

Abstract
At excitatory synapses onto hippocampal CA1 pyramidal cells, activation of cyclic AMP-dependent protein kinase and subsequent down-regulation of protein phosphatases has a crucial role in the induction of long-term potentiation by low-frequency patterns of synaptic stimulation. Because the second messenger cyclic guanosine 3′,5′monophosphate can regulate the activity of different forms of the cyclic AMP degrading enzyme phosphodiesterase, we examined whether increases in cyclic guanosine 3′,5′monophosphate can modulate long-term potentiation induction in the mouse hippocampal CA1 region through effects on cyclic AMP signaling. Using the cyclic guanosine 3′,5′monophosphate-specific phosphodiesterase inhibitor zaprinast or the nitric oxide donor S-nitroso-D,L-penicillamine to elevate cyclic guanosine 3′,5′monophosphate levels we found that increases in cyclic guanosine 3′,5′monophosphate strongly inhibit the induction of long-term potentiation by low-frequency patterns of synaptic stimulation where protein kinase A activation is required for long-term potentiation induction. In contrast, zaprinast and S-nitroso-D,L-penicillamine had no effect on the induction of long-term potentiation by high-frequency patterns of synaptic stimulation that induce long-term potentiation in a protein kinase A-independent manner. Directly activating protein kinase A with the phosphodiesterase-resistant cyclic AMP analog 8-Br-cAMP, blocking all phosphodiesterases with 3-isobutyl-1-methylxanthine, or inhibiting protein phosphatases rescued long-term potentiation induction in zaprinast-treated slices. Together, these results suggest that increases in cyclic guanosine 3′,5′monophosphate inhibit long-term potentiation by activating phosphodiesterases that interfere with the protein kinase A-mediated suppression of protein phosphatases needed for long-term potentiation induction. Consistent with the notion that this cyclic guanosine 3′,5′mono-phosphate-mediated inhibitory pathway is recruited by some patterns of synaptic activity, blocking cyclic guanosine 3′,5′monophosphate production strongly facilitated the induction of long-term potentiation by long trains of theta-frequency synaptic stimulation. Together, our results indicate that increases in cyclic guanosine 3′,5′monophosphate can act as a long-term potentiation suppressor mechanism that selectively constrains the induction of protein kinase A-dependent forms of long-term potentiation induced by low-frequency patterns of synaptic stimulation.
Keywords: hippocampus, cyclic nucleotides, phosphodiesterase, protein phosphatase
The cyclic AMP (cAMP)/protein kinase A (PKA) signaling pathway has a crucial role in learning in animals as diverse as Aplysia, Drosophila, and mammals (Bailey et al., 1996; Burrell and Sahley, 2001; Selcher et al., 2002) and is importantly involved in activity-dependent forms of synaptic plasticity, such as long-term potentiation (LTP) (Nguyen and Woo, 2003). At excitatory synapses onto hippocampal CA1 pyramidal cells PKA has multiple roles in LTP. For instance, PKA activation is not only involved in the transcription-dependent processes responsible for the later stages of LTP maintenance but, for certain patterns of synaptic activity, also enables the induction of LTP by suppressing protein phosphatases that would otherwise oppose the protein kinase mediated processes underlying LTP induction (Blitzer et al., 1995, 1998; Thomas et al., 1996; Makhinson et al., 1999). Moreover, activation of PKA by neurotransmitters acting through G protein-coupled receptors can strongly facilitate LTP induction (Thomas et al., 1996; Otmakhova and Lisman, 1996; Katsuki et al., 1997; Winder et al., 1999; Brown et al., 2000; Lin et al., 2003). Thus, the cAMP/PKA pathway also provides a mechanism through which modulatory neurotransmitters can regulate LTP induction.
The cAMP/PKA pathway is not only activated by G protein-coupled receptors but is also regulated by a variety of second messengers acting through different isoforms of adenylyl cyclase and phosphodiesterase, the enzymes responsible for cAMP production and degradation respectively (Hanoune and Defer, 2001; Mehats et al., 2002). An important example of this is the second messenger cyclic guanosine 3′,5′monophosphate (cGMP) which, by regulating different forms of the enzyme phosphodiesterase, can either facilitate or inhibit cAMP signaling by attenuating or enhancing cAMP degradation (Mehats et al., 2002). One phosphodiesterase expressed by hippocampal neurons, PDE3 (Reinhardt and Bondy, 1996), is inhibited by cGMP (Mehats et al., 2002). This suggests that increases in cGMP might facilitate LTP induction by inhibiting cAMP degradation. However, hippocampal neurons also express PDE2, a phosphodiesterase that is strongly stimulated by cGMP (Repaske et al., 1993; Van Staveren et al., 2003). Thus, cGMP production also has the potential to suppress LTP induction by facilitating cAMP degradation and inhibiting PKA activation. Although previous findings suggest that activation of protein kinase G (PKG) by cGMP has an important role in hippocampal LTP and long-term depression (LTD) (Zhuo et al., 1994a; Wu et al., 1998; Santschi et al., 1999; Arancio et al., 2001; Monfort et al., 2002), the possibility that cGMP regulates LTP induction through phosphodiesterase-mediated alterations in cAMP signaling has not been examined.
Here we investigated how the induction of LTP in the hippocampal CA1 region might be regulated by interactions between the cAMP and cGMP signaling pathways. We find that activation of cGMP-stimulated phosphodiesterases strongly inhibits the induction of LTP in a protein phosphatase-dependent manner. Our results thus indicate that cGMP can act as a potent suppressor of LTP induction by opposing the PKA-dependent inhibition of protein phosphatases required for LTP induction.
EXPERIMENTAL PROCEDURES
Slice preparation and extracellular recordings
Standard techniques approved by the UCLA Institutional Animal Care and Use Committee were used to prepare 400 μm thick hippocampal slices from tissue obtained from halothane anesthetized, 6–8 week-old male C57BL/6 mice (Charles River Laboratories, Wilmington, MA, USA). Slices were maintained (at 30 °C) in an interface-type chamber perfused (2–3 ml/min) with an oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (ACSF) containing 124 mM NaCl, 4.4 mM KCl, 25 mM NaHCO3, 1 mM NaH2PO4, 2 mM CaCl2, 1.2 mM MgSO4, and 10 mM glucose. A biopolar, nichrome wire stimulating electrode placed in stratum radiatum of the CA1 region of the slice was used to stimulate Schaffer collateral/commissural fibers (stimulation rate=0.02 Hz) and a low resistance glass microelectrode filled with ACSF (resistance=5–10 Mohm) was used to record field excitatory postsynaptic potentials (fEPSPs) in stratum radiatum. At the start of each experiment the maximal fEPSP that could be evoked in each slice was determined by gradually increasing the stimulation intensity until the evoked fEPSP amplitude reached a saturating level. The stimulation intensity was then adjusted to elicit fEPSPs that were approximately 50% of the maximal fEPSP amplitude. In some experiments an extracellular recording electrode was placed in stratum pyramidale to record population spikes evoked by Schaffer collateral/commissural fiber stimulation. In these experiments the stimulation intensity was adjusted to evoke baseline population spike amplitudes of approximately 2 mV (measured from the midpoint of a tangent connecting the two positive peaks of the fEPSP to the peak negativity of the population spike). All extra-cellular recordings were performed in slices maintained under interface conditions except those shown in Fig. 1A and B and Fig. 2, which were done using slices maintained in a submerged-slice recording chamber. In most experiments LTP was induced using a theta-pulse stimulation (TPS) protocol consisting of 150 pulses of presynaptic stimulation delivered at 5 Hz. High-frequency stimulation (HFS)-induced LTP was elicited using two trains of 100 Hz stimulation (100 pulses each, inter-train interval=10 s). The average slopes of fEPSPs evoked 40–45 min post-TPS or 55–60 min-post HFS were used for statistical comparisons (paired and unpaired t-tests or, where appropriate, one-way ANOVAs followed by Student-Newman-Keuls tests for multiple pairwise comparisons).
Fig. 1.
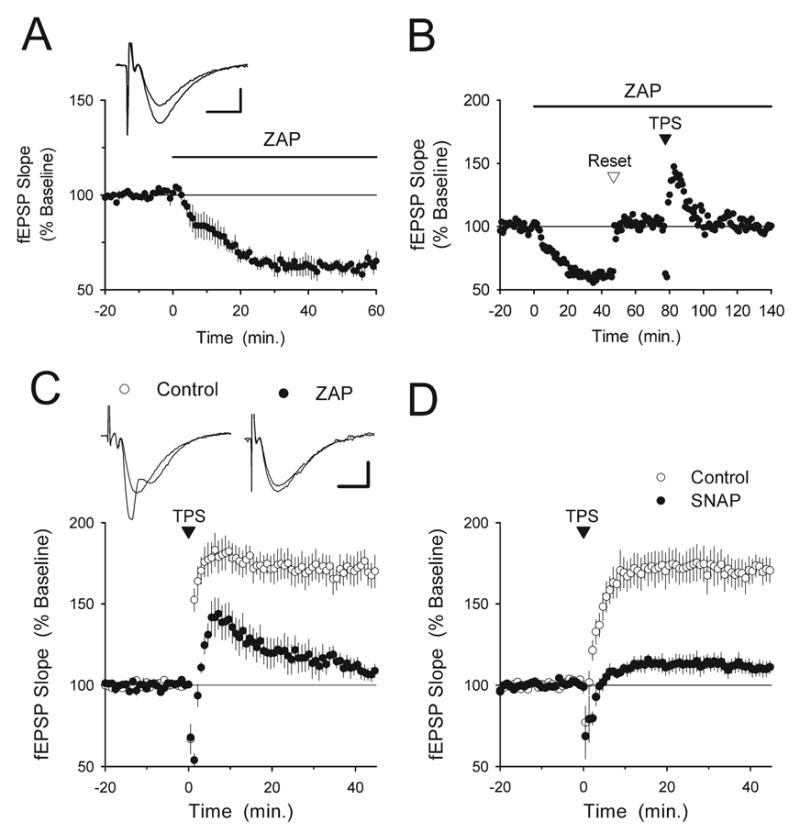
Increases in cGMP inhibit TPS-induced LTP. (A) Bath application of the cGMP-specific phosphodiesterase inhibitor ZAP (50 μM, indicated by the bar) induces a persistent inhibition of basal synaptic transmission (n=5). The inset shows fEPSPs recorded during baseline and 60 min after ZAP application in a representative experiment. Scale bars=5 ms and 1 mV. (B) A representative experiment illustrating the experimental protocol used to examine the effect of ZAP on TPS-induced LTP. After a stable 20 min period of baseline recording, 50 μM ZAP was applied for the remainder of the experiment (indicated by the bar). Once the ZAP-induced inhibition of synaptic transmission reached steadystate the intensity of presynaptic fiber stimulation was increased to restore fEPSPs to the pre-ZAP baseline level (reset). Following a second 20 min baseline, TPS was delivered at the indicated time in an attempt to induce LTP. (C) Summary of five experiments like those shown in (B) Note that TPS fails to induce LTP in ZAP-treated slices (filled symbols) while robust LTP is induced in interleaved vehicle control experiments (0.2% DMSO, open symbols, n=5). The inset shows fEPSPs recorded before and 45 min post-TPS in a control experiment (left set of traces) and in a ZAP-treated slice (right set of traces). Scale bars=5 ms and 1 mV. (D) The nitric oxide donor SNAP inhibits TPS-induced LTP. In vehicle control experiments (open symbols, slices bathed in 1 mM acetylpenicillamine+2 μM CuSO4), fEPSPs were potentiated to 171± 7% of baseline, n=5). In contrast, TPS had little lasting effect on synaptic strength in slices continuously bathed in ACSF containing 1 mM SNAP (filled symbols, post-TPS fEPSPs were 110±4% of baseline, n=6, P<0.001 compared with control). Like ZAP, SNAP inhibited basal synaptic transmission and experiments were performed using the protocol shown in B.
Fig. 2.
