

Abstract
Neonatal administration of the synthetic glucocorticoid, dexamethasone (DEX) retards brain growth, alters adult behaviors and induces cell death in the rat brain, thereby implicating glucocorticoids as developmentally neuroendangering compounds. Glucocorticoids also increase expression of pro-apoptotic Bcl-2 family members and exacerbate expression of hypoxic responsive genes. Bnip3 is a pro-apoptotic Bcl-2 family member that is upregulated in response to hypoxia. In these studies, we investigated the interactions of glucocorticoid receptor and hypoxia in the regulation of Bnip3 mRNA in cortical neurons. Using quantitative real time RT-PCR, we found that DEX treatment of postnatal day 4–6 rat pups caused a significant increase in Bnip3 mRNA expression compared to vehicle controls. A significant increase in Bnip3 mRNA was also measured in primary cortical neurons 72 hours after treatment with RU28362, a glucocorticoid receptor selective agonist. In primary cortical neurons, hypoxia increased Bnip3 mRNA expression and this was exacerbated with RU28362 treatment. To elucidate the mechanism of glucocorticoid and hypoxia mediated regulation of Bnip3 transcription, a Bnip3 promoter - luciferase reporter construct was utilized in primary cortical neurons. Upregulation of the Bnip3 promoter was mediated by a single glucocorticoid response element and a hypoxic response element. Bnip3 overexpression in primary cortical neurons significantly increased cell death, which is dependent on the Bnip3 transmembrane domain. However, despite the increased expression of Bnip3 following glucocorticoid and hypoxia treatment, corresponding decreases in cell survival were minimal. These studies identify a novel pathway in the developing cortex through which glucocorticoids may enhance a metabolic insult, such as hypoxia.
Keywords: Bcl-2, glucocorticoid receptor, dexamethasone, primary cortical neurons, postnatal, apoptosis
Corticosterone, the endogenous glucocorticoid secreted by the rat adrenal gland, binds with high affinity to two different receptors, the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR) (Reagan and McEwen, 1997). MR activation is associated with a neuroprotective phenotype (Almeida et al., 2000), whereas GR activation is implicated in the induction of an endangered neural phenotype (Sapolsky et al., 1986, Reagan and McEwen, 1997). For example, in the adult, GR activation by the synthetic glucocorticoid dexamethasone (DEX) reduces anterior cingulate cortex volume through dendritic atrophy (Cerqueira et al., 2005a, Cerqueira et al., 2005b).
Elevated glucocorticoid levels also endanger cells in the neonatal brain as evidenced by decreased hippocampal volume (Coe et al., 2003) and retarded overall brain growth and neurological development (Flagel et al., 2002). Postnatal DEX treatment decreases both the number and size of cortical neurons (Kreider et al., 2006), which persists into adulthood as these rats show altered social behavior and learning and memory deficits (Kamphuis et al., 2004, Neal et al., 2004).
In juvenile rats, GR activation increases the incidence of apoptosis in the hippocampus and striatum (Hassan et al., 1996, Haynes et al., 2001). This is consistent with their ability to regulate Bcl-2 family member expression. In the juvenile rat hippocampus, DEX downregulates the anti-apoptotic genes Bcl-2 and Bcl-XL; while upregulating the pro-apoptotic gene Bax (Almeida et al., 2000). In contrast, in the neonatal rat hippocampus, DEX does not alter Bcl-2 or Bax (Tan et al., 2002). Taken together, these data indicate that glucocorticoid regulation of apoptosis is mediated through a novel mechanism in the neonatal rat brain.
Pro and anti-apoptotic members of the Bcl-2 family initiate apoptotic events. Bnip3 is a unique pro-apoptotic member of the Bcl-2 homology domain 3 (BH3) subfamily (Boyd et al., 1994, Yasuda et al., 1998) in that Bnip3 induced cell death is independent of the BH3 domain (Ray et al., 2000), and is not prevented by Bcl-2 and Bcl-XL overexpression (Ray et al., 2000). Bnip3 gene regulation has been predominantly investigated following hypoxia. During hypoxic events, hypoxia inducible factor-1 (HIF-1) mediates the transcription of Bnip3 via interaction with a hypoxic response element (HRE) in the promoter region (Bruick, 2000, Sowter et al., 2001). Furthermore, hypoxia induces Bnip3 expression in adult rat hippocampus, cortex and striatum which correlates with increases in cell death (Schmidt-Kastner et al., 2004, Althaus et al., 2006). Notwithstanding, Bnip3 expression in response to hypoxia, its regulation by mechanisms independent of hypoxia, or interactions between the two have not been investigated in the developing brain. Consequently, the following studies examined glucocorticoid regulation of Bnip3 during both normoxic and hypoxic conditions in cortical neurons.
Experimental Procedures
Animals
Timed pregnant female Sprague-Dawley rats were purchased from Charles River Laboratories (Wilmington, MA, USA) and housed at Colorado State University’s laboratory animal research facility. For in vivo experiments that used postnatal offspring, pregnant dams were monitored on a daily basis in order to record the litter’s birth date and time. The day of birth was considered as postnatal day (PND) 0. Upon birth all litters were culled into groups of 10 pups, 5 males and 5 females, and returned to the dam. Male and female neonates were subcutaneously injected with a daily dose of DEX (0.2mg/kg in 100μl safflower oil) from PND 4 through 6. Controls received vehicle at the same injection regimen. Twenty four hours after receiving the third injection (PND7) animals were halothane anesthetized and killed. For in vitro studies using primary cortical neurons, pregnant dams were halothane anesthetized on either gestational day 17 or 18 and the rat fetuses were delivered by cesarean section. The dam was subsequently sacrificed by decapitation. All animal protocols were previously approved by the Colorado State University Animal Care and Use Committee and carried out in accordance with the National Institutes of Health and Institutional Animal Care and Use Guidelines.
Primary Cortical Neurons
The primary cortical neurons were harvested from embryonic day 17–18 rat fetuses using protocols modified from Banker and Cowen, (1977) and Brewer et al., (1993). During the dissection, brain tissue was kept in a CMF Ringers-glucose solution (0.155M NaCl, 5.0mM KCl, 10.0mM Hepes, 11.0mM D-glucose) on ice. Dissected cortical tissue was incubated at room temperature in a solution consisting of 50% CMF Ringers-glucose plus 50% trypsin (62,000units/ml) for 15 minutes. Cortical tissue was washed once with Dulbecco’s Modified Eagles Medium (Invitrogen, Carlsbad, CA, USA [DMEM]) containing 15% Fetal Bovine Serum then dissociate by titration in the same media. Cortical neurons were plated in Neurobasal medium without phenol red (Invitrogen, Carlsbad, CA, USA) supplemented with 1.0x B27 (Invitrogen, Carlsbad, CA, USA), 100μg/ml penicillin-streptomycin (Invitrogen, Carlsbad, CA, USA), 0.5mM L-glutamine (Mediatach, Inc. Herndon, VA, USA) and 0.025mM L-glutamic acid (Sigma-Aldrich Co. St. Louis, MO, USA). Primary cortical neurons were plated at a density of 5x105 cells/9.4cm2 for RNA isolation studies, 3x105 cells/1.9cm2 for MTT, lactate dehydrogenase (LDH), and Bnip3 promoter studies, and .3x105 cells/0.3cm2 for Bnip3 expression plasmid studies. All cultures were maintained for 3 days in vitro (DIV) in plating media. Experiments were begun after 4–5 DIV and concluded by 8 DIV so that results were from primary cortical neurons of an immature phenotype.
Experiments that involve RNA isolation and cell death as a result of glucocorticoid and hypoxia treatment were conducted in cells grown in Neurobasal media without phenol red, and supplemented with 0.1x B27, 100μg/ml penicillin-streptomycin and 0.5mM L-glutamine. Transfection studies using the Bnip3 promoter constructs were conducted in cells grown in the above media minus penicillin-streptomycin. The B27 was diluted in order to obtain a final concentration of 2.8nM corticosterone in the treatment media. Primary cortical neurons treated with glucocorticoids received either vehicle (0.005% of 95%EtOH in the previously described media) or doses of the serially diluted GR agonist RU28362 (0.1, 1.0, 5.0 or 10.0nM; gift from Roussel-UCLAF, Romainville, France). Throughout the experiment cultures were maintained at 37°C in ambient air infused with 5% CO2, or in Modulator Incubator Chambers (Billups-Rothenberg Inc. Del Mar, CA, USA) filled with 20% O2, 5% CO2, N2 balanced (normoxic) or 1% O2, 5% CO2, N2 balanced (hypoxic) for either 48 or 72 hours. Transfection of primary cortical neurons with the Bnip3 and Bnip3ΔTM expression plasmids were conducted in Neurobasal medium minus phenol red, supplemented with 1x B27, and 0.5mM L-glutamine.
Bnip3 cDNA and expression plasmids
A Bnip3 cDNA plasmid was designed for use in in situ hybridization (ISH) studies and for generating the Bnip3 cDNA standards for real time RT-PCR. The Bnip3 cDNA plasmid was designed as previously described by Sandau and Handa, 2006. The Bnip3 and Bnip3ΔTM expression plasmids were kindly provided by Dr. Don Dubik (Manitoba Institute of Cell Research, Manitoba, CA). The expression plasmids were synthesized as previously described (Chen et al., 1997). The pcDNA 3.0 (Invitrogen, Carlsbad, CA) expression plasmid was transfected as a negative control.
