

Abstract
In stomatal guard cells of higher-plant leaves, abscisic acid (ABA) evokes increases in cytosolic free Ca2+ concentration ([Ca2+]i) by means of Ca2+ entry from outside and release from intracellular stores. The mechanism(s) for Ca2+ flux across the plasma membrane is poorly understood. Because [Ca2+]i increases are voltage-sensitive, we suspected a Ca2+ channel at the guard cell plasma membrane that activates on hyperpolarization and is regulated by ABA. We recorded single-channel currents across the Vicia guard cell plasma membrane using Ba2+ as a charge-carrying ion. Both cell-attached and excised-patch measurements uncovered single-channel events with a maximum conductance of 12.8 ± 0.4 pS and a high selectivity for Ba2+ (and Ca2+) over K+ and Cl−. Unlike other Ca2+ channels characterized to date, these channels rectified strongly toward negative voltages with an open probability (Po) that increased with [Ba2+] outside and decreased roughly 10-fold when [Ca2+]i was raised from 200 nM to 2 μM. Adding 20 μM ABA increased Po, initially by 63- to 260-fold; in both cell-attached and excised patches, it shifted the voltage sensitivity for channel activation, and evoked damped oscillations in Po with periods near 50 s. A similar, but delayed response was observed in 0.1 μM ABA. These results identify a Ca2+-selective channel that can account for Ca2+ influx and increases in [Ca2+]i triggered by voltage and ABA, and they imply a close physical coupling at the plasma membrane between ABA perception and Ca2+ channel control.
Keywords: plasma membrane Ca2+ flux, cytosolic free Ca2+ oscillations, K+ channel, inward rectifier, voltage clamp
Calcium ions are ubiquitous second messengers in living cells and contribute to physiological and developmental events in plants and animals (1–3). In plants, changes in cytosolic free Ca2+ concentration ([Ca2+]i) are associated with mechanical and thermal disturbances (3, 4), pathogen attack (5), and nodulation (6) and are central to hormonal physiology (7–9). Changes in [Ca2+]i influence ion channel gating (1, 9, 10), light-mediated gene expression (11), cell differentiation, elongation, and tip growth (3).
In stomatal guard cells, one of the best-characterized plant cell models, increasing [Ca2+]i is known to inactivate inward-rectifying K+ channels and to activate Cl− channels, biasing the plasma membrane for solute efflux, which drives stomatal closure (9). Changes in [Ca2+]i have been associated with stimuli that lead to stomatal closure, notably abscisic acid (ABA) and CO2 (9, 12). These changes in [Ca2+]i depend on Ca2+ release from intracellular stores (13–16) and on Ca2+ entry across the plasma membrane (17, 18). Nonetheless, direct evidence for channels that could mediate Ca2+ influx has been lacking. Indeed, little evidence has come forth for Ca2+ channels at the plasma membrane of higher-plant cells until recently (19–23).
One clue to a major pathway for Ca2+ entry into guard cells has come from measurements of [Ca2+]i and its elevation by ABA under voltage clamp (17). These studies indicated a voltage dependence to the [Ca2+]i rise, suggesting that ABA stimulated a Ca2+ channel, but that its activity also required membrane hyperpolarization. We have recorded single-channel currents from Vicia guard cell protoplasts under conditions that eliminate the background of current through K+ and Cl− channels. The results reported here demonstrate the presence of Ca2+ channels at the plasma membrane that open on membrane hyperpolarization and are activated by ABA.
Materials and Methods
Plant Material.
Epidermal strips of Vicia faba L., cv. Bunyard Exhibition, were obtained and protoplasts were prepared as described (24, 25). All operations were carried out on a Zeiss Axiovert microscope with 40× LWD Nomarski DIC optics at 20–22°C. Solution was added (≃20 chamber vol/min) by gravity feed and removed by aspiration.
Electrophysiology.