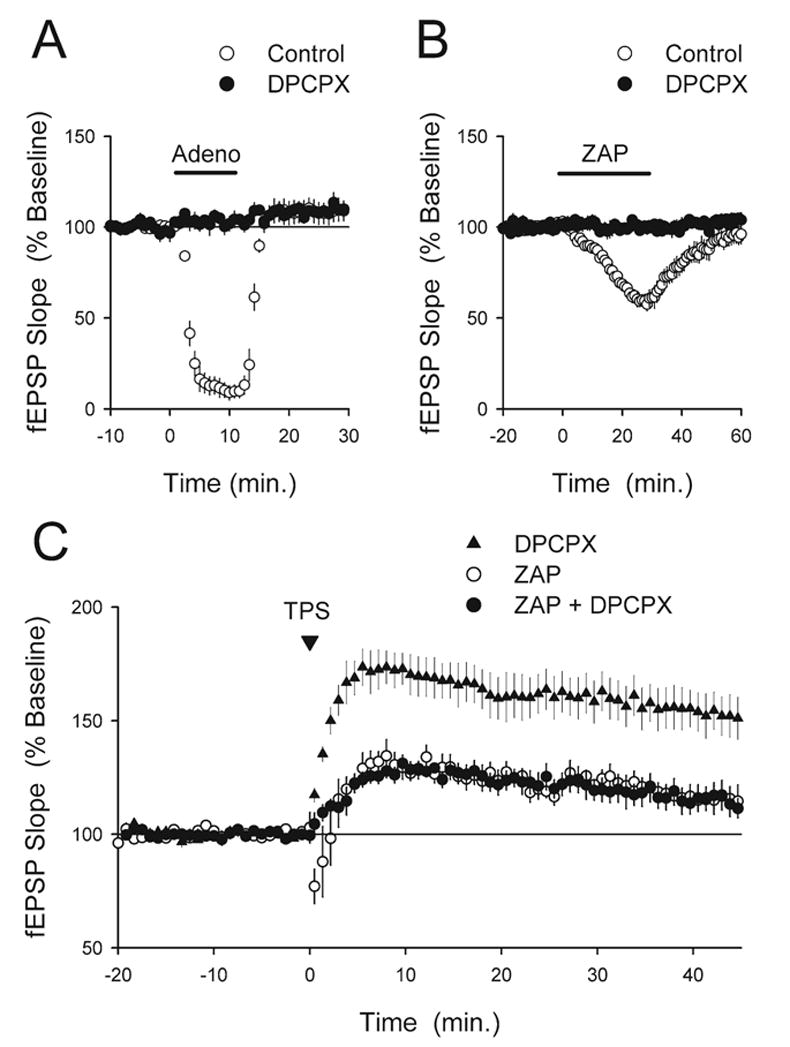
ZAP does not inhibit LTP via indirect effects mediated by A1 adenosine receptors. (A) The A1 receptor antagonist DPCPX (200 nM) completely blocks the inhibition of synaptic transmission caused by bath application of adenosine. Bath application of 100 μM adenosine for 10 min strongly inhibited synaptic transmission in control experiments (open symbols, at the end of the adenosine application fEPSPs were reduced to 11±4% of baseline, n=5) but had no effect on synaptic transmission in slices continuously bathed in ACSF containing 200 nM DPCPX (filled symbols, n=6). (B) Blocking A1 receptors inhibits the effects of ZAP on basal synaptic transmission. Following at 30 min bath application of ZAP (50 μM, indicated by the bar) fEPSPs were depressed to 60±4% of baseline in control experiments (open symbols, n=4). Field EPSPs recovered to near baseline levels following ZAP washout (fEPSPs recovered to 95±5% of baseline 30 min after ZAP washout). As shown by the filled symbols, ZAP had no effect on synaptic transmission in slices continuously bathed in ACSF containing 200 nM DPCPX (fEPSPs were 100±2% of baseline at the end of the ZAP application, n=4). (C) Blocking A1 receptors with DPCPX does not rescue LTP in ZAP-treated slices. Continuous bath application of DPCPX (200 nM) by itself had no apparent effect on TPS-induced LTP (triangles, fEPSPs were potentiated to 153±9% of baseline, n=6). As shown by the filled circles, LTP was still inhibited by 50 μM ZAP applied in the presence of DPCPX (fEPSPs were 115±5% of baseline, n=5, P<0.02 compared with TPS in DPCPX alone). Results from interleaved experiments testing the effects of TPS on synaptic transmission in slices bathed in normal ACSF containing 50μM ZAP are shown for comparison (open circles, fEPSPs were 116±5% of baseline, n=5). TPS was delivered at time=0.
Whole-cell current-clamp recordings
Low resistance electrodes (Relectrode=3–7 Mohm, Rseries=10–30 Mohm) filled with a solution containing 122.5 cesium gluconate, 17.4 CsCl, 2 mM MgCl2, 10 mM TEA-Cl, 0.2 mM EGTA, 2 mM ATP, 0.3 mM GTP, and 10 mM HEPES (pH=7.2, osmolarity=290–300 mOsm, adjusted with cesium gluconate) were used to perform whole-cell current-clamp recordings. In these experiments slices were maintained in a submerged-slice recording chamber and 100 μM picrotoxin was added to the ACSF to block inhibitory synaptic transmission. To prevent the spontaneous bursting that can occur when inhibition is blocked the hippocampal CA3 region was removed and slices were bathed in a modified ACSF containing 2.4 mM KCl, 4 mM CaCl2 and 4 mM MgSO4. The intensity of presynaptic fiber stimulation was adjusted to evoke 5–10 mV EPSPs and constant current injection through the recording electrode was used to hyperpolarize membrane potentials to −80 to −85 mV throughout the experiment. A brief (40–50 ms) hyperpolarizing pulse of current (50–100 pA) injected through the recording electrode either 90 ms before or 150 ms after each presynaptic fiber stimulation pulse was used to monitor input and series resistance throughout the experiment. In these experiments LTP was induced by pairing 100 pulses of presynaptic fiber stimulation (at 2 Hz) with tonic depolarization of the postsynaptic cell’s membrane potential to approximately −10 mV. This pairing protocol was delivered after 17–20 min of whole-cell recording to avoid the potential loss of LTP induction associated with prolonged whole-cell recordings yet allow as much time as possible for compounds in the electrode solution to diffuse into the cell. Unless indicated otherwise, the average slope of EPSPs evoked between 25 and 30 min post-pairing was used for statistical comparisons.
Rp-cAMPS (Alexis Biochemicals, San Diego, CA, USA) and PKI(6–22)amide (Biomol, Plymouth Meeting, PA, USA) were dissolved directly into the electrode-filling solution. H89 (N-[2-(p-bromo-cinnamylamino)ethyl]-5-isoquinoline sulfonamide) (Biomol), calyculin A, microcystin LR, and nodularin (Alexis Biochemicals) were prepared as a concentrated stock solutions in dimethyl-sulfoxide (DMSO) and stored at −20 °C. S-nitroso-D,L-penicillamine (SNAP) was synthesized according to the reaction developed by Field et al. (1978) or purchased from Tocris Cookson (Ellisville, MO, USA) and co-applied with 2 μM CuSO4 to facilitate nitric oxide release. DPCPX (1,3-dipropyl-8-cyclopentyl xanthine) was obtained from Tocris and prepared as a concentration stock solution in DMSO. All other compounds were obtained from Sigma (St. Louis, MO, USA).
RESULTS
Increases in cGMP inhibit TPS-induced LTP
In the hippocampal CA1 region the induction of LTP by TPS is strongly suppressed by PKA inhibitors and enhanced by pharmacological manipulations that increase intracellular levels of cAMP (Thomas et al., 1996; Winder et al., 1999; Brown et al., 2000). Thus, to determine whether cGMP signaling can regulate the induction of PKA-dependent forms of LTP we examined whether TPS-induced LTP was altered in slices bathed in ACSF containing the cGMP-specific phosphodiesterase inhibitor zaprinast (ZAP), a compound previously shown to elevate cGMP levels in hippocampal slices (Boulton et al., 1994). Because hippocampal pyramidal cells express two distinct cGMP-specific phosphodiesterases (PDE5 and PDE9) (Andreeva et al., 2001; Van Staveren et al., 2003) with different sensitivities to ZAP (IC50s of 0.76 and 29 μM, respectively) (Soderling et al., 1998), ZAP was bath applied at a concentration of 50 μM to effectively inhibit both isoforms.
Consistent with previous reports (Boulton et al., 1994; Broome et al., 1994; Wu et al., 1998; Santschi et al., 1999), ZAP induced a significant depression of basal synaptic transmission that persisted for as long as ZAP was present in the bath (Fig. 1A, 60 min after the start of ZAP application fEPSPs were reduced to 63±3% of baseline, n=5, P<0.001 compared with baseline). Importantly, this effect of ZAP on basal synaptic transmission could inhibit LTP induction by decreasing the postsynaptic depolarization needed for NMDA receptor activation. To control for this we continuously bathed slices in ACSF containing ZAP and, after the ZAP-induced depression reached steady state, increased the intensity of presynaptic stimulation to restore fEPSPs to pre-ZAP baseline levels. A new 20 min baseline in the presence of ZAP was then recorded prior to attempting to induce LTP with TPS (Fig. 1B). As shown in Fig. 1C, TPS-induced LTP was strongly inhibited in ZAP-treated slices (post-TPS fEPSPs were just 109±8% of baseline n=6, compared with 171±8% of baseline in interleaved vehicle control experiments, 0.2% DMSO, n=5, P<0.001). Consistent with the notion that the effects of ZAP on LTP induction are due to increases in intracellular cGMP, the nitric oxide donor SNAP, which elevates cGMP levels in hippocampal slices via stimulation of soluble guanylyl cyclases (Boulton et al., 1994), also inhibited basal synaptic transmission and TPS-induced LTP (Fig. 1D).
Increases in cGMP enhance adenosine release in hippocampal slices (Fallahi et al., 1996; Saransaari and Oja, 2004) and activation of presynaptic adenosine receptors are thought to be responsible for the inhibitory effects of ZAP and SNAP on basal synaptic transmission (Boulton et al., 1994; Broome et al., 1994). Thus, as an additional control for the possibility that the effects of ZAP on TPS-induced LTP might be due to changes in basal synaptic transmission we examined the effects of ZAP on basal synaptic transmission and LTP induction in slices where A1 type adenosine receptors were blocked with the antagonist DPCPX. As shown in Fig. 2, bath application of 200 nM DPCPX completely blocked the inhibition of basal synaptic transmission by both adenosine (100 μM) and ZAP (Fig. 2A and 2B) but had no effect on the inhibition of TPS-induced LTP in ZAP-treated slices (Fig. 2C). These results, which demonstrate that TPS-induced LTP is strongly suppressed by ZAP even under experimental conditions where the effects of ZAP on basal synaptic transmission are prevented, indicate that the inhibition of LTP in ZAP-treated slices cannot be attributed to the effects of ZAP on basal synaptic transmission.
While brief trains of TPS reliably induce LTP in the hippocampal CA1 region, longer trains of TPS consisting of 900 stimulation pulses have little lasting effect on synaptic strength (Thomas et al., 1996, 1998; Winder et al., 1999). Long trains of TPS can induce a PKA-dependent form of LTP, however, when delivered in the presence of reagents that elevate intracellular cAMP levels, such as the adenylyl cyclase activator forskolin or β-adrenergic receptor agonists (Thomas et al., 1996; Winder et al., 1999 Brown et al., 2000). Thus, as an additional test of the effects of cGMP on PKA-dependent forms of LTP we examined whether ZAP and SNAP inhibit the induction of LTP by a long train of TPS delivered in the presence of the β-adrenergic receptor agonist isoproterenol (ISO). As shown in Fig. 3A, 900 pulses of TPS delivered at the end of a 10 min bath application of 1.0 μM ISO induced a two-fold potentiation of synaptic transmission in control slices but had relatively little persistent effect on synaptic transmission in slices continuously bathed in ACSF containing 50 μM ZAP or 1 mM SNAP. In these experiments we also observed that the small, PKA-mediated increase in basal synaptic transmission induced by β-adrenergic receptor activation (Thomas et al., 1996) was completely blocked by ZAP and SNAP (7 min after the start of ISO application fEPSPs were 119±4% of baseline in control slices, n=5, but were just 99±3% and 101±3% of baseline in slices bathed in ZAP or SNAP, respectively, n=5 for each, P<0.05 compared with control). In other experiments we found that ZAP also strongly suppressed the cAMP-dependent, persistent facilitation of EPSP-evoked population spikes induced by β-adrenergic receptor activation (Heginbotham and Dunwiddie, 1991; Dunwiddie et al., 1992) (Fig. 3B). Thus, increases in cGMP not only inhibit the induction of PKA-dependent forms of LTP but also strongly suppress the cAMP and PKA-mediated effects of β-adrenergic receptor activation on excitatory synaptic transmission and neuronal excitability.
