and inactive forms of the phenol hydroxylase stimulatory protein DmpM. Biochemistry. 1999;38:10714–10722. doi: 10.1021/bi990835q. [DOI] [PubMed] [Google Scholar]
- 52.Krutz DM., Jr. Oxo- and hydroxo-bridged diiron complexes: A chemical perspective on a biological unit. Chem. Rev. 1990;90:585–606. [Google Scholar]
- 53.Lindqvist Y, Huang W, Schneider G, Shanklin J. Crystal structure of Δ9 stearoyl-acyl carrier protein desaturase from castor seed and its relationship to other di-iron proteins. EMBO J. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 54.Logan DT, Su X-D, Åberg A, Regnström K, Hajdu J, Eklund H, Nordlund P. Crystal structure of reduced protein R2 of ribonucleotide reductase: The structural basis for oxygen activation at a dinuclear iron site. Structure. 1996;4:1053–1064. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- 55.Eriksson M, Jordan A, Eklund H. Structure of Salmonella typhimurium nrdF ribonucleotide reductase in its oxidized and reduced forms. Biochemistry. 1998;37:13359–13369. doi: 10.1021/bi981380s. [DOI] [PubMed] [Google Scholar]
- 56.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Nitrogenase MoFe-protein at 1.16 Å resolution: A central ligand in the FeMo-cofactor. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 57.McCormick MS, Sazinsky MH, Condon KL, Lippard SJ. X-ray crystal structures of manganese(II) reconstituted and native toluene/o-xylene monooxygenase hydroxylase reveal rotomer shifts in conserved residues and an enhanced view of the protein interior. J. Am. Chem. Soc. 2006;128:15108–15110. doi: 10.1021/ja064837r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pikus JD, Studts JM, McClay K, Steffan RJ, Fox BG. Changes in the regiospecificity of aromatic hydroxylation produced by active site engineering in the diiron enzyme toluene 4-monooxygenase. Biochemistry. 1997;36:9283–9289. doi: 10.1021/bi971049t. [DOI] [PubMed] [Google Scholar]
- 59.Newman LM, Wackett LP. Purification and characterization of toluene 2-monooxygenase from Burkholderia cepacia G4. Biochemistry. 1995;34:14066–14076. doi: 10.1021/bi00043a012. [DOI] [PubMed] [Google Scholar]
- 60.Pikus JD, Mitchell KH, Studts JM, McClay K, Steffan RJ, Fox BG. Threonine 201 in the diiron enzyme toluene 4-monooxygenase is not required for catalysis. Biochemistry. 2000;39:791–799. doi: 10.1021/bi992187g. [DOI] [PubMed] [Google Scholar]
- 61.Frolow F, Kalb AJ, Yariv J. Structure of a unique two fold symmetric heme-binding site. Nat. Struct. Biol. 1994;1:453–460. doi: 10.1038/nsb0794-453. [DOI] [PubMed] [Google Scholar]
- 62.deMare F, Kurtz DM, Jr., Nordlund P. The structure of Desulfovbrio vulgaris rubrerythrin reveals a unique combination of rubredoxin-like FeS4 and ferritin-like diiron domains. Nat. Struct. Biol. 1996;3:539–546. doi: 10.1038/nsb0696-539. [DOI] [PubMed] [Google Scholar]
- 63.Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical initiation in the Class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem. Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 64.Sobrado P, Lyle KS, Kaul SP, Turco MM, Arabshahi I, Marwah A, Fox BG. Identification of the binding region of the [2Fe-2S] ferredoxin in stearoyl-acyl carrier protein desaturase: Insight into the catalytic complex and mechanism of action. Biochemistry. 2006;45:4848–4858. doi: 10.1021/bi0600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blazyk JL, Gassner GT, Lippard SJ. Intermolecular electron-transfer reactions in soluble methane monooxygenase: A role for hysteresis in protein function. J. Am. Chem. Soc. 2005;127:17364–17376. doi: 10.1021/ja0554054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu KE, Lippard SJ. Redox properties of the hydroxylase component of methane monooxygenase from Methylococcus capsulatus (Bath): Effects of protein B, reductase, and substrate. J. Biol. Chem. 1991;266:12836–12839. [PubMed] [Google Scholar]