Pipettes were pulled with a Narishige (Tokyo) PP-81 puller modified for three-stage pulls (input resistances, 30–50 MΩ) to reduce the number of channels under a patch. Pipettes were coated with Sigmacote (Sigma) to reduce capacitance. Connections to amplifier and bath were by a 0.1 M KCl/Ag–AgCl liquid junctions, and junction potentials were taken into account (26). Single-channel currents were recorded with an Axopatch 200B patch amplifier (Axon Instruments, Foster City, CA) after filtering at 5 kHz and sampled at 44 kHz for analysis. Data were filtered at 1 kHz (Kemo, Beckenham, U.K.) offline and analyzed with n-pro (Wye Science, Wye, Kent, U.K.), p/v clamp v. 6 (CED, Cambridge, U.K.) software. Channel amplitudes were calculated from point-amplitude histograms estimated from open events ≥5-ms duration (Fig. 1) beyond closed levels determined from periods of no channel activity (27). Channel numbers were estimated from the maximum number of concurrent openings and from binomial distributions of open events (28). Channel openings were taken as transitions above thresholds of 60% of the single-channel amplitudes and open probability, Po, was estimated from the open-time fraction corrected for the number of channels. Voltages quoted are referenced to the physiological orientation of the membrane, the voltage on the cytosolic side relative to the extracellular side.
Figure 1.

Ca2+ channels at the plasma membrane of Vicia guard cells uncovered at negative voltages. (A) Representative whole-cell record with voltage clamped to a 30-s ramp from +50 to −200 mV with 30 mM Ba2+ in bath and 30 mM Ba2+ in the pipette. (Inset) Cell-attached records with voltages (in mV) calculated assuming that a protoplast voltage of −50 mV, close to the voltage obtained in whole-cell recordings with the current clamped to zero. (B) Current from one outside-out patch with 30 mM Ba2+ in the pipette (inside) and 10 mM Ba2+ in the bath. Voltages (Left) in mV. (Insets, Right) Point-amplitude histograms (27) used to estimate open-channel amplitudes (see Methods; note the log scale) and single-channel events at −150 mV shown on an expanded time scale. (C) Current from the same patch as in B after adding 100 μM LaCl3 to the bath. Scale (A–C): vertical, 2 pA (A, Inset: 4 pA); horizontal, 1 s (A, Inset: 2 s; B, Inset: 150 ms). (D) Means ± SEM of open-channel currents from outside-out patches (n = 9) with 30 mM Ba2+ inside and 2 (■), 10 (▴), and 30 mM Ba2+ (●) outside. Currents from cell-attached patches (○, n = 8) as in A but with voltage uncorrected for comparison of γBa. Regression analyses (lines) of these data extrapolated to the voltage axis to determine Erev. Means ± SEM of currents from separate measurements with addition of 20 mM Cl− as BaCl2 (▵, n = 5) and with 10 mM Ca2+ (◊, n = 4) in place of Ba2+ outside included for comparison. (Inset) γBa plotted against [Ba2+] and fitted to a simple hyperbolic function.
Chemicals and Solutions.
We use the terms “inside” and “outside” with reference to the physiological sidedness of the membrane. Protoplasts were normally bathed in 2, 10, and 30 mM Ba2+-Hepes or Ca2+-Hepes (pH 7.5) [Hepes buffer titrated to its pKa with Ba(OH)2 or Ca(OH)2] adjusted to 200 mOsM with sorbitol, and pipettes were filled with similar solutions adjusted to 240 mOsM with sorbitol. Mg2ATP at 1 mM was included in solutions on the inside of the membrane. K+ was added as K+-Hepes (pH 7.5) [Hepes buffer adjusted to its pKa with KOH] and Cl− was added as BaCl2. Ca2+ on the inside of the membrane was buffered with EGTA, and the final concentration was calculated according to Foehr et al. (29). ABA was prepared as a stock in ethanol and diluted ≥1,000-fold for use. Ethanol alone (0.1%) had no effect on the channels. Buffers, salts, and ABA were from Sigma.