Fig. 3.
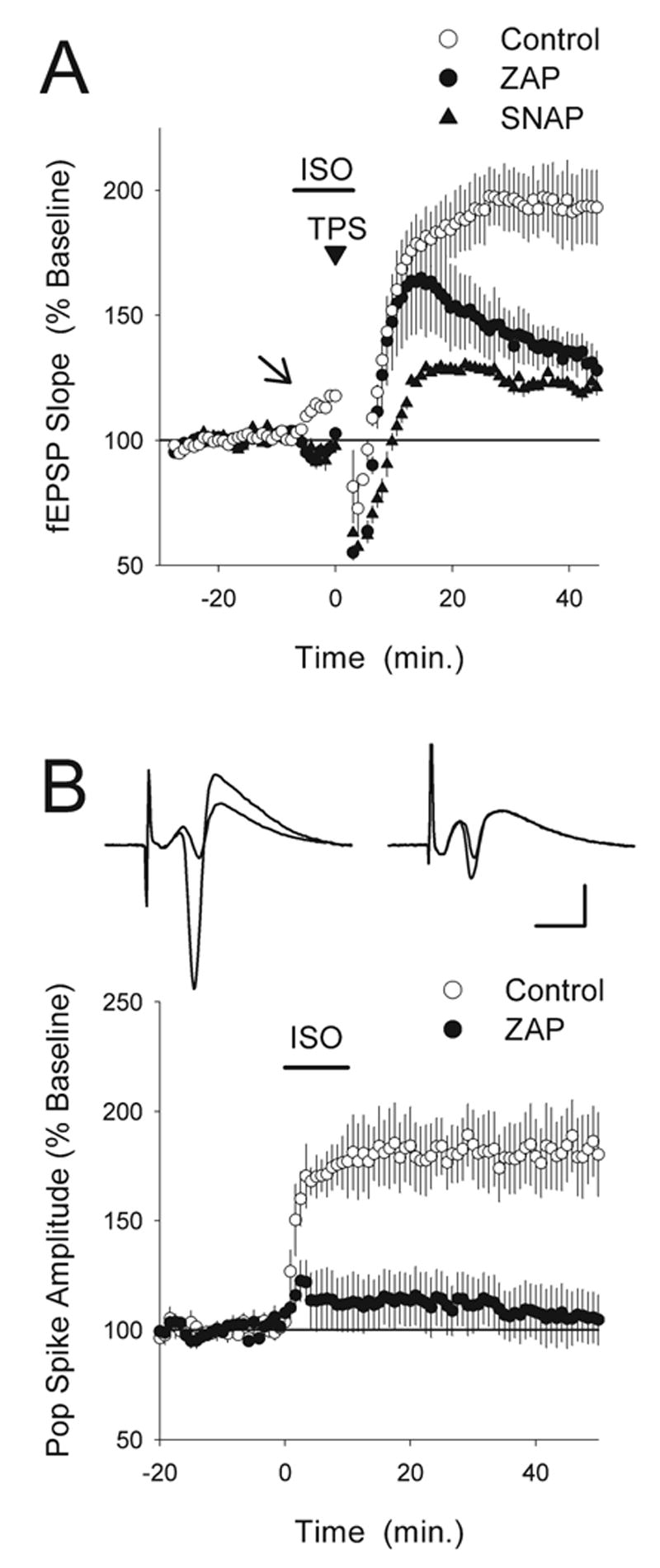
ZAP inhibits the effects of β-adrenergic receptor activation on synaptic transmission, LTP induction, and neuronal excitability. (A) The induction of LTP by a long-train of TPS in the presence of the β-adrenergic receptor agonist ISO is inhibited in ZAP and SNAP-treated slices. In control experiments (open symbols, n=5), EPSPs were potentiated to 206±17% of baseline following TPS (900 pulses) delivered at the end of a 10 min bath application of ISO (1 μM, application indicated by the bar). TPS in the presence of ISO induced significantly less LTP in slices continuously bathed in ACSF containing 50 μM ZAP (filled circles, fEPSPs were 128±8% of baseline, n=5) or 1.0 mM SNAP (triangles, fEPSPs were 122±8% of baseline, n=5, P<0.001 for both). Note that the ISO-induced enhancement of synaptic transmission seen prior to TPS in control slices (indicated by the arrow) was blocked in ZAP and SNAP-treated slices. (B) ZAP inhibits the persistent enhancement of EPSP-evoked population spikes induced by β-adrenergic receptor activation. In control experiments (open symbols) a 10 min bath application of 1.0 μM ISO (indicated by the bar) induced a nearly two-fold, persistent increase in the amplitude of population spikes elicited by presynaptic fiber stimulation (40 min after ISO washout population spike amplitudes were increased to 183±17% of baseline, n=7). β-Adrenergic receptor activation had no lasting effect on EPSP-evoked population spikes in slices continuously bathed in ACSF containing ZAP (50 μM, filled circles, 40 min after ISO washout population spike amplitudes were 106±12% of baseline, n=7, P<0.005 compared with control). The inset shows population spikes recorded before and 40 min after ISO application in a control experiment (left set of traces) and in a ZAP-treated slice (right set of traces). Scale bars=5 ms and 2 mV.
ZAP-induced increases in cGMP suppress LTP induction by inhibiting cAMP/PKA signaling
Based on our observations that ZAP and SNAP inhibit both PKA-dependent forms of LTP and the PKA-mediated effects of β-adrenergic receptor activation on synaptic strength and neuronal excitability we investigated whether the effects of ZAP and SNAP on LTP might be due to a cGMP-induced inhibition of cAMP signaling. Hippocampal neurons strongly express the cGMP-stimulated phosphodiesterase PDE2 (Repaske et al., 1993; Van Staveren et al., 2003) and cGMP stimulation of phosphodiesterases has been shown to inhibit cAMP-dependent modulation of voltage-activated calcium channels in hippocampal neurons (Doerner and Alger, 1988). Thus, one way that increases in cGMP could inhibit cAMP signaling is by activating PDE2 and enhancing cAMP degradation. Importantly, the hypothesis that increases in cGMP disrupt LTP induction through a phosphodiesterase-mediated inhibition of cAMP signaling makes a number of readily testable predictions. First, if the inhibitory effects of ZAP and SNAP on TPS-induced LTP are due to alterations in cAMP and PKA signaling then the induction of LTP by patterns of synaptic stimulation that induce LTP in a PKA-independent manner should not be affected by either compound. Consistent with this, ZAP and SNAP had no effect on the induction of LTP by two trains of 100 Hz stimulation (Fig. 4A), a stimulation protocol that induces a PKA-independent early phase of LTP (Frey et al., 1993; Weisskopf et al., 1994; Blitzer et al., 1995; Thomas et al., 1996). Second, if increases in cGMP inhibit cAMP signaling by activating phosphodiesterases, then application of phosphodiesterase-resistant cAMP analogues should rescue TPS-induced LTP in ZAP-treated slices. To test this prediction we bath applied the hydrolysis-resistant cAMP analog 8-Br-cAMP (1 mM) for 20 min prior to TPS in control and ZAP-treated slices. As shown in Fig. 4B and 4C, 8-Br-cAMP had no effect on LTP induction in slices bathed in normal ACSF (fEPSPs were potentiated to 170±7% of baseline) but completely rescued LTP in slices continuously bathed in ACSF containing 50 μM ZAP (fEPSPs were potentiated to 181±10% of baseline, n=11). In other experiments we found that a 20 min bath application of 1 mM 8-Br-cAMP by itself had no significant lasting effect on synaptic transmission (n=3, data not shown). This indicates that the ability of 8-Br-cAMP to rescue LTP in ZAP-treated slices is not due to an 8-Br-cAMP-induced potentiation of synaptic transmission that masks the inhibitory effects of ZAP. Finally, the hypothesis that increases in cGMP inhibit LTP induction by facilitating cAMP degradation predicts that inhibiting all phosphodiesterases should rescue LTP in ZAP-treated slices by inhibiting cAMP degradation even when cGMP levels are elevated. Consistent with this, bath application of the nonselective phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX, 100 μM) by itself had no effect on TPS-induced LTP (fEPSPs were potentiated to 160±11% of baseline in IBMX-treated slices, n=6) but completely prevented the suppression of LTP by ZAP (fEPSPs were potentiated to 158±5% of baseline in slices exposed to both IBMX and ZAP, n=6) (Fig. 4D).
Fig. 4.
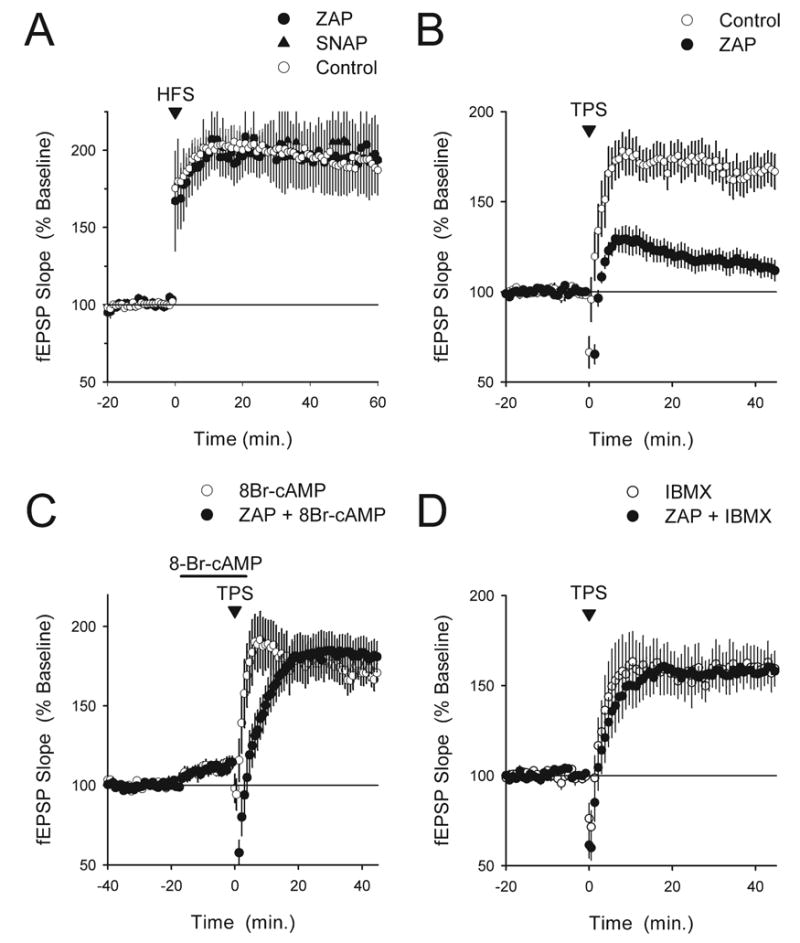
Increases in cGMP inhibit TPS-induced LTP by opposing cAMP/PKA signaling. (A) HFS-induced LTP is not inhibited in slices continuously bathed in ACSF containing 50 μM ZAP or 1 mM SNAP. HFS was delivered at time=0. Sixty minutes post-100 Hz stimulation fEPSPs were potentiated to 190±11% of baseline in ZAP-treated slices (filled circles, n=5) and were potentiated to 197±26% of baseline in SNAP-treated slices (triangles, n=5). The open circles show results from interleaved control experiments (fEPSPs were potentiated to 192±12% of baseline, n=10). (B) The induction of LTP by TPS is strongly inhibited in slices maintained in interface-slice type recording chambers and continuously bathed in ACSF containing 50 μM ZAP (filled symbols, fEPSPs were 113±5% of baseline, n=9). The open symbols show results from interleaved control experiments (0.2% DMSO, fEPSPs were potentiated to 168±9% of baseline, n=6). These experiments are the untreated and ZAP-treated controls for experiments shown in panels C and D. (C) The hydrolysis-resistant cAMP analog 8-Br-cAMP rescues LTP in ZAP-treated slices. A 20 min bath application of 1 mM 8-Br-cAMP (indicated by the bar) prior to TPS by itself had no effect on LTP induction (open symbols, n=6) but completely rescued LTP in slices exposed to 50 μM ZAP (filled symbols, n=11). In both experiments the A1 adenosine receptor antagonist 8-cyclopentyltheophylline (200 nM) was applied throughout the experiment to block the adenosine receptor-mediated inhibitory effects of 8-Br-cAMP on synaptic transmission. (D) Blocking phosphodiesterases with the non-selective inhibitor IBMX rescues TPS-induced LTP in ZAP-treated slices. Slices were continuously bathed in ACSF containing either 100 μM IBMX alone or IBMX plus 50 μM ZAP. IBMX alone had no effect on TPS-induced LTP (open symbols, n=6) but completely restored TPS-induced LTP in ZAP-treated slices (filled symbols, n=6). In panels B–D TPS was delivered at time=0.