Promoter Reporter Plasmids
The Bnip3 and mutHRE promoter luciferase reporter plasmids were kindly provided by Dr. Richard Bruick (University of Texas Southwestern Medical Center, Dallas, TX, USA). The promoter/reporter gene plasmids were generated as previously described (Bruick, 2000). The Bnip3 and mutHRE promoters were inserted into the pGL3-Basic plasmid (Promega Corp. Madison, WI, USA). The Bnip3 promoter luciferase reporter plasmid was used to generate the GRE1 (GRE1-luc), GRE2 (GRE2-luc), GRE3 (GRE3-luc) and Bnip3ΔGRE2 promoter luciferase reporter plasmids. The location of the three putative GREs within the Bnip3 promoter (GenBank accession nos. AF283504) are −528 to −514 (GRE1), −367 to −353 (GRE2) and −211 to −197 (GRE3). To generate the GRE1-luc and GRE 2-luc plasmids, forward and reverse primer pairs were designed to individually isolate the Bnip3 promoter regions containing the putative GRE1 and GRE2 sites. The forward (5′ CAC AGG TAC CGC AGG AGG AGG TCC CCA ACC C 3′) and reverse (5′ CAC AGC TCA GCG AGC AAC ACT GAG GCG CTA GG 3′) primer pairs for amplifying GRE1 are at positions −500 and −455. The forward (5′ CAC AGG TAC CGC TCA GTG TTG CTC GCA CCT CG 3′) and reverse (3′ CAC AGC TCA GCG GGG TGG AGC CTG GTT GG 3′) primer pairs for amplifying GRE2 are at position −499 to −263. The forward primers include a 5′ KpnI restriction site; while the reverse primers have a 5′ BlpI restriction site. PCR amplification of GRE1 and GRE2 was conducted using the Bnip3 promoter as template. Amplified PCR product was digested with the restriction enzymes KpnI and BlpI. Double digest with KpnI and BlpI was also performed on the full length Bnip3 promoter to excise the three putative GREs. The size of the GRE1 and GRE2 cDNAs and digested plasmid was confirmed by 1% agarose gel electrophoresis. GRE1 and GRE2 were subsequently directional cloned into the digested Bnip3 plasmid using the Rapid DNA Ligation Kit (Roche Inst. Indianapolis, IN, USA). The GRE3-luc plasmid was generated by double digest of the full length Bnip3 promoter luciferase reporter plasmid with BstXI and KpnI to excise the promoter region containing the putative GRE1 and GRE2 sites. The digested plasmid was blunt end ligated with the Rapid DNA Ligation Kit. The Bnip3ΔGRE2-luc plasmid was generated by site directed mutagenesis using the QuikChangeII XL Site-Directed Mutagenesis Kit (Stratagene,La Jolla, CA, USA) per the manufacturer’s specifications. Forward (5′ CGT GCA GGT CCC GGC TAG CCT CAA GG 3′) and reverse (5′ CCT GAG GCT AGC CGG GAC CTG CAC G 3′) primers were designed to eliminate GRE2 within the Bnip3 promoter without introducing nucleotide mutations. PCR amplification of the Bnip3ΔGRE2 plasmid was conducted using the Bnip3 promoter luciferase reporter plasmid as template. Sequencing reactions were performed with the GRE1-luc, GRE2-luc, GRE3-luc and Bnip3ΔGRE2-luc plasmids to confirm the cDNA sequence for each plasmid.
In Situ Hybridization
To examine the localization of Bnip3 mRNA in brain, we performed ISH on paraformaldehyde fixed neonatal rat brain tissue using a Bnip3 35S-labeled cRNA probe. The Bnip3 cDNA containing plasmid was linearized with either Not1 (antisense) or Kpn1 (sense). 35S-UTP (1000 ci/mmole) radiolabeled sense and antisense Bnip3 cRNA probes were transcribed in vitro using either T7 or SP6 RNA polymerase, respectively. The sense and antisense probes were used for ISH.
Brain tissue was collected from rat pups that were halothane anesthetized and subsequently perfused via the left ventricle initially with 0.9% saline plus heparin, followed by 4% neutral buffered paraformaldehyde. Brains were harvested and placed in 30% sucrose phosphate buffered saline until the brains were saturated. 25μm coronal sections were taken with a Leitz 1720 Digital Kryostat (VWR Scientific, West Chester, PA, USA). ISH was subsequently performed with the Bnip3 cRNA probe on the brain sections as previously described (Sandau and Handa, 2006). Radiolabeled tissues were exposed to Kodak Biomax MR Autoradiographic Film (Eastman Kodak Company, Rochester, NY, USA) for 3 days.
RNA Isolation for Quantitative Real Time RT-PCR
Brain tissue utilized for real time RT-PCR was collected from rat pups that were halothane anesthetized and subsequently sacrificed by decapitation. Brains were harvested and immediately frozen in −20°C 3-methylbutane. The anterior and posterior cingulate cortices and hippocampus were dissected from frozen 150μm coronal sections. Total RNA was also isolated from primary cortical neurons for real time RT-PCR. Prior to RNA isolation treatment media was removed and the cortical neurons were washed once with PBS. Cortical neurons were collected into PBS by manually removing the cells from the culture dish with a rubber policeman. PBS was removed by centrifugation at 2000rpm for 2 minutes. The neurons were resuspended then homogenized in GIT extraction buffer (4M guanidinium thiocyanate, 25mM sodium citrate, 0.5% N-laurel sarcosine, 0.1M β-mercaptoethanol). Total RNA was isolated from the rat brain tissue and primary cortical neurons using the protocols of Chomczynski and Sacchi, (1987). Concentration of total RNA was determined using spectrophotometry (O.D. 260/280).
Quantitative Real Time RT-PCR
0.5μg of total RNA was reverse transcribed with MMLV reverse transcriptase and oligoDT primers (Invitrogen, Carlsbad, CA, USA). The concentration of reverse transcribed cDNA was measured in the primary cortical neuron studies using Oligreen ssDNA Quantitation Reagent and Kit (Molecular Probes, Inc. Eugene, OR, USA). Quantification of Bnip3 mRNA expression was determined using real time RT-PCR with the LightCycler 2.0 system (Roche Inst. Indianapolis, IN, USA). Real time RT-PCR reactions were performed on the cDNA samples with the Bnip3 primers previously described. PCR reactions included 3mM MgCl2, 100mM Tris-Cl, 0.5U Taq polymerase, 2μl of 10X stock of Faststart SYBR green I (Roche Inst. Indianapolis, IN, USA) and 0.5 μM of forward primer and reverse Bnip3 primer. The initial melting step for the reaction was 92°C for 10 minutes followed by 45 cycles of 95°C (melting) for 1 second, 62°C (annealing) for 5 seconds and 72°C (elongation) for 15 seconds. Samples were assayed alongside a Bnip3 cDNA standard curve for determination of the absolute concentration of the experimental Bnip3 mRNAs. Bnip3 cDNA was used to generate the standard curve. The Bnip3 cDNA used was obtained by digesting the previously described Bnip3 cDNA plasmid with BamHI. The concentration of the digested Bnip3 cDNA was calculated from an absorbance reading at wavelength 260 (OD260) using a Beckman D6530 Spectrophotometer (Beckman Coulter, Inc., Fullerton, CA, USA). The cDNAs were diluted to a stock concentration of 1ng/ml then subsequently serially diluted from 1ng/ml to 10fg/ml in PCR grade sterile water (Roche Inst. Indianapolis, IN) to generate six working standards. The absolute concentration of unknown Bnip3 mRNA was determined by the LightCycler Data Analysis Software 4.0 (Roche Inst. Indianapolis, IN, USA). Studies measuring Bnip3 mRNA in brain tissue were reported as an absolute concentration (pg/μl of cDNA); while Bnip3 mRNA levels in primary cortical neurons were reported as pg of Bnip3/ng of cDNA. In all experiments, samples that included water in place of template were used as negative controls.
Transfection of Primary Cortical Neurons
Primary cortical neurons were dual transfected with either Bnip3, Bnip3ΔTM, or the empty pcDNA expression plasmids in conjunction with a β-galactosidase reporter. The β-galactosidase reporter is a β-actin LacZ reporter construct that is regulated by the human actin promoter and expresses the β-galactosidase gene. Prior to transfection, media was changed to a Neurobasal medium minus phenol red (1x B27, and 0.5mM L-glutamine). Expression plasmids were transfected using the FuGENE 6 Transfection reagent (Roche Applied Science, Indianapolis, IN, USA as per the manufacturer’s recommendations). The transfection reagent:DNA concentration ratio for each of the expression plasmids was 3:1 (0.18μl FuGENE 6:0.05μg expression plasmid and 0.01μg β-galactosidase). Cortical neurons were incubated with the transfection reagent for 2 hours at 37°C. Culture media was changed to Neurobasal medium minus phenol red (1x B27, and 0.5mM L-glutamine) and neurons were incubated at 37°C for an additional 70 hours. After 72 hours, cell viability of the transfected neurons was determined using the CellTiter-Glo Luminescent Cell Viability Assay (Promega Corp. Madison, WI, USA). β-galactosidase activity was measured in the transfected neurons using the Tropix GalactoLight kit assay system (Applied Biosystems, Foster City, CA, USA) according to manufacturer’s recommendations. Luciferase and β-galactosidase activity were measured with the 20/20 TD luminometer (Turner Designs, Sunnyvale, CA, USA). Neuron viability following transfection was normalized to β-galactosidase activity for each transfection paradigm to account for variations in transfection efficiency. The normalized data was reported as percent change in viability from 0.3x105 cortical neurons that were not transfected. The viability of the non-transfected cortical neurons was measured using the CellTiter-Glo Luminescent Cell Viability Assay.
Primary cortical neurons were dual transfected with either the empty pGL3-basic, Bnip3, mutHRE, GRE1, GRE2, GRE3 or Bnip3ΔGRE2 promoter luciferase reporter plasmids and the β-galactosidase reporter. Prior to transfection, culture media was changed to Neurobasal minus phenol red containing 0.1x B27 and 0.5mM L-glutamine. Transfection was performed using the FuGENE 6 Transfection Reagent. The transfection reagent:DNA concentration ratio for each of the plasmids was 3:1 (1.5μl FuGENE 6:0.4μg promoter reporter plus 0.1μg β-galactosidase). Cultures were incubated in transfection reagent for 2 hours at 37°C. After 2 hours, transfected cultures were treated with either vehicle (0.005% 95% EtOH) or doses of RU28362 (0.1, 1.0, 5.0 or 10.0nM) in Neurobasal minus phenol red (0.1x B27 and 0.5mM L-glutamine) for 40 hours. Cultures were maintained in normoxic, hypoxic or ambient air conditions depending on the experimental design. Forty two hours after transfection the cells were lysed. Luciferase activity was measured by adding 20μl of lysate into 100μl of luciferan substrate (Promega Corp. Madison, WI, USA). β’-galactosidase activity was measured by using 40μl of lysate with the Tropix-GalactoLight kit. Luciferase and β-galactosidase activity were measured with the 20/20 TD luminometer and reported as relative light units (RLU); luciferase/β-galactosidase activity.