Results
Guard Cells Harbor a Low-Conductance Channel Selective for Ca2+ and Permeable to Ba2+.
The activity of guard cell Cl− channels is suppressed when the external anion concentration is reduced (30, 31). Guard cell K+ channels are blocked by millimolar Ba2+, and extracellular K+ is required for channel activity of inward-rectifying K+ channels and as a substrate (32). Because Ba2+ also permeates many Ca2+ channels (19, 33, 34), we reasoned that Ca2+ channels might be identified if initially Ba2+ was the only charge-carrying ion. Fig. 1 shows measurements obtained from guard cell protoplasts at different stages of patch excision. Driving the membrane to inside negative voltages uncovered channel events of low amplitude and flickering characteristic in cell-attached measurements and excised patches, and a strongly inward-rectifying current in whole-cell recordings (Fig. 1A). No channel openings or whole-cell currents were observed when the membrane was held near 0 mV or at voltages to +100 mV (not shown). Similar results were obtained in excised patches with voltage under direct experimental control, and on replacing Ba2+ with Ca2+ in the bath (Fig. 1 B and D). No evidence of rundown was observed in these experiments and during recordings of up to 82 min. Channel activity (and whole-cell current, not shown) was blocked by 100 μM Gd3+ or La3+ (Fig. 1C), regardless of whether Ba2+ or Ca2+ was present as the charge-carrying ion.
Open-channel current–voltage curves indicated that the divalent cations were the principal charge-carrying species passing through the channels (Fig. 1D). Because the channels opened only at negative voltages, the current reversal (equilibrium) voltage, Erev, was obtained by extrapolation. These estimates were nonetheless in good agreement with the predicted equilibria and showed the expected positive-going shift in voltage with increasing Ba2+ in the bath. With 30 mM Ba2+ inside and 2, 10, and 30 mM Ba2+ outside, Erev was −34 ± 2, −12 ± 3, and −1 ± 3 mV, respectively. Increasing [Ba2+] outside also increased the single-channel conductance for Ba2+ (γBa), for a maximum γBa of 12.8 ± 0.4 pS and a K1/2 for γBa of 1.6 ± 0.2 mM Ba2+ (Fig. 1D, Inset), and a similar conductance was obtained in cell-attached recordings (Fig. 1D). Replacing Ba2+ with Ca2+ gave virtually identical results (Fig. 1D, ⋄), and replacing Ba2+-Hepes with BaCl2 (Fig. 1D, ▵) or including 5 mM K+ as K+-Hepes in the bath and pipette (not shown) had no measurable effect on the single-channel conductance or the current reversal voltage. With 10 mM Ca2+ outside, Erev was −10 ± 4 mV, and including 20 mM Cl− against a background of 10 mM Ba2+ gave Erev of −9 ± 4 mV. With 5 mM K+ and 2 mM Ba2+ outside, Erev was −36 ± 5 mV, respectively. These results indicate roughly equal permeabilities to Ca2+ and Ba2+, and a high selectivity (>10:1) for these ions over K+ and Cl−.
Ca2+ Channels Are Activated by Membrane Hyperpolarization and Extracellular Divalent Cations.
Previous measurements of [Ca2+]i (17) led us to suspect a steep dependence of channel opening on membrane voltage beyond about −100 mV. Calculated in consecutive 0.2- to 5-s segments showed Po rose immediately (τ ≤ 100 ms) on negative voltage steps (see Fig. 5, trace f). Therefore, to assess channel open probabilities (Po), single channel events were recorded from excised, outside-out patches after stepping the membrane to selected voltages for periods of 100 s.† Fig. 2 summarizes results from experiments, including data from Fig. 1B and measurements with patches transferred between 2, 10, and 30 mM Ba2+ outside (n = 6). In 2 mM Ba2+ outside, Po rose from values near 0.002 at −100 mV to 0.18 at −160 mV. Po also rose with [Ba2+] outside at any one voltage with the effect that the voltage sensitivity for channel opening appeared displaced to the right along the voltage axis. The effect was most pronounced at [Ba2+] outside above 10 mM. A similar voltage dependence was observed with Ca2+ substituting for Ba2+ outside, although Po was reduced in Ca2+ (Fig. 2), compared with the same concentration of Ba2+.