Fig. 8.
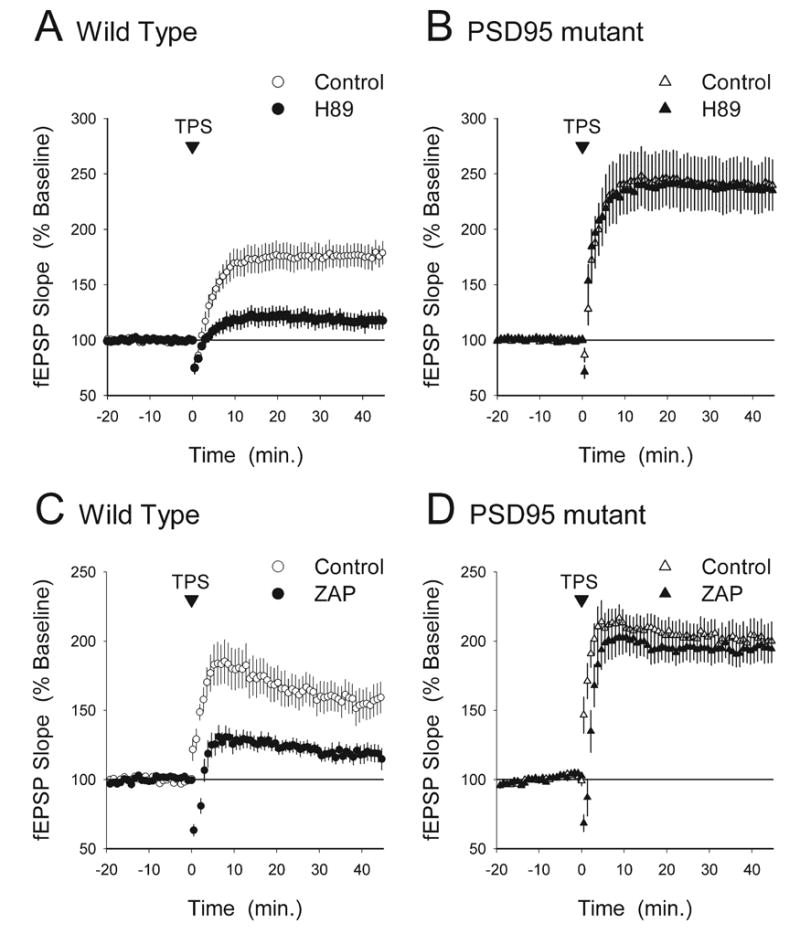
TPS induces a PKA-independent and ZAP-insensitive form of LTP in the CA1 region of hippocampal slices from PSD-95 mutant mice. (A) The PKA inhibitor H89 suppresses the induction of LTP by 150 pulses of TPS in hippocampal slices from wild type mice. In vehicle control experiments (0.1% DMSO) fEPSPs were potentiated to 176±11% of baseline 45 min post-TPS (open symbols, n=7). Significantly less LTP was induced in slices continuously bathed in ACSF containing 10 μM H89 (filled symbols, 45 min post-TPS fEPSPs were 117±8% of baseline, n=9, P<0.02 compared with vehicle controls). (B) H89 has no effect on TPS-induced LTP in slices from PSD-95 mutant mice. Forty-five minute post-TPS fEPSPs were potentiated to 238±22% in vehicle control slices (open symbols, n=8) and were potentiated to 237±17% of baseline in slices continuously bathed in ACSF containing 10 μM H89 (filled symbols, n=9). (C) In slices obtained from wild type littermates ZAP strongly inhibited TPS-induced LTP. Forty-five minute post-TPS fEPSPs were potentiated to 157±11% of baseline in vehicle control experiments (open symbols, n=8) but were 121±6% of baseline in ZAP-treated slices (filled symbols, n=8, P<0.05 compared with vehicle control). (D) ZAP has no effect on TPS-induced LTP in slices obtained from PSD-95 mutant mice. Forty-five minute post-TPS fEPSPs were potentiated to 200±12% of baseline in vehicle control experiments (open symbols, n=7) and were potentiated to 196±11% of baseline in ZAP-treated slices (filled symbols, n=8).
PKA inhibitors suppress pairing-induced LTP
Because the PKA-dependent processes important for the induction of LTP are thought to be postsynaptic (for review see Nguyen and Woo, 2003), our results suggest that increases in postsynaptic cGMP are primarily responsible for the inhibitory effects of ZAP on TPS-induced LTP. Previous studies have found, however, that presynaptic cGMP signaling has an important role in both LTP (Arancio et al., 1995, 2001; Blitzer et al., 1995) and LTD (Santschi et al., 1999; Stanton et al., 2003). Thus, to determine whether ZAP-induced increases in cGMP inhibit LTP through effects on postsynaptic cAMP signaling we examined the effects of introducing ZAP and other compounds into CA1 pyramidal cells on the induction of LTP by a pairing protocol where a brief train of low-frequency pre-synaptic fiber stimulation was paired with postsynaptic depolarization to −10 mV.
As noted above, the induction of LTP by some patterns of synaptic stimulation is PKA-independent. Thus, we first examined whether PKA activation is required for pairing-induced LTP under our experimental conditions by introducing various PKA inhibitors into the postsynaptic CA1 pyramidal cells via the recording electrode. As can be seen in Fig. 5A, pairing-induced LTP was strongly suppressed in cells where the PKA inhibitor Rp-cAMPs (1 mM) was present in the recording electrode solution (post-pairing EPSPs were 116±11% of baseline, n=11 compared with 277±16% of baseline in interleaved control experiments, n=13, P<0.001). Importantly, some cAMP analogues, including Rp-cAMPs, may inhibit LTP through PKA-independent mechanisms (Otmakhova and Lisman, 2002). Thus, we also examined the effects of two additional PKA inhibitors on pairing-induced LTP. As shown in Fig. 5B and 5C, postsynaptic infusion of the PKA inhibitors PKI(6–22) amide and H89 also significantly suppressed, but unlike Rp-cAMPs did not completely block, pairing-induced LTP. In a series of control experiments we introduced PKA inhibitors into CA1 pyramidal cells via the recording electrode and, after an amount of time in the whole-cell recording mode equivalent to that used in our LTP experiments, depolarized the postsynaptic cell to −10 mV for 50 s in the absence of presynaptic fiber stimulation. This protocol is identical to that used in our LTP experiments except that no LTP is induced due to the absence of presynaptic activity. Thirty minutes after postsynaptic depolarization alone EPSPs were not significantly different from baseline, (Rp-cAMPS: EPSPs were 110±9% of baseline, n=3; PKI(6–22)amide: EPSPs were 107±4% of baseline, n=4; H89: EPSPs were 93±5% of baseline, n=3). This indicates that the effects of PKA inhibitors on LTP induction cannot be attributed to nonselective, inhibitory effects on basal synaptic transmission. Thus, under our experimental conditions pairing-induced LTP is at least partially dependent on PKA activity (also see Otmakhova et al., 2000).
Fig. 5.
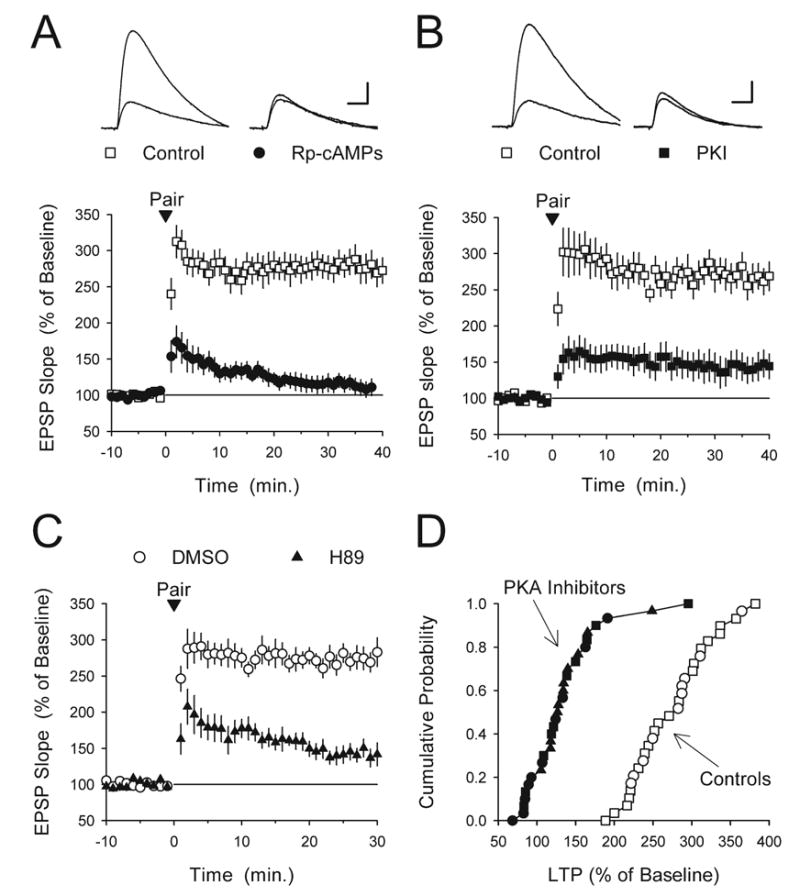
Postsynaptic delivery of PKA inhibitors suppresses pairing-induced LTP. (A) Pairing 100 pulses of 2 Hz presynaptic fiber stimulation (delivered at time=0) with postsynaptic depolarization to −10 mV induced more than two-fold LTP in control cells (open symbols, n=13) but had no lasting effect on synaptic transmission in cells where the recording electrode solution contained 1 mM Rp-cAMPs (filled symbols, n=11, EPSPs were not significantly different from baseline, P=0.27). (B) Postsynaptic delivery of the PKA inhibitory peptide PKI(6–22)amide (PKI, 500 μM) strongly suppresses, but does not completely block, pairing-induced LTP (filled symbols, EPSPs were potentiated to 144±19% of baseline, n=10, P<0.05 compared with baseline, P<0.001 compared with interleaved control experiments). The open symbols show interleaved control experiments (normal recording electrode-filling solution, EPSPs were potentiated to 272±19% of baseline, n=8). (C) Postsynaptic infusion of the PKA inhibitor H89 suppresses pairing-induced LTP. Post-pairing, EPSPs were potentiated to 143±12% of baseline in cells where the recording electrode contained 100 μM H89 (filled symbols, n=11, P<0.01 compared with baseline, P<0.001 compared with interleaved control experiments). In control experiments (open symbols, electrode-filling solution contained 0.2% DMSO) EPSPs were potentiated to 276±14% of baseline (n=10). (D) Cumulative probability distribution plot of results shown in panels A–C. Filled symbols correspond to recordings performed with PKA inhibitors in the electrode solution (Rp-cAMPS: circles; PKI(6–22)amide: squares; H89: triangles) while the open symbols correspond to interleaved control experiments (normal electrode-filling solution: squares; 0.2% DMSO: circles).