Lactate Dehydrogenase and MTT Assays
The percent of cell death was measured in primary cortical neurons treated with 1 of 4 doses of RU28362 (0.1, 1.0, 5.0 or 10.0nM) or vehicle (0.005% 95%EtOH) and incubated at 37°C for 48 and 72 hours in ambient air conditions. Cell death was determined by measuring LDH release into the culture media after RU28362 treatment using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega Corp. Madison, WI, USA). Data was reported as the percent cell death, experimental LDH release as a result of treatment relative to maximal LDH release.
The percent survival was measured after 48 or 72 hours in primary cortical neurons maintained in either normoxic (20% O2) or hypoxic (1% O2) conditions and neurons treated with 1 of 3 doses of RU28362 (0.1, 1.0 and 10.0nM) or vehicle and maintained in hypoxic (1% O2) conditions. Cell viability was measured using an MTT assay. After treatment primary cortical neurons were washed once with sterile PBS and media was replaced with 250μl of a 0.25mg/ml solution of MTT (Thiazolyl Blue Tetrazolium Bromide; Sigma-Aldrich, St. Louis, MO, USA) in Neurobasal media minus phenol red, containing 0.1x B27, 100μg/ml penicillin-streptomycin and 0.5mM L-glutamine. Cortical neurons were incubated at 37°C for 1.5 hours, the MTT solution was removed and 200μl of 0.04M HCL in absolute isopropanol was added to each experimental sample. 100ml of each sample was transferred to a 96 well plate and the absorbance of the converted dye was read at 570nm on a Bio-Rad Model 680 micro plate reader (Bio-Rad Laboratories, Hercules, CA, USA). The percent survival in hypoxic versus normoxic conditions was calculated relative to the normoxic controls for each time. For each time point the percent change in viability for the RU28362 treated cortical neurons under hypoxic conditions was calculated relative to the vehicle hypoxic controls.
Statistical analysis
Data were analyzed by a two way ANOVA using the Statview data analysis software (Abacus Concepts, Inc., Berkeley, CA, USA). Student-Newman-Keul’s and t-test procedures were used posthoc for individual comparisons. Differences were considered significant when p<0.05. Data were expressed as group means ± SEM.
Results
Bnip3 mRNA expression is upregulated by dexamethasone in the neonatal rat brain
Using ISH we localized Bnip3 mRNA expression in the brain of vehicle (Fig. 1A–B) or DEX- treated PND 7 rats (Fig. 1C–D). Bnip3 mRNA was expressed in the same brain regions of animals from both treatment groups with greatest hybridization density throughout the cortex, including the anterior and posterior cingulate cortices and piriform cortex. Additionally, Bnip3 mRNA was expressed in the CA1 through CA3 regions of the hippocampus, as well as in the dentate gyrus, septum, striatum medial thalamus and habenula (Fig. 1A–D).
Figure 1.
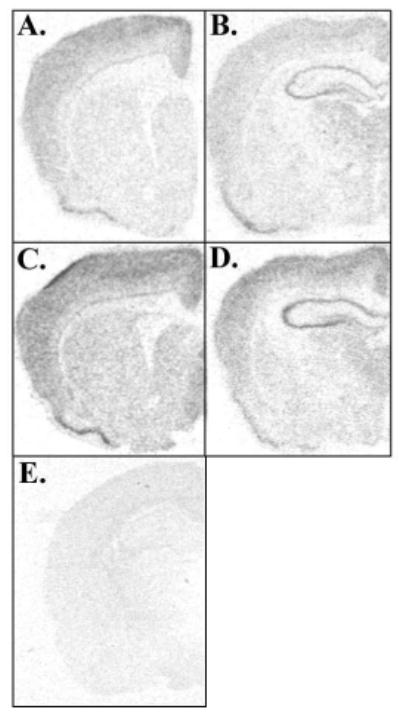
Photomicrographs showing in situ hybridization analysis of Bnip3 mRNA expression in DEX treated rats. Bnip3 mRNA expression in the PND 7 (A–B) vehicle and (C–D) DEX treated rat brain. (E) Sense directed probes show no hybridization.
Visual inspection of the Bnip3 labeled photomicrographs suggests that DEX upregulates Bnip3 mRNA expression. To investigate this further we quantified Bnip3 mRNA levels using real time RT-PCR in the postnatal rat brain. Bnip3 mRNA expression levels were quantified in the anterior and posterior cingulate cortices and hippocampus. On PND 7, female rat pups treated with DEX had significantly elevated levels of Bnip3 mRNA in the anterior (F1,40=7.72, *p<0.01) and posterior (F1,40=8.29, *p<0.01) cingulate cortices compared to vehicle treated controls (Fig. 2A–B). There was no effect of DEX in male rats (Fig. 2A–B). Additionally, we detected a significant sex by treatment effect in the anterior cingulate cortex (F1,40=7.27, *p<0.05). In the PND 7 hippocampus DEX treatment significantly (F1,40=9.57, *p<0.005) upregulated Bnip3 mRNA expression in both the male and female rat compared to vehicle treated controls (Fig. 2C).
Figure 2.
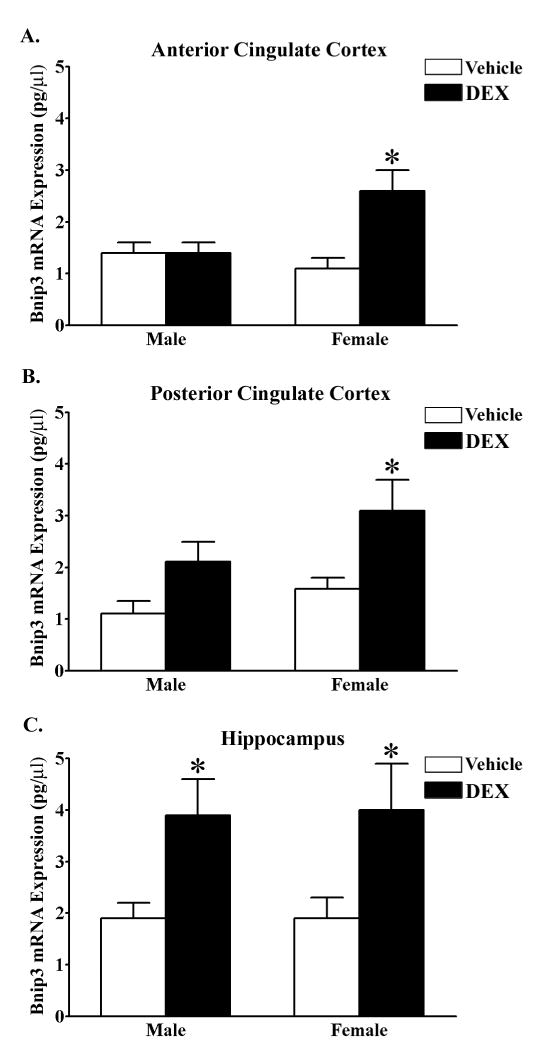
DEX upregulates Bnip3 mRNA expression in the neonatal anterior and posterior cingulate cortices and hippocampus. Quantitative real time RT-PCR was utilized to measure Bnip3 expression levels (pg/μl cDNA) in the (A) anterior cingulate cortex, (B) posterior cingulate cortex and (C) hippocampus of PND 7.0 male and female rat pups treated with either vehicle (white bars) or DEX (black bars). Each bar represents the mean ± S.E.M. of 10–14 determinations. Data were analyzed by two-way ANOVA (sex by treatment). Multiple t-test comparisons was used to indicate groups that were significantly different (*p<0.05) in Bnip3 mRNA expression as a result of DEX treatment.
Glucocorticoids endanger cortical neurons by increasing Bnip3 mRNA expression
To determine if GR regulates Bnip3 gene expression in developing neurons, we treated primary cortical neurons with the selective GR agonist, RU28362. Bnip3 mRNA expression was quantified using real time RT-PCR. Bnip3 mRNA levels were not changed after treatment with RU28362 for 48 hours compared to vehicle treated controls (Fig. 3A). However, after 72 hours there was a significant (F3,32=5.26, *p<0.05) increase in Bnip3 mRNA levels in neurons treated with 10.0nM RU28362 compared to neurons treated with vehicle and 0.1nM RU28362 (Fig. 3A).
Figure 3.
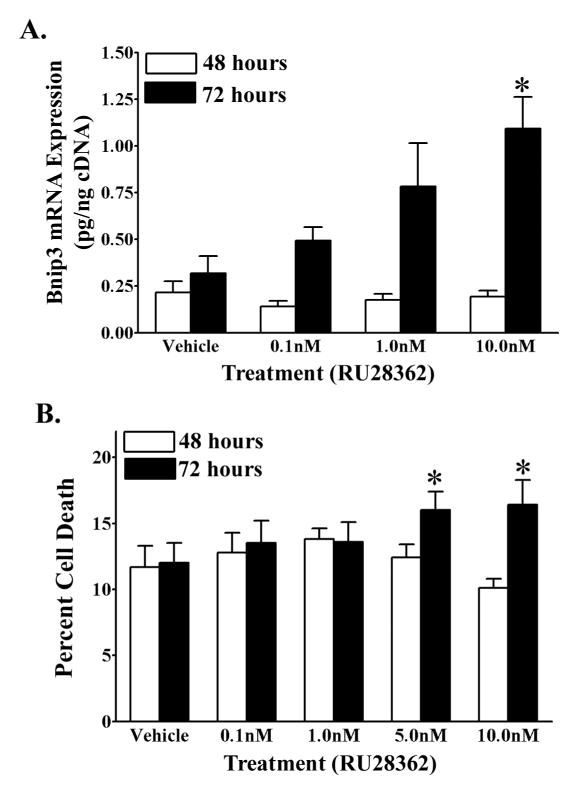
A GR selective agonist increases Bnip3 mRNA expression and induces cell death in primary cortical neurons. (A) Quantitative real time RT-PCR of Bnip3 mRNA levels (pg/ng cDNA) in primary cortical neurons treated with either RU28362 or vehicle and maintained in normoxic (20% O2) conditions for 48 (white bars) or 72 (black bars) hours. Each bar represents the mean ± S.E.M. of 8–10 determinations. (B) Lactate dehydrogenase assay determination of percent cell death in primary cortical neurons treated with either RU28362 or vehicle and maintained in ambient air for 48 (white bars) and 72 (black bars) hours. Each bar represents the mean ± S.E.M. of 13–21 determinations. Data were analyzed by two-way ANOVA (treatment by time). (A) Student-Newman Keul’s posthoc test indicates significant differences (*p<0.05) in Bnip3 mRNA levels relative to vehicle treated controls. (B) Multiple t-test comparisons were used to indicate groups with significant differences (*p<0.05) in percent cell death as a result of treatment at a given time.