Figure 5.

ABA, but not voltage steps, evokes oscillations in Ca2+ channel activity. Po (data points) and relative net charge, Q (solid lines), calculated at consecutive 5-s intervals in six independent experiments (a–f, three cell-attached, three excised, inside-out patches). Note the different Po scales (Right) for each experiment. Q calculated by summing all points above the zero current level during each 5-s interval and scaled to Po ranges. Segments of current traces c and d (including data from Fig. 4) with closed level (−c) as indicated. Scale: vertical, 2 pA; horizontal, 1 s. Times of ABA addition indicated (|, Left) in each experiment and aligned between c and e. Voltage stepped from −100 to −150 mV at 50 s in f.
Figure 2.

Ca2+ channel open probability (Po) is elevated by membrane hyperpolarization and [Ba2+] outside. Means ± SE of Po from mean open times of 100-s recordings including data of Fig. 1 with 2 (■), 10 (▴), and 30 mM Ba2+ (●) outside, and with 10 mM Ca2+ (◊) in place of Ba2+ outside. Mean ± SE of measurements at −100 mV (n = 3) with 10 mM Ba2+ inside and 30 mM Ba2+ outside (○, compare ●) show the effect of changing [Ba2+] inside. Curves are empirical fittings to a common exponential function.
Changing [Ba2+] on the inside of the membrane had a similar effect on Po (see Fig. 2, ○). However, increasing [Ca2+]i affected Po conversely. Fig. 3 shows the results of measurements at −120 mV from inside-out patches with 10 mM Ba2+ outside and Ca2+ added against a background of 30 mM Ba2+ inside. Raising [Ca2+]i from 200 nM to 2 μM reduced Po roughly 10-fold. Ba2+ often is a poor substitute for Ca2+ in binding/regulatory functions (33, 35, 36), and these results suggest that channel opening is subject to control by [Ca2+]i.
Figure 3.

Po is suppressed by micromolar [Ca2+]i. Means ± SE of Po from mean open times of 100-s recordings at −120 mV (n = 3). Ca2+ added on the cytosolic side (inside) during inside-out recordings against a background of 30 mM Ba2+ and with 10 mM Ba2+ outside. (Insets) Segments of traces at each [Ca2+]i. Data from one patch. Scale: vertical, 1 pA; horizontal, 1 s.
ABA Potentiates Ca2+ Channel Activity.
ABA alters the voltage sensitivity of evoked increases in [Ca2+]i (17), an observation that could be explained if the hormone affected the Po of Ca2+ channels mediating Ca2+ influx across the plasma membrane. To test this idea, we replaced the standard bathing medium with medium containing ABA during recordings. Fig. 4A (Left) shows sections of a continuous recording from one cell-attached patch clamped near −100 mV. Before ABA exposure, the current trace was punctuated by occasional channel openings (upper trace). Po calculated from the 100-s period immediately before adding ABA gave a value of 0.002. Within 60 s of adding 20 μM ABA, the channel activity increased dramatically, consistent with at least seven channels in the patch (lower trace). Po calculated from a 5-s period 60 s after ABA addition was 0.19, equivalent to a 93-fold rise over the value before ABA exposure. Similar results were obtained in six other independent experiments, giving a mean rise in Po of 125-fold from 0.002 ± 0.0005 before to 0.25 ± 0.08 after adding ABA.
Figure 4.

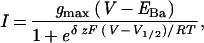 |
1 |
One difficulty with cell-attached measurements lies in the uncertainty about the membrane voltage (see Fig. 1, legend). In principle, the increase in Po might have resulted either directly from an effect of ABA on Ca2+ channel gating or indirectly through a change in voltage. However, whereas Ca2+ channel opening is promoted by hyperpolarization (Fig. 2), the effect of ABA, if any, is to depolarize the membrane (32, 37). Hyperpolarizing the membrane would also be expected to increase open-channel current by virtue of the increase in electrical driving force. Yet, comparing open-channel currents before and after ABA treatments showed no significant difference in current amplitude or single-channel conductance, arguing for a direct effect of ABA on Po rather than one mediated by a change in voltage.