Increases in intracellular cGMP suppress pairing-induced LTP by inhibiting cAMP signaling
To determine whether cGMP modulates pairing-induced LTP we first examined the effects of blocking cGMP-specific phosphodiesterases with ZAP on LTP induction (Fig. 6A). Compared with control experiments, where synaptic transmission was potentiated to 268±20% of baseline (n=13), pairing-induced LTP was completely blocked in cells where ZAP (50 μM) was present in the bath (EPSPs were 94±10% of baseline, n=6, P<0.001 compared with control). Pairing-induced LTP was also strongly inhibited when ZAP was introduced into postsynaptic CA1 pyramidal cells via the recording electrode (EPSPs were 126±10% of baseline, n=6, P<0.001 compared with control). Pairing-induced LTP was also significantly inhibited in cells were 8-Br-cGMP (1 mM) was present in the recording electrode (EPSPs were 152±14% of baseline, n=15, compared with 259±18% of baseline in interleaved control experiments, n=14, P<0.001).
Fig. 6.
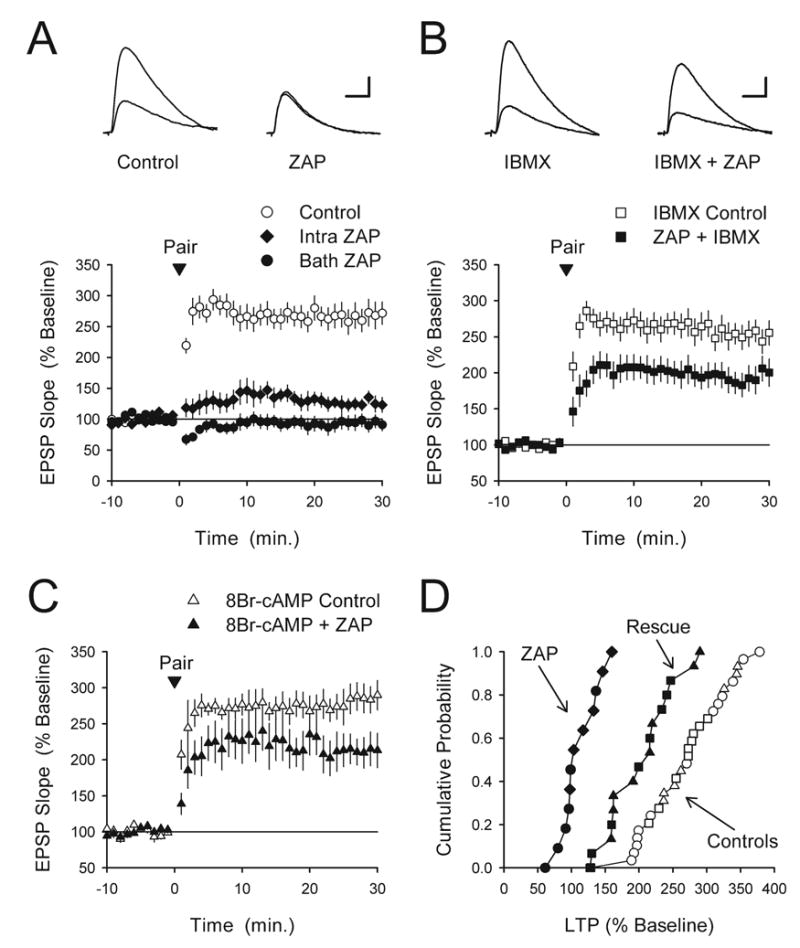
ZAP-induced increases in cGMP inhibit pairing-induced LTP by interfering with cAMP signaling. (A) Pairing-induced LTP was strongly inhibited when ZAP was applied in the bath (50 μM, filled circles, n=6) or when it was delivered postsynaptically via the recording electrode (500 μM, diamonds, n=6). Interleaved vehicle control experiments (0.2% DMSO, n=13) are shown for comparison. The inset shows EPSPs recorded during baseline and 30 min post-pairing in a control cell (left set of traces) and in a cell where ZAP was present in the bath (right set of traces). Scale bars=20 ms and 5 mV. (B) Blocking phosphodiesterases with IBMX rescues pairing-induced LTP in ZAP-treated slices. Slices were continuously bathed in ACSF containing either 100 μM IBMX (open symbols, n=10) or IBMX+50 μM ZAP (filled symbols, n=8). While IBMX alone had no effect on LTP it significantly reversed the inhibition of LTP in ZAP-treated slices (P<0.001 compared with bath applied ZAP results shown in panel A). The amount of LTP present 30 min post-pairing in the presence of ZAP+IBMX was still significantly less than that seen in cells recorded from slices bathed in IBMX alone (P<0.05). The inset shows EPSPs recorded during baseline or 30 min post-pairing in the presence of IBMX alone (left set of traces) or in the presence of IBMX+ZAP (right set of traces). Scale bars=5 mV and 20 ms. (C) Postsynaptic infusion of the hydrolysis-resistant cAMP analog 8-Br-cAMP rescues pairing-induced LTP in ZAP-treated cells. By itself, the presence of 8-Br-cAMP in the recording electrode filling solution (1.0 mM) had no effect on LTP (open symbols, n=7) but enabled the induction of LTP in cells were the recoding electrode also contained 500 μM ZAP (filled symbols, n=8). The amount of LTP present 30 min post-pairing in cells where 8-Br-cAMP and ZAP were present in the recording electrode was significantly larger than that seen in cells where the recording electrode solution contained ZAP alone (P<0.001 compared with intracellular ZAP results shown in panel A) but still significantly less than that seen in cells where the recording electrode solution contained 8-Br-cAMP alone (P<0.02). (D) Cumulative probability plot of the results shown in panels A–C. The results from control experiments were the same and are plotted together (open symbols correspond to those in panels A–C). The filled circles and diamonds correspond to the ZAP alone experiments shown in panel A while the filled squares and triangles correspond to the experiments shown in panels B and C where IBMX and 8-Br-cAMP were co-applied with ZAP (rescue experiments).
To test whether the ZAP inhibition of pairing-induced LTP, like TPS-induced LTP, involves cGMP-mediated alterations in cAMP signaling we examined whether IBMX could rescue LTP induction in cells where ZAP was present in the bath. As shown in Fig. 6B, bath application of IBMX (100 μM) by itself had no effect on pairing induced-LTP (EPSPs were 252±16% of baseline, n=10), but almost completely rescued LTP induction in ZAP-treated cells. (EPSPs were 194±17% of control in cells treated with both IBMX and ZAP, P<0.001 compared with bath applied ZAP alone, P<0.05 compared with control). Similarly, postsynaptic delivery of 8-Br-cAMP via the recording electrode had no effect on pairing-induced LTP (EPSPs were 286±19% of baseline, n=7) but enabled the induction of nearly normal levels of LTP when co-applied with ZAP via the recording electrode (EPSPs were 210±18% of baseline, n=8, P<0.001 compared with intracellular ZAP alone, P<0.02 compared with control) (Fig. 6C). Together, the results shown in Fig. 6 suggest that ZAP-induced increases in cGMP inhibit pairing-induced LTP via phosphodiesterase-mediated inhibition of cAMP signaling. Moreover, our observations that postsynaptic infusion of ZAP inhibits LTP and that LTP is largely rescued by postsynaptic delivery of 8-Br-cAMP suggest that ZAP-induced increases in cGMP inhibit LTP via effects on postsynaptic cAMP signaling. However, because both ZAP and 8-Br-cAMP are membrane permeable these results do not rule out the possibility that presynaptic effects are also involved.
Protein phosphatase inhibitors rescue LTP induction in the presence of ZAP
Activation of PKA is thought to have a crucial role in LTP of excitatory synapses onto hippocampal CA1 pyramidal cells because it provides a mechanism for suppressing the activity of protein phosphatases that would otherwise oppose LTP induction (Winder and Sweatt, 2001). Indeed, several studies have found that the effects of PKA inhibitors on LTP induction can be overcome by inhibitors of protein phosphatase 1 and 2A such as okadaic acid (Blitzer et al., 1995) and calyculin A (Thomas et al., 1996) (also see Brown et al., 2000). In addition, inhibitors of protein phosphatases rescue LTP in hippocampal slices from PKA mutant mice (Woo et al., 2002). These findings suggest that protein phosphatase inhibitors should rescue LTP in ZAP-treated slices if ZAP-induced increases in cGMP inhibit LTP by opposing PKA activation. To test this idea we pretreated slices for at least 45 min in ACSF containing 750 nM calyculin A, a concentration that by itself has no effect on TPS-induced LTP (Thomas et al., 1996) and determined whether ZAP still blocked LTP induction. Consistent with the hypothesis that ZAP-induced increases in cGMP inhibit LTP induction by interfering with cAMP signaling, blocking protein phosphatases with calyculin A completely prevented the effects of ZAP on TPS-induced LTP (Fig. 7A).
Fig. 7.