We next examined the putative neuroendangering affects that low doses of GR agonist have on cortical neuron survival. After 72 hours, a significant (F1,149=4.41, p<0.05) increase in the percent of cell death was found after treatment of cortical neurons with 5.0 and 10.0nM RU28362 compared to neurons treated with 5.0 and 10.0nM RU28362 for 48 hours (Fig. 3B). The increase in cell death following 72 hours of 5 .0 and 10.0nM RU28362 relative to 48 hours of steroid treatment was approximately 6% and 10%, respectively. However, RU28362 treatment for either 48 or 72 hours, at any of the doses tested, did not significantly increase levels of cell death compared to vehicle treated controls (Fig. 3B).
Hypoxia increases Bnip3 transcription through a hypoxic response element
To determine if hypoxia regulates Bnip3 transcription in cortical neurons, we maintained primary cortical neurons in either normoxic (20% O2) or hypoxic (1% O2) conditions for 48 and 72 hours and subsequently quantified Bnip3 mRNA levels. Bnip3 mRNA was significantly (F1,33=10.42, *p<0.05) elevated, to levels approximately 2 fold of that of normoxic controls when primary cortical neurons were maintained in hypoxic conditions for 72 hours (Fig. 4A). Bnip3 mRNA was not changed in cortical neurons after 48 hours of hypoxia compared to normoxic controls (Fig. 4A). In order to establish a mechanism for hypoxia-mediated Bnip3 gene transcription, we next transfected primary cortical neurons with a luciferase reporter construct that contained either the full length Bnip3 promoter or the Bnip3 promoter with a mutation in the HRE site (mutHRE luc). Promoter activity was assessed in transfected cortical neurons maintained in normoxic or hypoxic conditions. A significant (F1,76=18.96; *p<0.0001) increase in promoter activity was measured in cortical neurons transfected with Bnip3-luc and maintained in hypoxic conditions, compared to Bnip3-luc transfected neurons maintained in normoxic conditions (Fig. 4B). This effect of hypoxia was not seen when the mutHRE was transfected into cortical neurons and exposed to hypoxic conditions (Fig. 4B).
Figure 4.
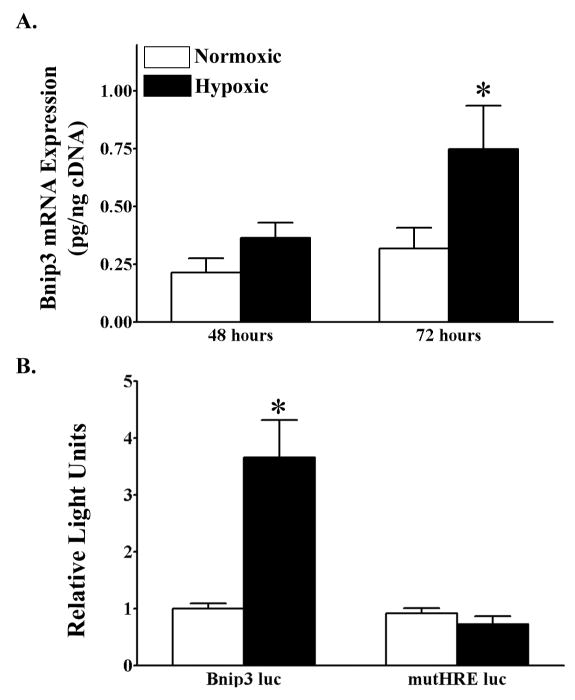
Hypoxia increases Bnip3 mRNA levels in primary cortical neurons through the hypoxic response element. (A) Quantitative real time RT-PCR was used to measure Bnip3 mRNA levels (pg/ng cDNA) in primary cortical neurons maintained in either normoxic (20% O2) or hypoxic (1% O2) conditions for 48 (white bars) or 72 (black bars) hours. Each bar represents the mean ± S.E.M. of 9–11 determinations. Data were analyzed by two-way ANOVA (treatment by time). (B) Luciferase reporter gene assay was used to measure promoter activity in primary cortical neurons dual transfected with either a Bnip3 luc or mutHRE luc reporter plasmid and a β-galactosidase reporter and maintained in either normoxic (white bars) or hypoxic (black bars) conditions. Relative light units (RLU) were calculated as a ratio of luciferase:β-galactosidase activity for each transfected culture and normalized to the promoter activity of the normoxic cultures. Each bar represents the mean ± S.E.M. of 11–15 determinations generated in 5 separate transfection assays. Data were analyzed by two-way ANOVA (plasmid type by oxygen condition). Multiple t-test comparisons were used to indicate significant differences (*p<0.05) from control groups.
Hypoxia induces primary cortical neuron death
We next established the percent survival of cortical neurons maintained in hypoxic conditions for either 48 or 72 hours. A significant (F1,31=76.63, *p<0.0001) decrease in cell survival (26%) was measured in neuronal cultures maintained in hypoxic conditions for 48 when compared to cultures maintained in normoxic conditions (Fig. 5A). Cortical neurons maintained in hypoxic conditions for 72 hours also had a significant (F1,33=195.79, *p<0.0001) decrease in cell survival (50%) compared to vehicle treated normoxic controls (Fig. 5B).
Figure 5.
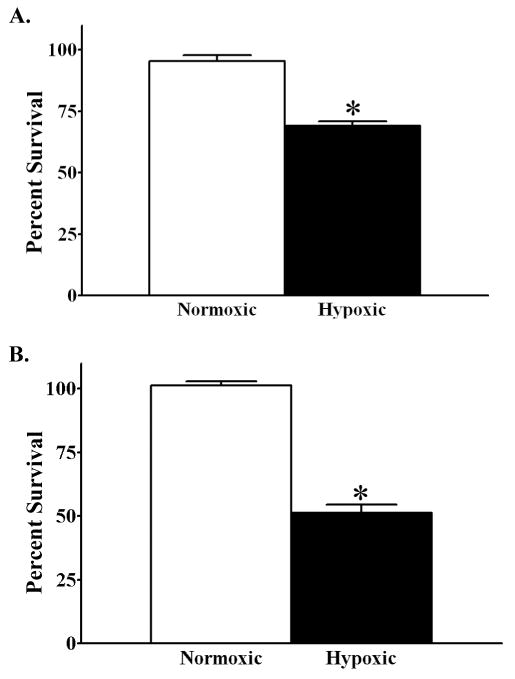
Hypoxia induces primary cortical neuron death. MTT assay to measure percent change in viability of vehicle treated primary cortical maintained in either normoxic or hypoxic conditions for (A) 48 and (B) 72 hours. Percent survival is relative to normoxic vehicle treated controls for each time point. Each bar represents the mean ± S.E.M. of 16–18 determinations. Data were analyzed by two-way ANOVA (air conditions by time). Multiple t-test comparisons indicate significant differences (*p<0.05) in percent change viability as a result of hypoxia at a given time.
Glucocorticoids exacerbate hypoxic induced Bnip3 mRNA expression in primary cortical neurons
The previous experiments established that Bnip3 mRNA expression levels are increased following administration of a GR selective agonist. Furthermore, we also established that hypoxia induces Bnip3 mRNA levels in cortical neurons through the HRE in the Bnip3 promoter. We subsequently investigated whether GR activation exacerbated the effects of hypoxia on Bnip3 mRNA expression. Treatment with RU28362 increased Bnip3 mRNA above that of cultures exposed to hypoxia alone. There was as significant (F3,35=3.51, *p<0.05) 2 fold increase in Bnip3 mRNA levels compared to hypoxic, vehicle treated controls when cortical neurons were treated with either 0.1, 1.0 or 10 nM RU28362 for 72 hours (Fig. 6A). Such a synergistic affect of glucocorticoids and hypoxia on Bnip3 expression was not present after 48 hours of RU28362 treatment (Fig. 6A).
Figure 6.
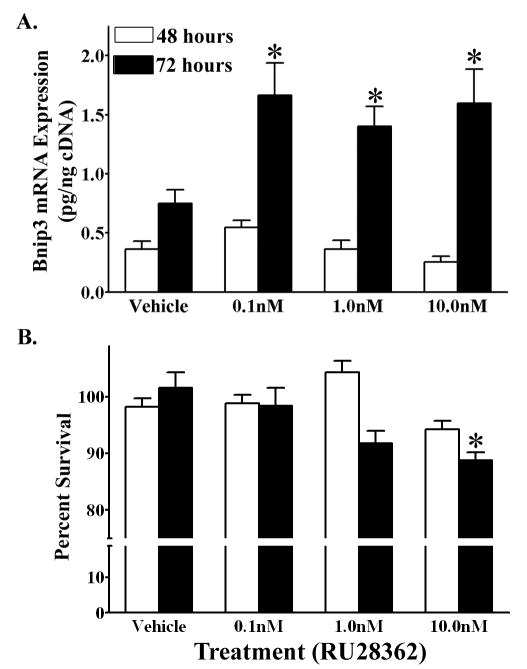
Glucocorticoids enhance hypoxia induced Bnip3 mRNA expression in primary cortical neurons. (A) Bnip3 mRNA expression (pg/ng cDNA) in primary cortical neurons treated with either RU28362 or vehicle and maintained in hypoxic (1% O2) conditions for 48 (white bars) or 72 (black bars) hours. Each bar represents the mean ± S.E.M. of 9–11 determinations. Data were analyzed by two-way ANOVA (treatment by time). (B) MTT assay to measure percent change in viability of RU28362 or vehicle treated primary cortical neurons maintained in hypoxic conditions for 48 (white bars) or 72 (black bars) hours. The percent survival for hypoxic cultures is relative to vehicle treated hypoxic controls. Each bar represents the mean ± S.E.M. of 12–18 determinations. Data were analyzed by two-way ANOVA (RU28362 by air conditions). Student-Newman Keul’s posthoc test was used to indicate groups with significant differences (*p<0.05) in (A) Bnip3 mRNA levels relative to vehicle treated controls and (B) percent survival relative to hypoxic vehicle- treated cultures.