To confirm this interpretation, we carried out recordings with excised, inside-out patches clamping the membrane to −100 mV. In each of four experiments, adding 20 μM ABA resulted in an immediate (<10-s lag time) increase in Ca2+ channel events (Fig. 4A, Right). Equivalent results were obtained in one recording from an outside-out patch, and in two inside-out patches with 0.1 μM ABA, with the difference that Po rose first 110–205 s after adding ABA. In every case, the increase in Po occurred without measurable change in γBa. For the data shown, Po calculated from the 100-s period immediately before adding ABA was 0.003. After 20 s in ABA, at least six channels could be identified in the patch (lower trace), and Po calculated from a 5-s period was 0.55, equivalent to a 183-fold rise over the value before ABA exposure. For all seven experiments, the mean rise in Po was 187-fold from 0.003 ± 0.0001 before to 0.56 ± 0.06 measured 30 s after adding ABA; γBa and Erev, respectively, were 9.8 ± 0.4 pS and 9 ± 3 mV before, and 9.3 ± 3 pS and 8 ± 2 mV after adding ABA.
We also clamped membrane patches driving the voltage in 5-s ramps between 0 and −200 mV, sufficiently long to ensure a Po was at quasi-steady state throughout (see above and Fig. 5). Ramps yielding no channel activity were subtracted from these records to eliminate the baseline (leakage) current of the patch to examine the effect of ABA on the voltage sensitivity of channel activation. The resulting current-voltage curves were fitted to a Boltzmann function (Fig. 4) to estimate the voltage giving half-maximal activation, V1/2. Fig. 4B shows results from one inside-out patch, 40 ramps sampled before and 10 ramps sampled immediately after adding 20 μM ABA. Before adding ABA, very little current was evident at voltages positive −120 mV, whereas in ABA an appreciable current was evident even at voltages near −50 mV. For the currents recorded in ABA, fitting gave a V1/2 of −83 ± 2 mV. Best fittings for currents recorded before ABA addition gave a V1/2 near −140 mV, but, because of the low signal level, this value should be seen as a rough estimate only. Comparable results were obtained in three other experiments. Thus, the analyses indicate a significant increase in current positive of −100 mV in the presence of ABA.
ABA Evokes Oscillations in Ca2+ Channel Open Probability.
Both cell-attached and excised patch recordings showed that Po rose transiently in ABA and that, even after washing 5–8 min without ABA, subsequent exposures failed to elicit a second rise in Po. We also noted a delay in response to ABA in cell-attached recordings, whereas in inside-out patches the rise in Po occurred roughly within the time required (≈5 s) for exchange of the bath solution. These observations suggested that elements necessary for Ca2+ channel activation and its regulation by ABA were retained on the excised membrane, and raised a question about the temporal characteristics of response. To detail the time course of Ca2+ channel activation, we calculated Po for consecutive 5-s segments of recordings before and during treatments with 20 μM ABA with the membrane clamped throughout at or near −100 mV. For comparison, similar calculations were carried out for voltage steps to −150 mV and using total charge, taken as the data-point amplitude sum above the closed-channel level. Fig. 5 shows measurements from six experiments (three cell-attached, three excised patches). In all but one case (Fig. 5, trace a), ABA exposures led to damped oscillations in Po. Equivalent oscillations were seen in total charge (solid lines), so discounting possible errors in estimating Po, channel number or γBa. Both cell-attached and excised, inside-out recordings showed a similar period of oscillation near 50 s [means of (n) patches: cell-attached, 53 ± 6 s (5); inside-out, 55 ± 8 s (4)]. No oscillations were observed following voltage steps alone (Fig. 5, trace f). The initial rise of Po in cell-attached measurements was delayed by 23–57 s (mean, 33 ± 4 s). For the inside-out patches, however, a significant increase in Po was evident in every case during the first 5-s time segment after adding ABA.