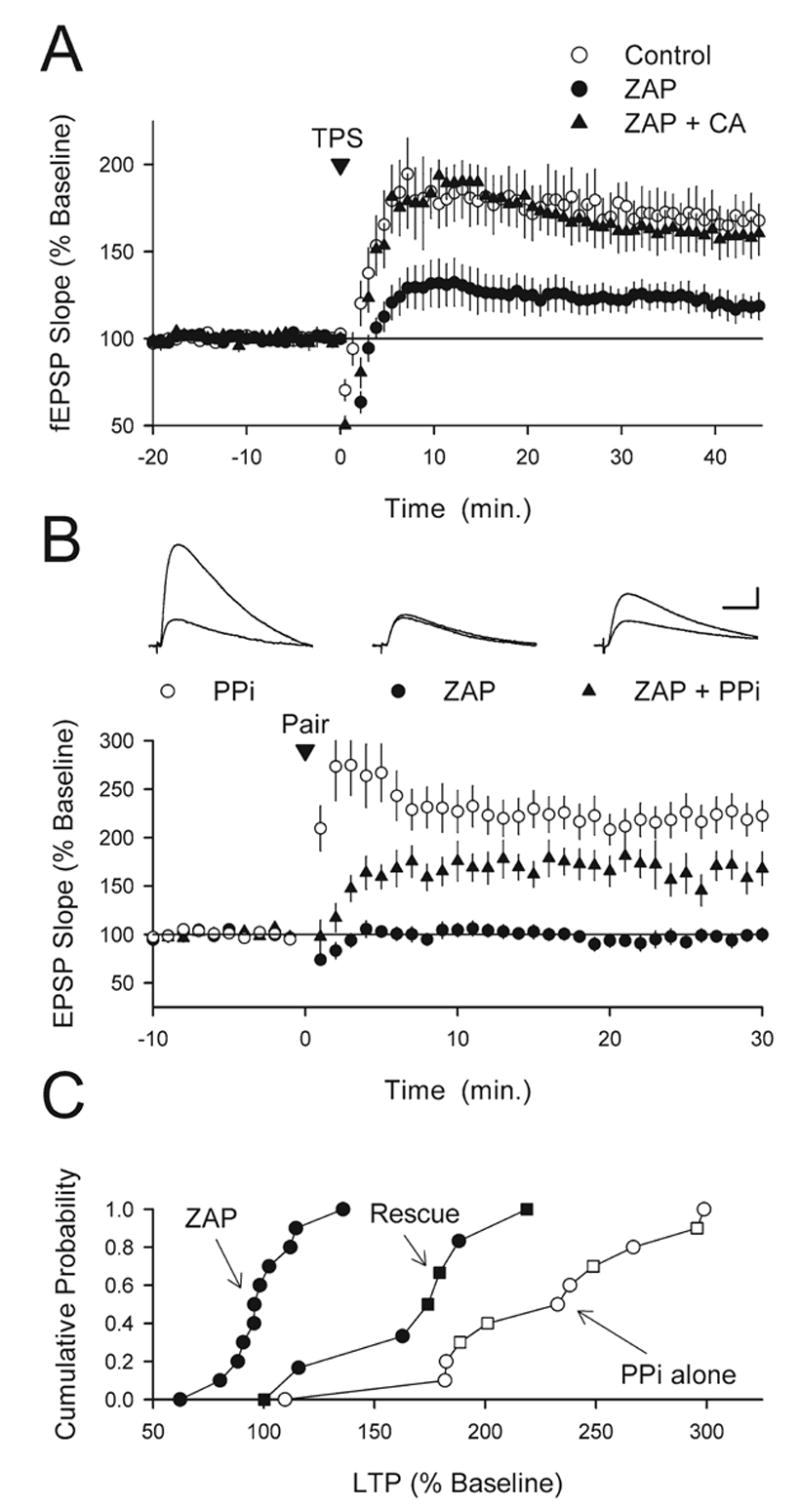
Protein phosphatase inhibitors rescue LTP induction in ZAP-treated slices. (A) Blocking protein phosphatases with calyculin A rescues TPS-induced LTP in ZAP-treated slices. Forty-five minutes post-TPS (delivered at time=0) fEPSPs were potentiated to 159±12% of baseline in slices continuously bathed in ACSF containing 50 μM ZAP plus 750 nM calyculin A (triangles, n=6) compared with 119±7% of baseline in ZAP-treated slices (filled circles, n=5, P<0.02). The open circles show results from interleaved, vehicle control experiments (0.36% DMSO, fEPSPs were potentiated to 168±11% of baseline, n=6). (B) Protein phosphatase inhibitors (PPi) partially rescue pairing-induced LTP induction in ZAP-treated slices. Intracellular delivery (via the recording electrode) of either microcystin LR (10 μM, n=4) or nodularin (50 μM, n=7) had no effect on pairing-induced LTP (open circles, similar results were obtained with both compounds and the results were combined). While LTP induction was completely blocked in cells continuously bathed in ACSF containing 50 μM ZAP (filled circles, n=11), significant LTP was induced in ZAP-treated cells when the recording electrode contained microcystin (n=3) or nodularin (n=4) (triangles, similar results were obtained with both compounds and the results were combined, n=7, P<0.01 compared with ZAP alone). The amount of LTP induced in ZAP-treated cells where postsynaptic protein phosphatases were blocked was, however, still significantly less that that seen in control cells (postsynaptic protein phosphatase inhibitors alone, P<0.01). The inset shows EPSPs recorded during baseline and 30 min post-pairing in a control cell (postsynaptic phosphatase inhibitors alone, left set of traces), a ZAP-treated cell recorded with normal electrode solution (middle set of traces), and a ZAP-treated cell where protein phosphatase inhibitors were present in the recording electrode (right set of traces). Scale bars=10 ms and 5 mV. (C) Cumulative probability plot of the results shown in (B) The open symbols show results from control experiments (PPi alone) performed in slices bathed in normal ACSF where the electrode-filling solution contained either microcystin-LR (squares) or nodularin (circles). The filled circles show results from ZAP alone experiments (normal electrode-filling solution) while the filled squares and triangles correspond to experiments where microcystin-LR (squares) or nodularin (triangles) was used to inhibit postsynaptic protein phosphatases in ZAP-treated slices (rescue experiments).
To examine whether blocking postsynaptic protein phosphatases is sufficient to rescue LTP induction in the presence of elevated cGMP levels, we examined the effects of ZAP on pairing-induced LTP in experiments where the membrane impermeant protein phosphatase inhibitors microcystin-LR or nodularin were introduced into the postsynaptic cell via the recording electrode. The effects of microcystin-LR and nodularin on LTP induction in these experiments were the same and the results were therefore combined. As shown in Fig. 7B and C, postsynaptic delivery of protein phosphatase inhibitors had no effect on LTP under control conditions (EPSPs were potentiated to 222±17% of baseline, n=11) but facilitated LTP induction in cells bathed in ACSF containing 50 μM ZAP (EPSPs were potentiated to 163±16% of baseline, n=7, compared with 98±6% of baseline in interleaved experiments where whole-cell recordings from ZAP-treated slices were done using normal electrode-filling solution, n=11, P<0.01). The amount of potentiation induced in the presence of ZAP when postsynaptic protein phosphatases were inhibited was, however, still significantly less than that seen in control cells (postsynaptic protein phosphatase inhibitors alone, P<0.01). Thus, although it does not completely rescue LTP, inhibiting postsynaptic protein phosphatases does enable the induction of significant pairing-induced LTP in ZAP-treated slices.
The induction of LTP by TPS is PKA-independent and ZAP-insensitive in PSD-95 mutant mice
The NMDA receptor scaffolding protein PSD-95 appears to have a crucial role in coupling synaptic NMDA receptors to signaling pathways involved in LTP (Migaud et al., 1998; Komiyama et al., 2002; Beique and Andrade, 2003; Kim et al., 2003; Opazo et al., 2003; Stein et al., 2003; for review see Kim and Sheng, 2004). Previously, we found that the induction of LTP by TPS, which is normally PKA-dependent, is dramatically facilitated in the hippocampal CA1 region of PSD-95 mutant mice (Migaud et al., 1998; Opazo et al., 2003). Thus, we examined whether the role of PKA in LTP might be altered in the hippocampal CA1 region of PSD-95 mutant mice and, if so, whether these changes might be associated with alterations in the ability of increases in cGMP to suppress LTP. In these experiments we first compared the effects of the PKA inhibitor H89 on TPS-induced LTP in hippocampal slices obtained from PSD-95 mutants and wild type littermates. Consistent with previous findings showing that TPS-induced LTP is PKA-dependent (Thomas et al., 1996; Winder et al., 1999), H89 strongly suppressed TPS-induced LTP in wild type slices (Fig. 8A). As expected from previous work (Migaud et al., 1998; Opazo et al., 2003), TPS induced a larger potentiation of synaptic transmission from PSD-95 mutant mice. Remarkably, H89 had no effect on TPS-induced LTP in slices from PSD-95 mutants (Fig. 8B), suggesting that in the absence of PSD-95 TPS induces a PKA-independent form of LTP. Although the molecular basis for this change in PSD-95 mutants is unclear, the fact that TPS-induced LTP is PKA-independent in PSD-95 mutants provides an additional way to test our hypothesis that increases in cGMP inhibit LTP induction by interfering with PKA activation. Specifically, this hypothesis predicts that ZAP-induced increases in cGMP should have no effect on TPS-induced LTP slices obtained from PSD-95 mutant mice. Consistent with this prediction, we found that while bath application of ZAP strongly impaired the induction of LTP in slices from wild type littermates (Fig. 8C) it had no effect on TPS-induced LTP in slices from PSD-95 mutant mice (Fig. 8D).
Increases in cGMP inhibit the induction of LTP by long trains of TPS
An unusual feature of TPS stimulation-induced LTP is that little or no LTP is induced by long trains of TPS containing 300 or more stimulation pulses even though robust LTP can be induce by brief trains containing 75–150 pulses (Thomas et al., 1996, 1998). Interestingly, activation of protein phosphatases appears to be responsible for the failure of long trains of TPS to induce significant LTP (Thomas et al., 1996) and our results suggest that increases in cGMP can inhibit LTP induction by opposing the PKA-dependent down-regulation of protein phosphatase activity needed for TPS-induced LTP. Thus, we examined whether increases in cGMP might have an important role in protein phosphatase-mediated suppression of LTP induction during longer trains of TPS. To test this idea we examined the effects of inhibiting cGMP production with the guanylyl cyclase inhibitor 1H-[1,2,4] oxadiazolo [4,3-a] quinoxalin-1-one (ODQ) on the induction of LTP by TPS. As shown in Fig. 9, bath application of ODQ for 20 min prior to TPS had no effect on the induction of LTP by a short train of TPS (150 pulses). In contrast, while a long train of TPS had little lasting effect on synaptic strength under control conditions (45 min post-TPS fEPSPs were 105±6% of baseline, n=10), it induced significant LTP in slices bathed in ACSF containing ODQ (fEPSPs were potentiated to 166±7% of baseline, n=10, P<0.01 compared with control). Thus, unlike HFS-induced LTP (Boulton et al., 1994; Son et al., 1998; Lu et al., 1999; Monfort et al., 2002, but see Kleppisch et al., 1999), cGMP production is not required for the induction of LTP by TPS. Instead, the results shown in Fig. 9 indicate that activity-dependent increases in cGMP inhibit the induction of LTP by long trains of TPS.
Fig. 9.
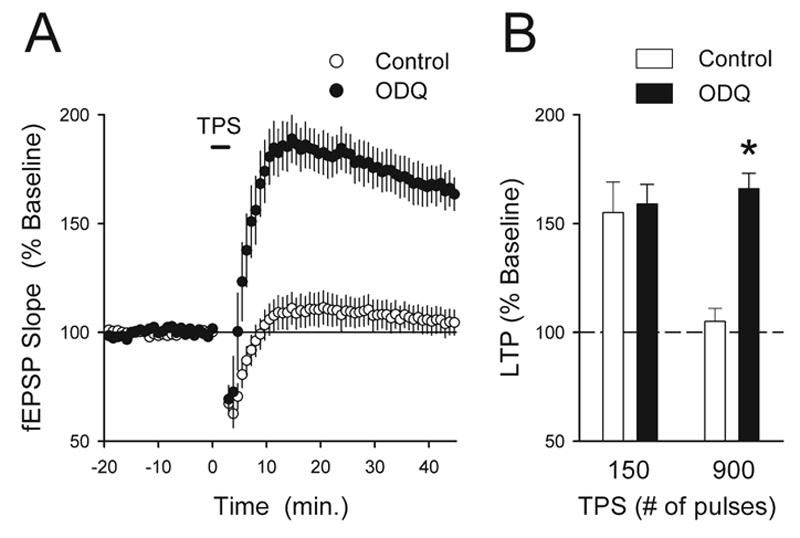
cGMP production inhibits the induction of LTP during long trains of TPS. (A) The guanylyl cyclase inhibitor ODQ enables the induction of LTP by a long train of TPS (900 pulses). While TPS had little lasting effect on synaptic strength in vehicle control experiments (open symbols, n=10), it induced significant LTP in slices pretreated in ACSF containing 10 μM ODQ for 20 min prior to TPS (filled symbols, n=10). (B) Summary of the effects of ODQ on TPS-induced LTP. Blocking guanylyl cyclase had no effect on the induction of LTP by brief trains of TPS (150 pulses, fEPSPs were potentiated to 155±14% of baseline in control experiments and were potentiated to 159±9% of baseline in slices bathed in ACSF containing 10 μM ODQ, n=5 for both) but significantly enhanced the induction of LTP by a long train of TPS (900 pulses, * P<0.01 compared with control).
DISCUSSION
Early studies investigating the role of cGMP signaling in LTP provided evidence for an appealingly simple mechanism through which cGMP might be involved in LTP. Specifically, results from several studies suggested that increases in transmitter release associated with LTP induction were due to a nitric oxide-dependent increase in presynaptic cGMP levels and subsequent activation of PKG (Zhuo et al., 1994a; Arancio et al., 2001). More recent findings, however, indicate that the role of cGMP in hippocampal synaptic plasticity is much more complex. For instance, under some experimental conditions guanylyl cyclase and PKG inhibitors have no effect on LTP (Gage et al., 1997; Kleppisch et al., 1999), suggesting that cGMP signaling through PKG is not required for LTP induction but instead has a modulatory role. Moreover, in addition to its presynaptic effects, PKG may also be involved in postsynaptic signaling events important for LTP, such as membrane insertion of AMPA-type glutamate receptors (Wang et al., 2005) and phosphorylation of the transcriptional regulator CREB (Lu et al., 1999; Lu and Hawkins, 2002). In contrast to these findings, our results indicate that cGMP signaling has a very different role in the induction of LTP by low-frequency patterns of synaptic stimulation. Specifically, we find that increases in cGMP are not required for the induction of LTP by TPS but instead act as an inhibitory constraint on the induction of PKA-dependent forms of LTP by low-frequency trains of synaptic stimulation.