We subsequently investigated the synergistic affects of glucocorticoids and hypoxia on cortical neuron survival. Primary cortical neurons treated with 10.0nM RU28362 and maintained in hypoxic conditions for 72 hours had a significant (F3,55=4.637, *p<0.01) decrease in cell survival relative to hypoxic cultures treated with vehicle, 0.1nM and 1.0nM RU28362. However, the decrease in cell survival following concomitant glucocorticoid and hypoxia treatment was minimal. We measured a 13% decrease in cell survival relative to hypoxic vehicle treated controls (Fig. 6B), which corresponds to an additional 8% decrease in neuron survival as a result of glucocorticoid and hypoxia treatment relative to vehicle normoxic treated controls (Fig. 5B). Primary cortical neurons treated with RU28362 and maintained in hypoxic conditions for 48 hours did not have a decrease in cell survival compared to vehicle treated hypoxic cultures (Fig. 6B).
Bnip3 protein decreases primary cortical neuron viability
Primary cortical neurons were transfected with a Bnip3 expression plasmid and cell viability was subsequently determined after 72 hours. Overexpression of Bnip3 protein significantly (F3,55=16.41, *p<0.0001) decreased cortical neuron viability by 38% relative to the non-transfected controls and controls transfected with the empty pcDNA plasmid (Fig. 7). Because the transmembrane (TM) domain of the Bnip3 protein is thought to be as essential for the induction of Bnip3 mediated cell death (Ray et al., 2000), we also used a plasmid that encodes for a Bnip3 protein with a deletion of the TM domain (Bnip3ΔTM). Deletion of the TM domain prevented the decrease in cell viability relative to the full length Bnip3 expression plasmid (Fig 7). Transfection of primary cortical neurons with an empty plasmid was not different from cell viability in non-transfected control neurons. These data confirm that our transfection protocol does not by itself, alter baseline cell numbers (Fig. 7).
Figure 7.
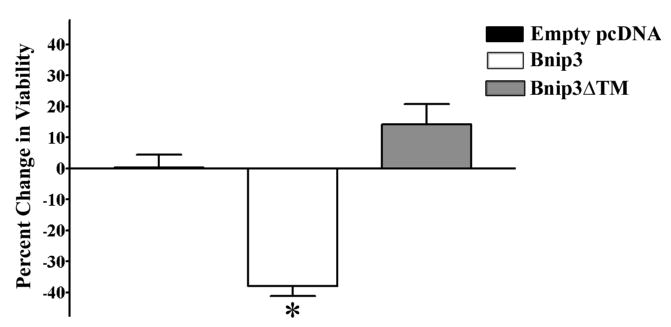
Overexpression of Bnip3 in primary cortical neurons induces cell death. Percent change in viability was measured in primary cortical neurons dual transfected with Bnip3, Bnip3ΔTM or empty PCDNA expression plasmid and a β-galactosidase reporter for 72 hours. Viability of transfected neurons was normalized to β-galactosidase activity and reported as percent change in viability relative to the viability of non-transfected controls. Each bar represents the mean ± S.E.M. of 18–24 determinations generated in 3 transfections. Data were analyzed by one-way ANOVA followed by Student-Newman Keul’s posthoc test. * indicates groups that were significantly different (p<0.05) in percent change in viability relative to non-transfected control.
GR regulation of Bnip3 promoter activity in primary cortical neurons is dependent on a GRE
Since the previous experiments indicated that GR increased levels of Bnip3 mRNA, we sought to determine whether GR acts to increase Bnip3 mRNA levels by inducing gene transcription. Primary cortical neurons were transiently transfected with a Bnip3 promoter - luciferase reporter containing plasmid and then treated with RU28362 or vehicle for 42 hours. Bnip3 promoter activity was significantly (F4,95=4.55, *p<0.05) elevated in transfected neurons treated with 1.0, 5.0 and 10.0nM RU28362 compared to vehicle-treated, transfected neurons (Fig. 8).
Figure 8.
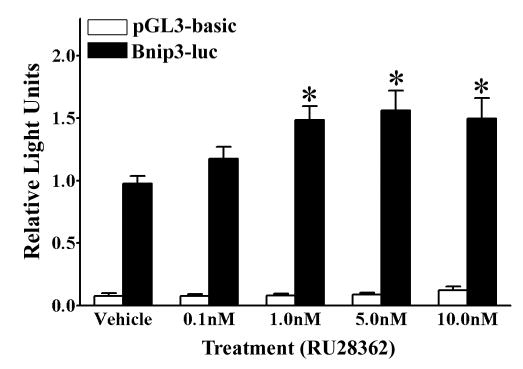
GR induces Bnip3 promoter activity in primary cortical neurons. A luciferase based reporter gene assay was used to measure promoter activity in primary cortical neurons dual transected with either the empty pGL3-basic luciferase reporter (white bars) or Bnip3 promoter luciferase reporter (black bars) containing plasmids and a β-galactosidase reporter. Transfected neurons were treated with either RU28362 or vehicle. Relative light units (RLU) were calculated as a ratio of luciferase:β-galactosidase activity for each transfected culture and normalized to the mean RLU of vehicle treated neurons transfected with the Bnip3 promoter. Each bar represents the mean ± S.E.M. of 18–24 determinations generated in 8 separate transfection assays. Data were analyzed by two-way ANOVA (treatment by plasmid) followed by Student-Newman Keul’s posthoc test. * indicates those groups with significant differences (*p<0.05) in promoter activity relative to vehicle treated controls.
We further examined the Bnip3 promoter using a computer based analysis, and identified 3 consensus GRE sites. Figure 9A illustrates the Bnip3 promoter with the locations of the 3 putative GRE sites relative to the transcription start site. In order to determine which of the three putative GREs mediates GR regulation of Bnip3 gene transcription, three mutant Bnip3 promoter - luciferase reporter constructs were generated to isolate the individual putative GREs (Fig. 9A bottom 3 illustrations). Primary cortical neurons were transfected with either GRE1-luc, GRE2-luc or GRE3-luc and treated with varying doses of RU28362 or vehicle for 42 hours. Basal activity for GRE1-luc, GRE2-luc and GRE3-luc were not significantly different from that of the full length Bnip3 promoter - luciferase reporter (data not shown). Primary cortical neurons transfected with GRE1-luc and GRE3-luc did not respond to RU28362 with a significant change in promoter activity relative to vehicle treated controls (Fig. 9B). However, cortical neurons transfected with GRE2-luc had a significant (F4,62=4.24, *p<0.05) increase in promoter activity in response to RU28362 treatment relative to vehicle treated transfected controls (Fig 9B). The magnitude of this increase was approximately 2- fold greater after RU28362 treatment (Fig. 9B), and achieved levels equivalent to the promoter activity of the full length Bnip3 promoter after RU28362 treatment (Fig. 8).
Figure 9.
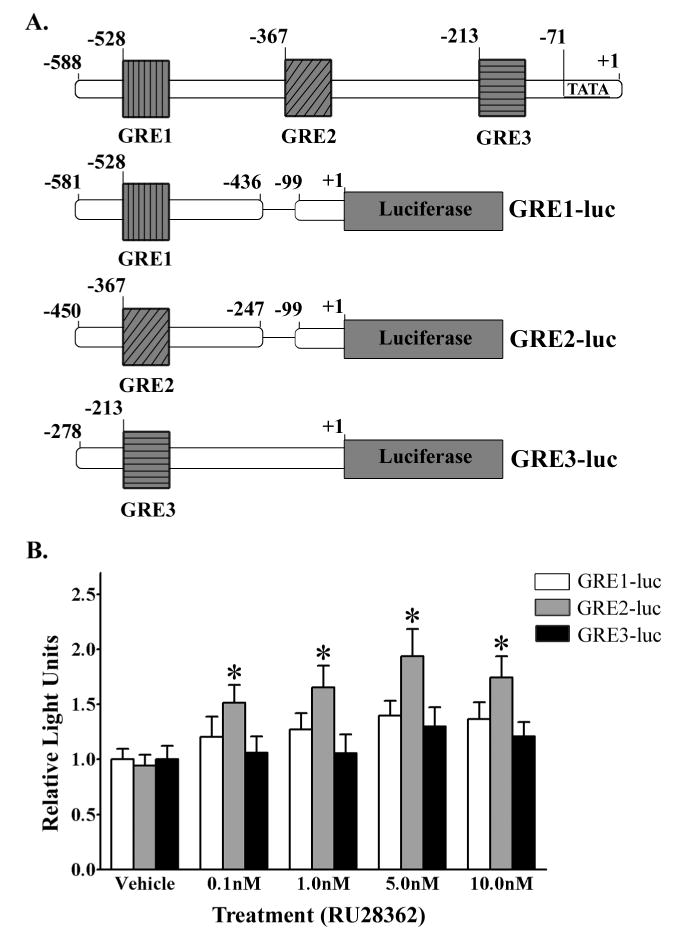
The Bnip3 promoter region containing the putative GRE2 is responsive to activated GR. (A) Illustration depicting the endogenous Bnip3 promoter with the relative locations of the three putative GREs and a schematic of the three mutant Bnip3 promoter luciferase reporters (GRE1-luc, GRE2-luc and GRE3-luc) generated to isolate each individual GRE. (B) Luciferase based reporter gene assay was used to measure promoter activity in primary cortical neurons dual transfected with either GRE1-luc (white bars), GRE2-luc (grey bars) or GRE3-luc (black bars) containing plasmids and a β-galactosidase reporter and subsequently treated with vehicle or RU28362. Relative Light Units (RLU) were calculated as a ratio of luciferase:β-galactosidase activity for each transfected culture and normalized to the mean vehicle treated RLUs for each promoter reporter tested. Each bar represents the mean ± S.E.M. of 11–15 determinations generated in 5 separate transfection assays. Data were analyzed by two-way ANOVA (treatment by plasmid type) followed by Student-Newman Keul’s posthoc test. * indicates those groups that were significantly different (*p<0.05) in promoter activity relative to vehicle treated controls.