Discussion
In guard cells, ABA evokes increases in [Ca2+]i that depend in part on Ca2+ entry across the plasma membrane. Although a role for Ca2+ channels has been suggested (18, 38–40)—most recently based on the voltage sensitivity of evoked [Ca2+]i increases (17)—direct evidence for such channels has been lacking. We now demonstrate that Vicia guard cells harbor a low-conductance Ca2+ channel at the plasma membrane that is selective for Ca2+ (and Ba2+) over K+ and Cl−, and is blocked by the Ca2+ channel antagonists Gd3+ and La3+ (Fig. 1). Channel open probability (Po) was increased by membrane hyperpolarization, both in cell-attached and excised-patch recordings, and was favored by increasing divalent concentration outside (Fig. 2), whereas increasing [Ca2+]i suppressed Po (Fig. 3). Channel opening was strongly and transiently enhanced during exposures to ABA, leading to a significant current at voltages as positive as −50 mV (Fig. 4) and oscillations in Po (Fig. 5). These results match the characteristics anticipated from measurements of [Ca2+]i (17), and point to a close physical association between the Ca2+ channel and the site of perception for ABA.
A key feature of the Ca2+ channel is its activation by negative membrane voltage. Channel openings were extremely rare at voltages above −50 mV both in excised and, to a best estimate, in cell-attached patches. Channel openings were generally evident at voltages negative of −100 mV in 2 and 10 mM Ba2+ or Ca2+ outside, with Po rising from values near 0.002 to 0.15–0.2 at −160 mV. The observations imply an appreciable Ca2+ influx only on membrane hyperpolarization and, thus, complement evidence of voltage-evoked changes in [Ca2+]i. Grabov and Blatt (17) reported increases in [Ca2+]i that were triggered when the guard cell plasma membrane was hyperpolarized beyond a threshold near −120 mV. They showed that the rise in [Ca2+]i depended on a voltage-triggered influx of Ca2+ and was sensitive to Gd3+. Furthermore, they found that increasing [Ca2+] in the bath accelerated the rate of [Ca2+]i rise during negative voltage steps, consistent with our present observation that Po rose with [Ba2+] outside (Fig. 2). Thus, the characteristics of the Ca2+ channel match closely the properties anticipated from these earlier measurements of voltage-evoked [Ca2+]i increases.
In fact, the sensitivity to negative voltages may be a common feature of plant plasma-membrane Ca2+ channels that effect [Ca2+]i changes. Stoeckel and Takeda (41) reported an inactivation of K+ channels in Mimosa that was relieved by Gd3+, and proposed that Ca2+ channel activation at the plasma membrane triggered a rise in [Ca2+]i to mediate this effect. Hyperpolarization-activated Ca2+-permeable channels occur in tomato (19) and have been implicated in response to pathogen attack (20, 42). However, the acute voltage-sensitivity, high selectivity for Ca2+ over K+ and Cl−, and activation by ABA distinguish the present data from reports of a nonselective, Ca2+-permeable channel (43) and of Ca2+ entry through K+ channels (44). The guard cell Ca2+ channel also differs from depolarization-activated Ca2+ channels found in wheat roots (22) and carrot cell cultures (21).
A significant feature of the Ca2+ channel was its sensitivity to [Ca2+]i. Increasing [Ca2+]i to 200 nM had little influence on Po, but at 2 μM [Ca2+]i reduced Po by roughly 10-fold (Fig. 3). Because [Ca2+]i normally lies near 200 nM in vivo, but on stimulation can rise to values above 1 μM (12, 15–17), this sensitivity of Po implies a critical role of [Ca2+]i in feedback regulation of the channel. Similar characteristics of [Ca2+]i regulation are known for mammalian L-type Ca2+ channels and are associated with Ca2+ binding sites on the channel protein (45, 46).