How might increases in cellular levels of cGMP inhibit LTP induction? The effects of SNAP and ZAP on the induction of LTP by the four different stimulation protocols used in our experiments provide a number of important clues regarding the underlying mechanisms. First, the suppression of pairing-induced LTP in ZAP-treated slices indicates that the inhibition of LTP by increases in cGMP is not due to changes in postsynaptic excitability and/or inhibitory synaptic transmission because in these experiments inhibitory synaptic transmission was blocked with picrotoxin and postsynaptic depolarization was directly induced by injecting depolarizing current through the recording electrode. Second, the fact that HFS-induced LTP is not inhibited by ZAP or SNAP suggests that increases in cGMP do not directly alter signaling processes, such as NMDA receptor activation or calcium/calmodulin-dependent kinase II activity, that are required for the induction of LTP by all patterns of synaptic stimulation. Finally, our observation that ZAP and SNAP only inhibit the induction of LTP by patterns of synaptic activation that induce a PKA-dependent early phase of LTP suggests that cGMP may inhibit LTP induction by disrupting cAMP signaling. Consistent with this notion, ZAP has no effect on TPS-induced LTP in hippocampal slices from PSD-95 mutant mice where LTP induction is PKA-independent.
In most cells cGMP signaling involves three main downstream targets: PKG, cyclic nucleotide-gated ion channels, and cGMP-regulated phosphodiesterases (Lucas et al., 2000). It seems unlikely that activation of PKG is responsible for the inhibition of TPS-induced LTP by ZAP-induced increases in cGMP because, as discussed above, PKG activation is thought be a positive modulator of LTP induction. Similarly, activation of cyclic nucleotide-gated ion channels is thought to facilitate, rather than inhibit, LTP induction (Parent et al., 1998) and it is thus unlikely that activation of these channels can account for the inhibition of LTP by ZAP and SNAP. In contrast, activation of the cGMP-stimulated phosphodiesterase PDE2 could readily account for the inhibitory effects of ZAP and SNAP on both LTP and β-adrenergic receptor signaling.
Importantly, the hypothesis that increases in cGMP suppress LTP induction by stimulating phosphodiesterases and inhibiting PKA activation predicts that manipulations that facilitate cAMP signaling should rescue LTP in the presence of ZAP and SNAP. In agreement with this prediction, we found that pharmacological activation of PKA with the phosphodiesterase-resistant cAMP analog 8-Br-cAMP or blocking all phosphodiesterases with IBMX completely rescued TPS-induced LTP in ZAP-treated slices. This hypothesis can also be tested by examining the effects of pharmacological manipulations that act downstream of PKA activation. In the hippocampal CA1 region, PKA activation has a crucial role in LTP induction because it provides a mechanism for inhibiting the activity of protein phosphatases that would otherwise inhibit LTP induction. Thus, pharmacological inhibition of protein phosphatases should obviate the need for PKA activation and render LTP induction insensitive to inhibitors that act via the cAMP/PKA pathway. Consistent with this prediction we found that three different inhibitors of protein phosphatases 1 and 2A significantly rescued LTP in ZAP-treated slices. These findings indicate that increases in cGMP inhibit LTP by opposing activation of the PKA-dependent, gate-like mechanism that normally enables LTP induction by inhibiting protein phosphatases. Moreover, the rescue of pairing-induced LTP by postsynaptic infusion of membrane-impermeant protein phosphatase inhibitors suggests that the inhibition of LTP by increases in cGMP is due, at least in part, to effects on postsynaptic PKA signaling pathways. Although our results are consistent with the notion that increases in cGMP inhibit LTP by stimulating the activity of phosphodiesterases, we have not directly tested whether PKG and/or cyclic nucleotide-gated ion channels might also be involved. Thus, additional experiments will be required to determine whether activation of PKG or cyclic-nucleotide-gated ion channels might also contribute to inhibitory effects of cGMP on low-frequency stimulation-induced LTP.
In our experiments we observed that 8-Br-cAMP, IBMX, and protein phosphatase inhibitors completely rescued TPS-induced LTP in ZAP-treated slices but only partially rescued the induction of LTP by low-frequency presynaptic fiber stimulation paired with postsynaptic depolarization. One possible explanation for this difference is that the potentiation induced by the pairing protocol used in our experiments is due to both PKA-dependent and independent mechanisms. If both components are inhibited by increases in cGMP, then manipulations that restore cAMP signaling would be expected to only rescue the PKA-dependent component and thus only partially rescue LTP. Consistent with the idea that the pairing protocol used in our experiments induces LTP through both PKA-dependent and independent mechanisms we found that the PKA inhibitors H89 and PKI(6–22)-amide only partially inhibit pairing-induced LTP. It is difficult, however, to reconcile the notion that increases in cGMP can inhibit LTP through PKA-independent mechanisms with the fact that ZAP and SNAP have no effect on HFS-induced LTP. Another possibility is that increases in cGMP not only inhibit the PKA-dependent component of pairing-induced LTP but also facilitate the induction of a phosphodiesterase-independent, presynaptic form of LTD that partially masks LTP. If so, then neither bath application of IBMX nor postsynaptic infusion of 8-Br-cAMP or membrane impermeant protein phosphatase inhibitors would be able to prevent this depression and thus completely rescue pairing-induced LTP. Consistent with this notion, increases in cGMP can induce a PKG-dependent, presynaptic form of LTD in the hippocampal CA1 region (Santschi et al., 1999; Reyes-Harde et al., 1999; Stanton et al., 2003) and can facilitate the induction of LTD by low-frequency presynaptic fiber stimulation (Zhuo et al., 1994b; Gage et al., 1997).
Numerous findings support the notion that PKA activation acts as a kind of biochemical gate that enables LTP induction by suppressing protein phosphatase activity (see Winder and Sweatt, 2001 for review). Although activation of the PKA gate has a pivotal role in the induction of LTP by some patterns of synaptic stimulation, it also provides a means by which modulatory neurotransmitters that increase cAMP production can facilitate LTP induction (Thomas et al., 1996; Winder et al., 1999; Brown et al., 2000). Our results demonstrate, however, that the PKA gate is not a unidirectional mechanism only engaged to facilitate LTP induction. Instead, by opposing cAMP signaling, increases in cGMP can close the PKA gate, thereby allowing protein phosphatases to remain active and inhibit LTP induction (Fig. 10). Activation of cGMP-stimulated phosphodiesterases may thus act as a kind of “plasticity filter” that strongly suppresses the ability of low-frequency patterns of synaptic activity to induce LTP while allowing stronger, high-frequency bursts of presynaptic activity to induce LTP through PKA-independent mechanisms. Indeed, along with the facilitatory, PKG-mediated effects of cGMP on HFS-induced LTP (Zhuo et al., 1994a; Lu et al., 1999; Arancio et al., 2001; Monfort et al., 2002; Wang et al., 2005), the suppression of low-frequency stimulation-induced LTP by cGMP-activated phosphodiesterases indicates that cGMP can engage multiple downstream mechanisms that convert synapses into an altered state where LTP induction is strongly biased toward higher frequency patterns of synaptic activity.
Fig. 10.
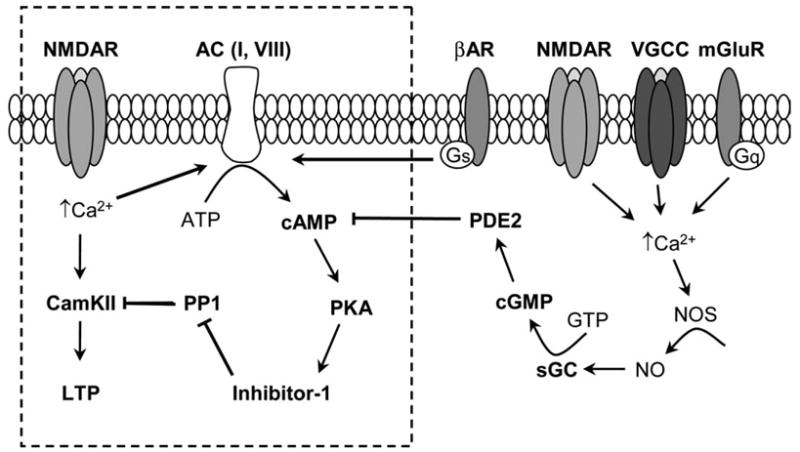
Increases in cGMP inhibit LTP by closing the PKA gate. As depicted by the signaling pathways outlined in the box, increases in intracellular Ca2+ due to influx through NMDA type glutamate receptors (NMDAR) induce LTP by not only activating calcium/calmodulin-dependent kinase II (CamKII) but also by activating calcium/calmodulin-dependent forms of the enzyme adenylyl cyclase (AC), such as AC I and AC VIII. The subsequent increase in cAMP and activation of PKA lead to phosphorylation of inhibitor 1 which, when phosphorylated, inhibits protein phosphatase 1 (PP1) and prevents it from opposing the activity of CamKII and other protein kinases needed for LTP induction. Neurotransmitters acting through receptors coupled to Gs type G proteins, such as β-adrenergic receptors (βAR), can facilitate LTP induction by stimulating AC and opening the PKA gate. Activation of soluble guanylyl cyclases (sGC) and subsequent increases in cGMP can inhibit LTP induction by activating the cGMP-stimulated phosphodiesterase PDE2, facilitating cAMP degradation, and thus inhibiting PKA activation. As shown on the right side of the figure, possible upstream signals that might lead to sGC activation and closing of the PKA gate include nitric oxide synthase (NOS) activation by increases in intracellular Ca2+ due to activation of NMDARs, voltage-gated calcium channels (VGCC) and/or metabotropic glutamate receptors (mGluRs).
A limitation of our experiments is that direct effects of SNAP and ZAP on basal synaptic transmission necessitated the use of prolonged applications of these compounds. Moreover, the magnitude of the increase in cGMP produced by SNAP and ZAP under out experimental conditions is unknown. Because of this we cannot rule out the possibility that the cGMP-induced closing of the PKA gate occurs only in response to prolonged, and perhaps unphysiological, elevations in cGMP. We found, however, that while blocking cGMP production with the guanylyl cyclase inhibitor ODQ had no effect on the induction of LTP by short trains of TPS it strongly enhanced the induction of LTP by long trains of TPS. Thus, activity-dependent increases in cGMP levels appear to have a potent, inhibitory effect on the induction of LTP by some patterns of synaptic activity. Indeed, a recent study has found that selective inhibition of the cGMP-activated phosphodiesterase PDE2 enhances learning on a number of different behavioral tasks (Boess et al., 2004), suggesting that cGMP-activated phosphodiesterases have an important, inhibitory role in the neuronal mechanisms underlying memory formation. Our results reveal a molecular mechanism that could underlie this role of cGMP in memory formation and suggest that cross-talk between cAMP and cGMP signaling pathways mediated by cGMP-stimulated phosphodiesterases is a key, modulatory component of the network of postsynaptic signaling pathways regulating LTP induction.
Acknowledgments
We are grateful to Jary Delgado, Ann Fink, and members of the UCLA Learning and Memory Project for helpful comments. T.J.O. is a member of the UCLA Brain Research Institute. This work was supported by grants from the NIMH (T.J.O.) and the Wellcome Trust (S.G.N.G).