Finally, to confirm that the putative GRE2 is the region that mediates GR regulation of Bnip3 gene transcription we transfected primary cortical neurons with a Bnip3 promoter - luciferase reporter plasmid, with a disrupted GRE2 site (Bnip3ΔGRE2-luc; Fig. 10A). The Bnip3ΔGRE2-luc transfected cortical neurons had basal promoter activity that was not different from the basal promoter activity in Bnip3-luc transfected cortical neurons (data not shown). Cortical neurons transfected with Bnip3-luc and treated with 10.0nM RU28362 had significantly (F2,149=3.96; *p<0.05) increased promoter activity compared to vehicle and 0.1nM RU28362 treated Bnip3-luc transfected cultures (Fig. 10B). Conversely, RU28362 did not induce luciferase activity in cortical neurons transfected with Bnip3ΔGRE2-luc when compared to vehicle-treated Bnip3ΔGRE2-luc transfected cultures (Fig 10B).
Figure 10.
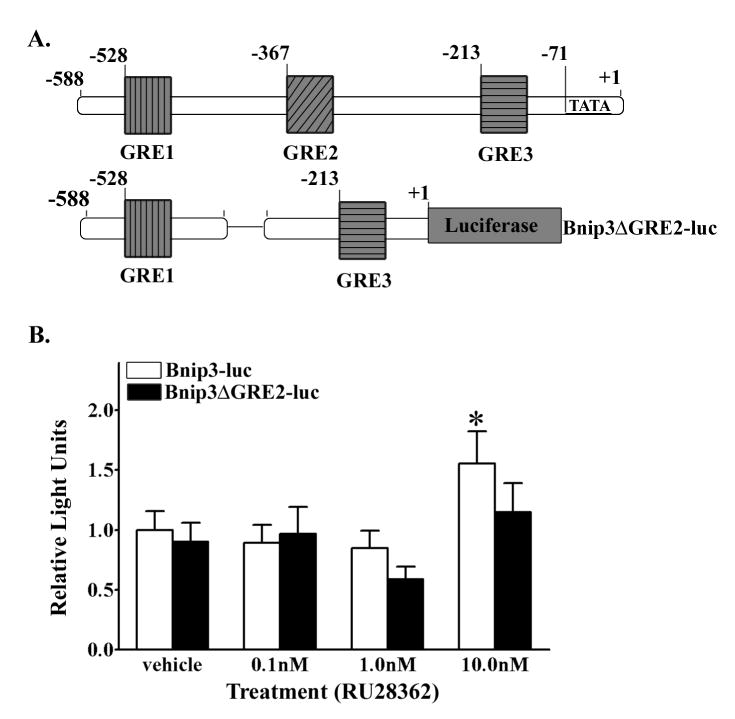
GR regulation of Bnip3 gene transcription is mediated through a GRE. (A) Upper illustration depicting the endogenous Bnip3 promoter with the relative locations of the three putative GREs. The lower illustration is a schematic of the mutant Bnip3ΔGRE2 promoter luciferase / reporter (Bnip3ΔGRE2-luc) generated to eliminate the 2nd GRE site while leaving the remainder of the promoter is intact (B) Luciferase based reporter gene assay was used to measure promoter activity in primary cortical neurons dual transfected with either Bnip3-luc (white bars) or Bnip3ΔGRE2-luc (black bars) containing plasmids and a β-galactosidase reporter and subsequently vehicle or RU28362 treated. Relative light units (RLU) were calculated as a ratio of luciferase:β-galactosidase activity for each transfected culture and normalized to the vehicle treated mean RLUs for each promoter reporter. Each bar represents the mean ± S.E.M. of 10–15 determinations generated in 5 transfections. Data were analyzed by two-way ANOVA (treatment by plasmid) followed by Student-Newman Keul’s posthoc test. * indicates those groups with significant differences (*p<0.05) in Bnip3-luc promoter activity relative to vehicle and 0.1nM RU28362 treated cultures.
Discussion
The results of the experiments in this study identified the pro-apoptotic gene Bnip3 as a mediator of cortical neuron death and also as a glucocorticoid and hypoxia responsive gene in the rat cortex. DEX treatment of postnatal female rat pups increased Bnip3 mRNA expression in the anterior and posterior cingulate cortices and the hippocampus. However, in male rat pups, DEX increased Bnip3 mRNA levels in the hippocampus, but not the cingulate cortex. Additionally, in cortical neurons the activated glucocorticoid receptor induces Bnip3 transcription at a GRE site within the 5′ untranslated region of the Bnip3 promoter. Moreover, glucocorticoids were found to exacerbated hypoxia induced Bnip3 expression in cortical neurons. Despite the upregulation of Bnip3 mRNA expression in response to glucocorticoid treatment either in the presence or absence of hypoxia, this was insufficient to cause a large induction of neuronal death. However, increases in primary cortical neuron death were measured following overexpression of Bnip3 protein, independent of glucocorticoid and hypoxia treatment.
Previous studies have established that glucocorticoids can endanger the CNS by increasing levels of the pro-apoptotic protein Bax and decreasing levels of the anti-apoptotic proteins Bcl-2 and Bcl-XL, in the juvenile and adult rat hippocampus (Almeida et al., 2000) and in primary hippocampal neurons (Crochemore et al., 2005). Furthermore, DEX treatment results in an increase in apoptosis in the adult rat hippocampus and striatum (Hassan et al., 1996, Haynes et al., 2001). Consistent with these studies, our results indicate that DEX similarly induces expression of Bnip3 in the postnatal male and female rat hippocampus. This may be an additional component of the mechanism mediating DEX induced hippocampal cell death. However, Bnip3 interactions with the Bcl-2 family and subsequent implications for cell viability are only partially defined (Ray et al., 2000, Gao et al., 2005). It is known that Bnip3 protein heterodimerizes with the anti-apoptotic proteins Bcl-2 and Bcl-XL, but this does not seem to prevent Bnip3 mediated cell death. Furthermore, Bnip3 interactions with other proteins are mediated by the TM domain and not the BH3 domain (Chen et al., 1997, Ray et al., 2000), the site within BH3-only proteins that are implicated as essential to Bax activation (Wei et al., 2001). Regardless, the putative interaction of Bnip3 and Bax has not been reported in the current literature.
Despite the observations showing glucocorticoid neuroendangerment in the hippocampus, and DEX dependent decreases in cortical neuron numbers (Crochemore et al., 2005, Kreider et al., 2006), to the best of our knowledge, glucocorticoid endangerment of cortical neuron responses to a secondary metabolic insult, have not yet been described. The results of our studies have established that activated GR increases Bnip3 transcription in the anterior and posterior cingulate cortices of the postnatal female rat, but not in the male cingulate cortex. This sex difference suggests the possibility that the female cingulate cortex may be more susceptible to the neuroendangering aspects of glucocorticoids than the male cingulate cortex.
In a previous study, we investigated a potential mechanism governing sex differences in Bnip3 expression in the developing rat brain. It was determined that sex differences in postnatal gonadal steroid hormone levels are not responsible for the differing basal levels of Bnip3 mRNA expression in the male and female anterior and posterior cingulate cortices (Sandau and Handa, 2006). It is possible that this sex difference may be the result of genetic differences; however, additional experiments are necessary to understand the underlying mechanism mediating sex difference Bnip3 gene expression.
Even though we observed DEX induced changes in Bnip3 mRNA expression in the postnatal rat CNS the possibility exists that these affects may result from indirect steroid actions. Previous reports indicate that DEX entry into the CNS is hindered by P-glycoprotein product at the blood brain barrier (Meijer et al., 1998). Furthermore, it has been shown that DEX alters HPA axis regulation at the level of the anterior pituitary (de Kloet et al., 1974, De Kloet et al., 1975). Based on these reports and our experimental design we are unable to conclude if DEX in vivo regulates Bnip3 directly at the level of the cortex and hippocampus or indirectly through HPA axis dysregulation. However, the results of our in vitro studies suggest that activated GR directly regulates Bnip3 expression within the cortex. Regardless, additional studies employing neonatal adrenalectomy may elucidate the underlying mechanism.
Our in vitro studies indicate that glucocorticoids act directly on cortical neurons to upregulate Bnip3 mRNA expression. Furthermore, activated GR directly regulates Bnip3 gene transcription via a GRE site within the Bnip3 promoter. Notwithstanding, earlier studies show that glucocorticoids can act indirectly to regulate expression of the Bcl-2 gene family by modulating NF-κB (Ramdas and Harmon, 1998, Dhandapani et al., 2005) and p53 transcriptional activity (Miyashita et al., 1994, Miyashita and Reed, 1995, Maiyar et al., 1997, Crochemore et al., 2002, Iyer et al., 2003). Bnip3 expression has similarly been shown to be attenuated following inhibition of NF-κB (Baetz et al., 2005), a gene that is glucocorticoid regulated (Webster and Cidlowski, 1999). Therefore, indirect regulation of Bnip3 gene transcription through glucocorticoid modulation of NF-κB remains a possibility. Conversely, Bnip3 gene transcription appears to be independent of p53 activity (Guo et al., 2001, Fei et al., 2004), despite reports of glucocorticoid regulation of p53 (Crochemore et al., 2002).
Bnip3 regulation has been shown to be hypoxia responsive, both in peripheral cell types as well as in the CNS (Bruick, 2000, Althaus et al., 2006, Burton et al., 2006). Increases in hypoxia-induced cell death are associated with increases in Bnip3 expression, both in vivo and in vitro (Schmidt-Kastner et al., 2004, Althaus et al., 2006, Burton et al., 2006). Using in vivo approaches, Bnip3 protein levels were found to be increased following a hypoxic-ischemic insult in the adult rat hippocampus, cortex and striatum (Schmidt-Kastner et al., 2004, Althaus et al., 2006). Similarly, in a differentiated oligodendroglial cell line, Bnip3 protein levels are increased in response to hypoxia (Burton et al., 2006). Consistent with these studies, our data indicate that Bnip3 mRNA is upregulated by hypoxia in primary cortical neurons.