- 67.Paulsen KE, Liu Y, Fox BG, Lipscomb JD, Münck E, Stankovich MT. Oxidation-reduction potentials of the methane monooxygenase hydroxylase component from Methylosinus trichosporium OB3b. Biochemistry. 1994;33:713–722. doi: 10.1021/bi00169a013. [DOI] [PubMed] [Google Scholar]
- 68.Kazlauskaite J, Hill HAO, Wilkins PC, Dalton H. Direct electrochemistry of the hydroxylase of soluble methane monooxygenase from Methylococcus capsulatus (Bath) Eur. J. Biochem. 1996;241:552–556. doi: 10.1111/j.1432-1033.1996.00552.x. [DOI] [PubMed] [Google Scholar]
- 69.Whittington DA, Rosenzweig AC, Frederick CA, Lippard SJ. Xenon and halogenated alkanes track putative substrate binding cavities in the soluble methane monooxygenase hydroxylase. Biochemistry. 2001;40:3476–3482. doi: 10.1021/bi0022487. [DOI] [PubMed] [Google Scholar]
- 70.DeLano WL. The PyMol Molecular Graphics System. DeLano Scientific LLC; San Carlos, CA: 2002. [Google Scholar]
- 71.Zheng H, Lipcomb JD. Regulation of methane monooxygenase catalysis based on size exclusion and quantum tunneling. Biochemistry. 2006;45:1685–1692. doi: 10.1021/bi051605g. [DOI] [PubMed] [Google Scholar]
- 72.Brazeau BJ, Lipscomb JD. Key amino acid residues in the regulation of soluble methane monooxygenase catalysis by component B. Biochemistry. 2003;42:5618–5631. doi: 10.1021/bi027429i. [DOI] [PubMed] [Google Scholar]
- 73.Lippard SJ. Hydroxylation of C–H bonds at carboxylate-bridged diiron centres. Philos. Trans. R. Soc. London, Ser. A. 2005;363:861–877. doi: 10.1098/rsta.2004.1532. [DOI] [PubMed] [Google Scholar]
- 74.Canada KA, Iwashita A, Shim H, Wood TK. Directed evolution of toluene ortho-monooxygenase for enhanced 1-naphthol synthesis and chlorinated ethene degradation. J. Bacteriol. 2002;184:344–349. doi: 10.1128/JB.184.2.344-349.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cafaro V, Scognamiglio R, Viggiani A, Izzo V, Passaro I, Notomista E, Dal Piaz F, Amoresano A, Casbarra A, Pucci P, Di Donato A. Expression and purification of the recombinant subunits of toluene/o-xylene monooxygenase and reconstitution of the active complex. Eur. J. Biochem. 2002;269:5689–5699. doi: 10.1046/j.1432-1033.2002.03281.x. [DOI] [PubMed] [Google Scholar]
- 76.Cafaro V, Notomista E, Capasso P, Di Donato A. Regiospecificity of two multicomponent monooxygenases from Pseudomonas stutzeri OX1: Molecular basis for catabolic adaption of this microorganism to methylated aromatic compounds. Appl. Environ. Microbiol. 2005;71:4736–4743. doi: 10.1128/AEM.71.8.4736-4743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cafaro V, Notomista E, Capasso P, Di Donato A. Mutation of glutamic acid 103 of toluene o-xylene monooxygenase as a means to control the catabolic efficiency of a recombiant upper pathway for degradation of methylated aromatic compounds. Appl. Environ. Microbiol. 2005;71:4744–4750. doi: 10.1128/AEM.71.8.4744-4750.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baik M-H, Newcomb M, Friesner RA, Lippard SJ. Mechanistic studies on the hydroxylation of methane by methane monooxygenase. Chem. Rev. 2003;103:2385–2420. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.