Abbreviations
- ACSF
artificial cerebrospinal fluid
- cAMP
cyclic AMP
- cGMP
cyclic guanosine 3′, 5′monophosphate
- DMSO
dimethyl-sulfoxide
- DPCPX
1, 3-dipropyl-8-cyclopentyl xanthine
- fEPSP
field excitatory postsynaptic potential
- HFS
high-frequency stimulation
- H89
N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinoline sulfonamide
- IBMX
3-isobutyl-1-methylxanthine
- LTD
long-term depression
- LTP
long-term potentiation
- ODQ
1H-[1, 2, 4] oxadiazolo [4, 3-a] quinoxalin-1-one
- PKA
protein kinase A
- PKG
protein kinase G
- SNAP
S-nitroso-D, L-penicillamine
- TPS
theta-pulse stimulation
- ZAP
zaprinast
References
- Andreeva SG, Dikkes P, Epstein PM, Rosenberg PA. Expression of cGMP-specific phosphodiesterase 9A mRNA in the rat brain. J Neurosci. 2001;21:9068–9076. doi: 10.1523/JNEUROSCI.21-22-09068.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio O, Antonova I, Gambaryan S, Lohmann SM, Wood JS, Lawrence DS, Hawkins RD. Presynaptic role of cGMP-dependent protein kinase during long-lasting potentiation. J Neurosci. 2001;21:143–149. doi: 10.1523/JNEUROSCI.21-01-00143.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio O, Kandel ER, Hawkins RD. Activity-dependent long-term enhancement of transmitter release by presynaptic 3′,5′cyclic GMP in cultured hippocampal neurons. Nature. 1995;376:74–80. doi: 10.1038/376074a0. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci U S A. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beique JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol (Lond) 2003;546:859–867. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer R, Wong T, Nouranifar R, Iyengar R, Landau E. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GPL, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CamKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- Boess FG, Hendrix M, van der Staay FJ, Erb C, Schreiber R, van Staveren W, de Vente J, Pickaerts J, Blokland A, Koenig G. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology. 2004;47:1081–1082. doi: 10.1016/j.neuropharm.2004.07.040. [DOI] [PubMed] [Google Scholar]
- Boulton CL, Irving AJ, Southam E, Potier B, Garthwaite J, Collingridge GL. The nitric oxide-cyclic GMP pathway and synaptic depression in rat hippocampal slices. Eur J Neurosci. 1994;6:1528–1535. doi: 10.1111/j.1460-9568.1994.tb00543.x. [DOI] [PubMed] [Google Scholar]
- Broome MR, Collingridge GL, Irving AJ. Activation of the NO-cGMP signaling pathway depresses hippocampal synaptic transmission through an adenosine receptor-dependent mechanism. Neuropharmacology. 1994;33:1511–1513. doi: 10.1016/0028-3908(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Brown GP, Blitzer RD, Connor JH, Wong T, Shenolikar S, Iyengar R, Landau EM. Long-term potentiation induced by theta frequency stimulation regulated by a protein phosphatase-1-operated gate. J Neurosci. 2000;20:7880–7887. doi: 10.1523/JNEUROSCI.20-21-07880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell BD, Sahley CL. Learning in simple systems. Curr Opin Neurobiol. 2001;11:757–764. doi: 10.1016/s0959-4388(01)00281-1. [DOI] [PubMed] [Google Scholar]
- Doerner D, Alger BE. Cyclic GMP depresses hippocampal Ca2+ current through a mechanism independent of cGMP-dependent protein kinase. Neuron. 1988;1:693–699. doi: 10.1016/0896-6273(88)90168-7. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Taylor M, Heginbotham LR, Proctor WR. Long-term increases in excitability in the CA1 region of rat hippocampus induced by beta-adrenergic stimulation: possible mediation by cAMP. J Neurosci. 1992;12:506–517. doi: 10.1523/JNEUROSCI.12-02-00506.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallahi N, Broad RJ, Jin S, Fredholm BB. Release of adenosine from rat hippocampal slices by nitric oxide donors. J Neurochem. 1996;67:186–193. doi: 10.1046/j.1471-4159.1996.67010186.x. [DOI] [PubMed] [Google Scholar]
- Field L, Dilts RV, Ravichandran R, Lenhert PG, Carnahan GE. An unusually stable thionitrite from N-acetyl-D,L-penicillamine: X-ray crystal and molecular structure of 2-(acetylamino)-2-carboxy-1,1,-dimethylethyl thionitrite. J Chem Soc Chem Comm. 1978:210–211. [Google Scholar]
- Frey U, Huang Y-Y, Kandel ER. Effects of cAMP simulate a late phase of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Gage AT, Reyes M, Stanton PK. Nitric-oxide-guanylyl-cyclase-dependent and independent components of multiple forms of long-term synaptic depression. Hippocampus. 1997;7:286–295. doi: 10.1002/(SICI)1098-1063(1997)7:3<286::AID-HIPO4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Heginbotham LR, Dunwiddie TV. Long-term increases in evoked population spike in the CA1 region of rat hippocampus induced by beta-adrenergic receptor activation. J Neurosci. 1991;11:2519–2527. doi: 10.1523/JNEUROSCI.11-08-02519.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuki H, Izumi Y, Zorumski CF. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol. 1997;77:3013–3020. doi: 10.1152/jn.1997.77.6.3013. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee HK, Takamiya K, Huganir RL. The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci. 2003;23:1119–1124. doi: 10.1523/JNEUROSCI.23-04-01119.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Pfeifer A, Klatt P, Ruth P, Montkowski A, Fassler R, Hofmann F. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J Neurosci. 1999;19:48–55. doi: 10.1523/JNEUROSCI.19-01-00048.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Nomti J, Strathdee DJC, O’Carroll CM, Martin SJ, Morris RGM, O’Dell TJ, Grant SGN. SynGAP regulates ERK/MAPK signaling, synaptic plasticity and learning in the complex with PSD-95 and NMDA receptor. J Neurosci. 2002;22:9721–9732. doi: 10.1523/JNEUROSCI.22-22-09721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-W, Min MY, Chiu TH, Yang HW. Enhancement of associative long-term potentiation by activation of β-adrenergic receptors at CA1 synapses in rat hippocampal slices. J Neurosci. 2003;23:4173–4181. doi: 10.1523/JNEUROSCI.23-10-04173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;23:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YF, Hawkins RD. Ryanodine receptors contribute to cGMP-induced late-phase LTP and CREB phosphorylation in the hippocampus. J Neurophysiol. 2002;88:1270–1278. doi: 10.1152/jn.2002.88.3.1270. [DOI] [PubMed] [Google Scholar]
- Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- Makhinson M, Chotiner JK, Watson JB, O’Dell TJ. Adenylyl cyclase activation modulates activity-dependent changes in synaptic strength and Ca2+/calmodulin-dependent kinase II auto-phosphorylation. J Neurosci. 1999;19:2500–2510. doi: 10.1523/JNEUROSCI.19-07-02500.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehats C, Andersen CB, Filopanti M, Jin SLC, Conti M. Cyclic neucleotide phosphodiesterases and their role in endocrine cell signaling. Trends Edocrinol Metab. 2002;13:29–35. doi: 10.1016/s1043-2760(01)00523-9. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RGM, Morrison JH, O’Dell TJ, Grant SGN. Enhanced long term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Monfort P, Munoz MD, Kosenko E, Felipo V. Long-term potentiation in hippocampus involves sequential activation of soluble guanylate cyclase, cGMP-dependent protein kinase, and cGMP-degrading phophodiesterase. J Neurosci. 2002;22:10116–10122. doi: 10.1523/JNEUROSCI.22-23-10116.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SGN, O’Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 1996;16:7478–7486. doi: 10.1523/JNEUROSCI.16-23-07478.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova N, Lisman JE. Postsynaptic application of a cAMP analogue reverses long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2002;87:3018–3032. doi: 10.1152/jn.2002.87.6.3018. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE. Inhibition of the cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 2000;20:4446–4451. doi: 10.1523/JNEUROSCI.20-12-04446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A, Munger SD, Reed RR, Linden DJ, Ronnett GV. Synaptic transmission and hippocampal long-term potentiation in olfactory cyclic nucleotide-gated channel type I null mouse. J Neurophysiol. 1998;79:3295–3301. doi: 10.1152/jn.1998.79.6.3295. [DOI] [PubMed] [Google Scholar]
- Reinhardt RR, Bondy CA. Differential cellular pattern of gene expression for two distinct cGMP-inhibited cyclic nucleotide phosphodiesterases in developing and mature rat brain. Neuroscience. 1996;72:567–578. doi: 10.1016/0306-4522(95)00520-x. [DOI] [PubMed] [Google Scholar]
- Repaske DR, Corbin JG, Goy MF. A cyclic cGMP-stimulated phosphodiesterase gene is highly expressed in the limbic system of the rat brain. Neuroscience. 1993;56:673–686. doi: 10.1016/0306-4522(93)90364-l. [DOI] [PubMed] [Google Scholar]
- Reyes-Harde M, Potter BVL, Galione A, Stanton PK. Induction of hippocampal LTD requires nitric-oxide-stimulated PKG activity and Ca2+ release from cyclic ADP-ribose-sensitive stores. J Neurophysiol. 1999;82:1569 –1576. doi: 10.1152/jn.1999.82.3.1569. [DOI] [PubMed] [Google Scholar]
- Santschi L, Reyes-Harde M, Stanton PK. Chemically-induced, activity-independent LTD elicited by simultaneous activation of PKG and inhibition of PKA. J Neurophysiol. 1999;82:1577–1589. doi: 10.1152/jn.1999.82.3.1577. [DOI] [PubMed] [Google Scholar]
- Saransaari P, Oja SS. Involvement of nitric oxide in adenosine release in the developing and adult mouse hippocampus. Neurochem Res. 2004;29:219–225. doi: 10.1023/b:nere.0000010451.81201.f5. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Varga AW, Sweatt JD, Swank M. Protein kinase signal transduction cascades in mammalian associative conditioning. Neuroscientist. 2002;8:122–131. doi: 10.1177/107385840200800208. [DOI] [PubMed] [Google Scholar]
- Soderling SH, Bayuga SJ, Beavo JA. Identification and characterization of a novel family of cyclic nucleotide phosphodiesterases. J Biol Chem. 1998;273:15553–15558. doi: 10.1074/jbc.273.25.15553. [DOI] [PubMed] [Google Scholar]
- Son H, Lu YF, Zhuo M, Arancio O, Kandel ER, Hawkins RD. The specific role of cGMP in hippocampal LTP. Learn Mem. 1998;5:231–245. [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Bailey CP, Kyrozis A, Raginov I, Laube G, Veh RW, Nguyen CQ, Müller W. Long-term depression of pre-synaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J Neurosci. 2003;23:5936–5944. doi: 10.1523/JNEUROSCI.23-13-05936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DRC, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makhinson M, O’Dell TJ. Activity-dependent β-adrenergic modulation of low frequency stimulation-induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M, O’Dell TJ. Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci. 1998;18:7178–7126. doi: 10.1523/JNEUROSCI.18-18-07118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Staveren WCG, Steinbusch HWM, Ittersum MMV, Repaske DR, Goy MF, Kotera J, Omori K, Beavo JA, De Vente J. mRNA expression patterns of the cGMP-hydrolyzing phosphodiesterases types 2, 5, and 9 during development of the rat brain. J Comp Neurol. 2003;467:566–580. doi: 10.1002/cne.10955. [DOI] [PubMed] [Google Scholar]
- Wang HG, Lu FM, Jin I, Udo H, Kandel ER, de Vente J, Walter U, Lohmann SM, Hawkins RD, Antonova I. Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron. 2005;45:389–403. doi: 10.1016/j.neuron.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–727. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- Woo NH, Abel T, Nguyen PV. Genetic and pharmacological demonstration of a role for cyclic AMP-dependent protein kinase-mediated suppression of protein phosphatases in gating the expression of late LTP. Eur J Neurosci. 2002;16:1871–1876. doi: 10.1046/j.1460-9568.2002.02260.x. [DOI] [PubMed] [Google Scholar]
- Wu J, Wang Y, Rowan MJ, Anwyl R. Evidence for involvement of the cGMP-protein kinase G signaling system in the induction of long-term depression, but not long-term potentiation, in the dentate gyrus in vitro. J Neurosci. 1998;18:3589–3596. doi: 10.1523/JNEUROSCI.18-10-03589.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Yinghe H, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994a;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Kandel ER, Hawkins RD. Nitric oxide and cGMP can produce either synaptic depression or potentiation depending on frequency of presynaptic stimulation in the hippocampus. Neuro-report. 1994b;5:1033–1036. doi: 10.1097/00001756-199405000-00004. [DOI] [PubMed] [Google Scholar]