We have also determined that hypoxia regulates Bnip3 in cortical neurons via the HRE site within the promoter; a site that was previously only characterized in peripheral cell types (Bruick, 2000, Sowter et al., 2001). Accordingly, Bnip3 expression also decreased cortical neuron viability after hypoxia. We have also determined that removal of the TM domain of Bnip3 prevented the effects of Bnip3 and allowed partial recovery of primary cortical neuron viability following hypoxia. The Bnip3ΔTM has been previously reported to act as a dominant negative to the endogenous Bnip3 protein (Kanzawa et al., 2005, Burton et al., 2006) and negates hypoxia induced cell death in differentiated oligodendroglial cells (Burton et al., 2006). Taken together, these data suggest that Bnip3 mediates a component of hypoxia induced cell death in cortical neurons, but additional factors may also contribute to the hypoxic response.
Elevated glucocorticoids cause an altered metabolic state that is partially conferred by decreased glucose transporter expression levels and inhibition of glucose uptake in neurons (Sapolsky et al., 1986, Horner et al., 1990, Virgin et al., 1991, Booth et al., 1998). These changes subsequently endanger hippocampal neurons when challenged with a second metabolic insult such as glutamate excitoxicity or hypoxia-ischemia (Sapolsky and Pulsinelli, 1985, Roy and Sapolsky, 2003). Furthermore, it has been reported that activated GR stimulates or represses expression of additional hypoxic responsive genes including, glucose transporter 3 and vascular endothelial growth factor (Kodama et al., 2003, Leonard et al., 2005). In agreement with these reports, our studies demonstrate that low doses of GR agonist enhance hypoxia induced expression of the pro-apoptotic gene Bnip3. However, to our surprise, a corresponding additive increase in cortical neuron death was minimal following glucocorticoid and hypoxia treatment. A possible explanation may be due to the presence of low corticosterone levels in our culture system, which activates the MR. In primary rat hippocampal neurons, activated MR circumvents GR induced hippocampal neuron death (Crochemore et al., 2005). Furthermore, Crochemore et al, (2005) suggest that in culture, nanomolar concentrations of corticosterone are sufficient to inhibit GR’s killing activities. A second putative explanation for the lack of cortical neuron death following glucocorticoid and hypoxia treatment is our use of cortical neuron cultures obtained from both male and female rat fetuses. Our in vivo studies determined that DEX upregulates Bnip3 expression in the female, but not male, cortex. Unfortunately, in vitro studies were begun prior to knowing the outcome of the in vivo DEX studies. Therefore, additional studies utilizing primary cortical cultures obtained from only females and treated concomitantly with a GR agonist and MR antagonist both in the presence and absence of hypoxia may yield slightly different results.
Our experiments indicate that glucocorticoids and hypoxia cause delayed induction of Bnip3 expression in vitro. In agreement with this finding, previous studies show that in peripheral cell culture lines hypoxia causes delayed increases in Bnip3 with greatest levels achieved after 48 hours (Bruick, 2000). Similar to peripheral cell lines hypoxia induced increases in Bnip3 are also delayed in oligodendroglial cell lines (Burton et al., 2006). Additionally, the increases in Bnip3 expression closely correlate with cell death levels (Bruick, 2000, Burton et al., 2006). The delayed upregulation of Bnip3 following glucocorticoid exposure is more difficult to comment on as this is the first study which addresses GR regulation of Bnip3. However, studies have shown that following different insults, such as ischemia, Bnip3 increases are also delayed with greatest levels achieved after 48 hours in the rat cortex in vivo (Althaus et al., 2006). The underlying mechanism that regulates the delayed induction of Bnip3 has not yet been elucidated.
Our studies have also determined that overexpression of Bnip3 protein is independently sufficient to induce cortical neuron death. Cell death directly attributed to increases in Bnip3 protein has been demonstrated in multiple cell types including oligodendrocytes (Vande Velde et al., 2000, Burton et al., 2006). In each of the cell types tested, the Bnip3 protein TM domain is implicated as the region which confers its killing activity (Vande Velde et al., 2000, Yook et al., 2004, Burton et al., 2006). This aspect of Bnip3 protein function is conserved in cortical neurons as our data indicates that the TM domain, and not the BH3 domain, is essential to Bnip3 mediated cell death.
Limitations associated with our cell death assays prevent additional interpretation into the mechanism underlying Bnip3, glucocorticoid and hypoxia induced cell death. However, in recent studies Bnip3 has been implicated as a mediator of autophagic cell death (Daido et al., 2004, Kanzawa et al., 2005); while past studies have characterized it as ‘necrotic-like’ and independent of caspase activity, yet the protein is intimately associated with the mitochondrial membrane (Vande Velde et al., 2000). Based on these studies it would be beneficial to investigate autophagic, caspase-dependent and independent cell death markers following glucocorticoid and hypoxia treatment of cortical neurons.
In summary, the experiments conducted in these studies have identified a cellular mechanism by which glucocorticoids can endanger the cerebral cortex and may enhance hypoxia-induced cell death. These findings are in agreement with reports that identify glucocorticoids as compounds that enhance pathologies associated various secondary metabolic insults. Furthermore, our data have identified a candidate gene, Bnip3, as a mediator of these observed affects. However, additional studies are necessary to determine if Bnip3 acts in conjunction with the other Bcl-2 family members to mediate cell death. The results of these studies have potential clinical ramifications since synthetic glucocorticoids are commonly prescribed to prevent chronic lung disease in premature infants (Bar-Lev et al., 2004, Doyle et al., 2006). Furthermore, information gained in regards to GR and HIF-1 regulation of Bnip3 may serve as a therapeutic target for hypoxic insults in the developing and mature CNS.
Acknowledgments
The Bnip3 and Bnip3ΔTM expression plasmids were kindly provided by Dr. Don Dubik (Manitoba Institute of Cell Research, Manitoba, CA) and the Bnip3 and mutHRE promoter luciferase reporter plasmids were kindly provided by Dr. Richard Bruick (University of Texas Southwestern Medical Center, Dallas, TX, USA). The studies presented in this manuscript were supported by grants from the USPHS, National Research Service Award F31 NS046959 (USS) and RO1-NS039951 and AA014974 (RJH).
Comprehensive list of abbreviations
- BH3
Bcl-2 homology domain 3
- mutHRE luc
Bnip3 promoter with a mutated HRE site
- GRE1-luc
Bnip3 promoter with isolated putative GRE1 site
- GRE2-luc
Bnip3 promoter with isolated putative GRE2 site
- GRE2-luc
Bnip3 promoter with isolated putative GRE3 site
- Bnip3ΔGRE2-luc
Bnip3 promoter with a deletion of the GRE2 site
- Bnip3ΔTM
Bnip3 protein with a deletion of TM domain
- DIV
Days in vitro
- DEX
Dexamethasone
- GR
Glucocorticoid receptor
- GRE
Glucocorticoid response element
- HIF-1
Hypoxia inducible factor-1
- HRE
Hypoxic response element
- ISH
In situ hybridization
- MR
mineralocorticoid receptor
- PND
Postnatal day
- RLU
Relative light units
- TM
Transmembrane domain
Footnotes
Section Editor: Dr. Menahem Segal (Cellular Neuroscience)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almeida OF, Conde GL, Crochemore C, Demeneix BA, Fischer D, Hassan AH, Meyer M, Holsboer F, Michaelidis TM. Subtle shifts in the ratio between pro- and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. Faseb J. 2000;14:779–790. doi: 10.1096/fasebj.14.5.779. [DOI] [PubMed] [Google Scholar]
- Althaus J, Bernaudin M, Petit E, Toutain J, Touzani O, Rami A. Expression of the gene encoding the pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible factor-1alpha (HIF-1alpha) protein following focal cerebral ischemia in rats. Neurochem Int. 2006 doi: 10.1016/j.neuint.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Baetz D, Regula KM, Ens K, Shaw J, Kothari S, Yurkova N, Kirshenbaum LA. Nuclear factor-kappaB-mediated cell survival involves transcriptional silencing of the mitochondrial death gene BNIP3 in ventricular myocytes. Circulation. 2005;112:3777–3785. doi: 10.1161/CIRCULATIONAHA.105.573899. [DOI] [PubMed] [Google Scholar]
- Bar-Lev MR, Maayan-Metzger A, Matok I, Heyman Z, Sivan E, Kuint J. Short-term outcomes in low birth weight infants following antenatal exposure to betamethasone versus dexamethasone. Obstet Gynecol. 2004;104:484–488. doi: 10.1097/01.AOG.0000137351.71015.ac. [DOI] [PubMed] [Google Scholar]
- Booth C, Tian L, Shipston MJ. Dissociation of early glucocorticoid inhibition of ACTH secretion and glucose uptake in mouse AtT20 D16:16 corticotrophs. J Neuroendocrinol. 1998;10:447–452. doi: 10.1046/j.1365-2826.1998.00226.x. [DOI] [PubMed] [Google Scholar]
- Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, D’Sa-Eipper C, Chinnadurai G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell. 1994;79:341–351. doi: 10.1016/0092-8674(94)90202-x. [DOI] [PubMed] [Google Scholar]
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97:9082–9087. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton TR, Henson ES, Baijal P, Eisenstat DD, Gibson SB. The pro-cell death Bcl-2 family member, BNIP3, is localized to the nucleus of human glial cells: Implications for glioblastoma multiforme tumor cell survival under hypoxia. Int J Cancer. 2006;118:1660–1669. doi: 10.1002/ijc.21547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira JJ, Catania C, Sotiropoulos I, Schubert M, Kalisch R, Almeida OF, Auer DP, Sousa N. Corticosteroid status influences the volume of the rat cingulate cortex - a magnetic resonance imaging study. J Psychiatr Res. 2005a;39:451–460. doi: 10.1016/j.jpsychires.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Cerqueira JJ, Pego JM, Taipa R, Bessa JM, Almeida OF, Sousa N. Morphological correlates of corticosteroid-induced changes in prefrontal cortex-dependent behaviors. J Neurosci. 2005b;25:7792–7800. doi: 10.1523/JNEUROSCI.1598-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ray R, Dubik D, Shi L, Cizeau J, Bleackley RC, Saxena S, Gietz RD, Greenberg AH. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med. 1997;186:1975–1983. doi: 10.1084/jem.186.12.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe CL, Kramer M, Czeh B, Gould E, Reeves AJ, Kirschbaum C, Fuchs E. Prenatal stress diminishes neurogenesis in the dentate gyrus of juvenile rhesus monkeys. Biol Psychiatry. 2003;54:1025–1034. doi: 10.1016/s0006-3223(03)00698-x. [DOI] [PubMed] [Google Scholar]
- Crochemore C, Lu J, Wu Y, Liposits Z, Sousa N, Holsboer F, Almeida OF. Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol Psychiatry. 2005;10:790–798. doi: 10.1038/sj.mp.4001679. [DOI] [PubMed] [Google Scholar]
- Crochemore C, Michaelidis TM, Fischer D, Loeffler JP, Almeida OF. Enhancement of p53 activity and inhibition of neural cell proliferation by glucocorticoid receptor activation. Faseb J. 2002;16:761–770. doi: 10.1096/fj.01-0577com. [DOI] [PubMed] [Google Scholar]
- Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, van der Vies J, de Wied D. The site of the suppressive action of dexamethasone on pituitary-adrenal activity. Endocrinology. 1974;94:61–73. doi: 10.1210/endo-94-1-61. [DOI] [PubMed] [Google Scholar]
- De Kloet R, Wallach G, McEwen BS. Differences in corticosterone and dexamethasone binding to rat brain and pituitary. Endocrinology. 1975;96:598–609. doi: 10.1210/endo-96-3-598. [DOI] [PubMed] [Google Scholar]
- Dhandapani KM, Wade FM, Wakade C, Mahesh VB, Brann DW. Neuroprotection by stem cell factor in rat cortical neurons involves AKT and NFkappaB. J Neurochem. 2005;95:9–19. doi: 10.1111/j.1471-4159.2005.03319.x. [DOI] [PubMed] [Google Scholar]
- Doyle LW, Davis PG, Morley CJ, McPhee A, Carlin JB. Low-dose dexamethasone facilitates extubation among chronically ventilator-dependent infants: a multicenter, international, randomized, controlled trial. Pediatrics. 2006;117:75–83. doi: 10.1542/peds.2004-2843. [DOI] [PubMed] [Google Scholar]
- Fei P, Wang W, Kim SH, Wang S, Burns TF, Sax JK, Buzzai M, Dicker DT, McKenna WG, Bernhard EJ, El-Deiry WS. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell. 2004;6:597–609. doi: 10.1016/j.ccr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Flagel SB, Vazquez DM, Watson SJ, Jr, Neal CR., Jr Effects of tapering neonatal dexamethasone on rat growth, neurodevelopment, and stress response. Am J Physiol Regul Integr Comp Physiol. 2002;282:R55–63. doi: 10.1152/ajpregu.2002.282.1.R55. [DOI] [PubMed] [Google Scholar]
- Gao S, Fu W, Durrenberger M, De Geyter C, Zhang H. Membrane translocation and oligomerization of hBok are triggered in response to apoptotic stimuli and Bnip3. Cell Mol Life Sci. 2005;62:1015–1024. doi: 10.1007/s00018-005-4543-3. [DOI] [PubMed] [Google Scholar]
- Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K, Yu KT, Jaye M, Ivashchenko Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001;8:367–376. doi: 10.1038/sj.cdd.4400810. [DOI] [PubMed] [Google Scholar]
- Hassan AH, von Rosenstiel P, Patchev VK, Holsboer F, Almeida OF. Exacerbation of apoptosis in the dentate gyrus of the aged rat by dexamethasone and the protective role of corticosterone. Exp Neurol. 1996;140:43–52. doi: 10.1006/exnr.1996.0113. [DOI] [PubMed] [Google Scholar]
- Haynes LE, Griffiths MR, Hyde RE, Barber DJ, Mitchell IJ. Dexamethasone induces limited apoptosis and extensive sublethal damage to specific subregions of the striatum and hippocampus: implications for mood disorders. Neuroscience. 2001;104:57–69. doi: 10.1016/s0306-4522(01)00070-7. [DOI] [PubMed] [Google Scholar]
- Horner HC, Packan DR, Sapolsky RM. Glucocorticoids inhibit glucose transport in cultured hippocampal neurons and glia. Neuroendocrinology. 1990;52:57–64. doi: 10.1159/000125539. [DOI] [PubMed] [Google Scholar]
- Iyer R, Ding L, Batchu RB, Naugler S, Shammas MA, Munshi NC. Antisense p53 transduction leads to overexpression of bcl-2 and dexamethasone resistance in multiple myeloma. Leuk Res. 2003;27:73–78. doi: 10.1016/s0145-2126(02)00064-4. [DOI] [PubMed] [Google Scholar]
- Kamphuis PJ, Croiset G, Bakker JM, Van Bel F, Van Ree JM, Wiegant VM. Neonatal dexamethasone treatment affects social behaviour of rats in later life. Neuropharmacology. 2004;47:461–474. doi: 10.1016/j.neuropharm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene. 2005;24:980–991. doi: 10.1038/sj.onc.1208095. [DOI] [PubMed] [Google Scholar]
- Kodama T, Shimizu N, Yoshikawa N, Makino Y, Ouchida R, Okamoto K, Hisada T, Nakamura H, Morimoto C, Tanaka H. Role of the glucocorticoid receptor for regulation of hypoxia-dependent gene expression. J Biol Chem. 2003;278:33384–33391. doi: 10.1074/jbc.M302581200. [DOI] [PubMed] [Google Scholar]
- Kreider ML, Tate CA, Cousins MM, Oliver CA, Seidler FJ, Slotkin TA. Lasting effects of developmental dexamethasone treatment on neural cell number and size, synaptic activity, and cell signaling: critical periods of vulnerability, dose-effect relationships, regional targets, and sex selectivity. Neuropsychopharmacology. 2006;31:12–35. doi: 10.1038/sj.npp.1300783. [DOI] [PubMed] [Google Scholar]
- Leonard MO, Godson C, Brady HR, Taylor CT. Potentiation of glucocorticoid activity in hypoxia through induction of the glucocorticoid receptor. J Immunol. 2005;174:2250–2257. doi: 10.4049/jimmunol.174.4.2250. [DOI] [PubMed] [Google Scholar]
- Maiyar AC, Phu PT, Huang AJ, Firestone GL. Repression of glucocorticoid receptor transactivation and DNA binding of a glucocorticoid response element within the serum/glucocorticoid-inducible protein kinase (sgk) gene promoter by the p53 tumor suppressor protein. Mol Endocrinol. 1997;11:312–329. doi: 10.1210/mend.11.3.9893. [DOI] [PubMed] [Google Scholar]
- Meijer OC, de Lange EC, Breimer DD, de Boer AG, Workel JO, de Kloet ER. Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdr1A P-glycoprotein knockout mice. Endocrinology. 1998;139:1789–1793. doi: 10.1210/endo.139.4.5917. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- Neal CR, Jr, Weidemann G, Kabbaj M, Vazquez DM. Effect of neonatal dexamethasone exposure on growth and neurological development in the adult rat. Am J Physiol Regul Integr Comp Physiol. 2004;287:R375–385. doi: 10.1152/ajpregu.00012.2004. [DOI] [PubMed] [Google Scholar]
- Ramdas J, Harmon JM. Glucocorticoid-induced apoptosis and regulation of NF-kappaB activity in human leukemic T cells. Endocrinology. 1998;139:3813–3821. doi: 10.1210/endo.139.9.6180. [DOI] [PubMed] [Google Scholar]
- Ray R, Chen G, Vande Velde C, Cizeau J, Park JH, Reed JC, Gietz RD, Greenberg AH. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275:1439–1448. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- Reagan LP, McEwen BS. Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. J Chem Neuroanat. 1997;13:149–167. doi: 10.1016/s0891-0618(97)00031-8. [DOI] [PubMed] [Google Scholar]
- Roy M, Sapolsky RM. The exacerbation of hippocampal excitotoxicity by glucocorticoids is not mediated by apoptosis. Neuroendocrinology. 2003;77:24–31. doi: 10.1159/000068337. [DOI] [PubMed] [Google Scholar]
- Sandau US, Handa RJ. Localization and developmental ontogeny of the pro-apoptotic Bnip3 mRNA in the postnatal rat cortex and hippocampus. Brain Res. 2006 doi: 10.1016/j.brainres.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Pulsinelli WA. Glucocorticoids potentiate ischemic injury to neurons: therapeutic implications. Science. 1985;229:1397–1400. doi: 10.1126/science.4035356. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Aguirre-Chen C, Kietzmann T, Saul I, Busto R, Ginsberg MD. Nuclear localization of the hypoxia-regulated pro-apoptotic protein BNIP3 after global brain ischemia in the rat hippocampus. Brain Res. 2004;1001:133–142. doi: 10.1016/j.brainres.2003.11.065. [DOI] [PubMed] [Google Scholar]
- Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–6673. [PubMed] [Google Scholar]
- Tan CK, Yan J, Ananth C, Kaur C. Dexamethasone induces dendritic alteration but not apoptosis in the neurons of the hippocampus in postnatal rats. Neurosci Lett. 2002;326:206–210. doi: 10.1016/s0304-3940(02)00349-x. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin CE, Jr, Ha TP, Packan DR, Tombaugh GC, Yang SH, Horner HC, Sapolsky RM. Glucocorticoids inhibit glucose transport and glutamate uptake in hippocampal astrocytes: implications for glucocorticoid neurotoxicity. J Neurochem. 1991;57:1422–1428. doi: 10.1111/j.1471-4159.1991.tb08309.x. [DOI] [PubMed] [Google Scholar]
- Webster JC, Cidlowski JA. Mechanisms of Glucocorticoid-receptor-mediated Repression of Gene Expression. Trends Endocrinol Metab. 1999;10:396–402. doi: 10.1016/s1043-2760(99)00186-1. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Theodorakis P, Subramanian T, Chinnadurai G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem. 1998;273:12415–12421. doi: 10.1074/jbc.273.20.12415. [DOI] [PubMed] [Google Scholar]
- Yook YH, Kang KH, Maeng O, Kim TR, Lee JO, Kang KI, Kim YS, Paik SG, Lee H. Nitric oxide induces BNIP3 expression that causes cell death in macrophages. Biochem Biophys Res Commun. 2004;321:298–305. doi: 10.1016/j.bbrc.2004.06.144. [DOI] [PubMed] [Google Scholar]