Equally intriguing is the sensitivity of the Ca2+ channel gate to divalent cation concentration outside (Fig. 2), an effect that is distinct from any change in open-channel conductance. This action on Po differs from that of [Ca2+]i, and suggests a unique divalent cation binding site outside that interacts with the channel gate with a K1/2 above 10 mM. To our knowledge, these characteristics are without precedent among Ca2+ channels in plants and animals. For neuronal Ca2+ channels, limited increases in Po have been reported with external [Ca2+], but are the effect of surface-charge masking and saturate below 2 mM [Ca2+] (36). The action of external Ca2+ and Ba2+ on the guard cell Ca2+ channel does show similarities to K+ action on outward-rectifying K+ channels in yeast (47) and many plant cells, including guard cells (32, 48). Whether the mechanism(s) of action are analogous remains to be determined.
Most remarkable was our finding of Ca2+ channel activation by ABA. Channel open probability increased in ABA by up to 210-fold in cell-attached recordings, by up to 260-fold in excised patches, paralleling total charge flux, Q (Figs. 4 and 5). This activation was transient and was accompanied by a profound increase in current at voltages positive of −100 mV. Again, these characteristics are anticipated from measurements of [Ca2+]i, which showed that ABA treatments displace the voltage threshold necessary to raise [Ca2+]i (17). We noted also a significant delay before Po rose when ABA was added to the bath in the cell-attached recordings. By contrast, Po and Q rose without a measurable lag when ABA was added to the cytosolic side of the membrane in inside-out patches (Fig. 5). Little ABA in these measurements would have been present as the undissociated free acid (pKa = 4.7) and, therefore, membrane permeable at pH 7.5. So, the observation is consistent with, although not proof of, ABA acting at a binding site on the inside of the membrane.
The predicted Ca2+ flux through the channel is plausibly sufficient to raise or trigger a rise in [Ca2+]i in the presence of ABA. From the number of opening events evident in patches in the presence of ABA, we estimate that a Vicia guard cell harbors a minimum of 5,000 Ca2+ channels.† Given conditions of 1 mM Ca2+ outside, γCa could be reduced to approximately 2 pS (Fig. 1, Inset), although with 1 μM [Ca2+]i and a membrane voltage of −120 mV (17), the driving force for Ca2+ entry would be close to +220 mV. If we assume that Po rises to 0.1 in ABA, then the total Ca2+ current calculated is 200 pA and the Ca2+ flux is 2 fmol⋅s−1. For a guard cell with a cytosolic volume of 1 pl, this flux translates into a rate of rise in total cytosolic [Ca2+] close to 2 mM⋅s−1. Even allowing for a static Ca2+ buffer content of 50 μM (49), this value is well in excess of what would be required to elevate [Ca2+]i. At the same time, the current would be masked by the background of K+ and Cl− currents in whole-cell measurements (37, 50) and, in the absence of ABA, the Ca2+ current (<2 pA) would not be resolved under these conditions.
Because the Ca2+ channels responded to ABA even in excised patches (Figs. 4 and 5), our data also point to a close physical association between site of ABA perception and the Ca2+ channel. One intriguing discovery was that Po recovered to near-initial values through damped oscillations specific to the ABA stimulus (Fig. 5). ABA is known to evoke oscillations in [Ca2+]i of guard cells (16), although these events may be coupled secondarily to oscillations in membrane voltage (17, 51). In fact, the oscillations in Ca2+-channel Po occurred with a period roughly an order of magnitude shorter than free-running oscillations in [Ca2+]i reported in ABA and, therefore, alone cannot account for these phenomena. Nonetheless, the fact that the oscillations in Po were observed in excised patches, and that subsequent exposures to ABA failed to evoke a response, points to a subtle mechanism that regulates the Ca2+ channel and indicates that this mechanism, and other elements of the ABA signal cascade, remain intact associated with the plasma membrane. We can rule out a direct role for [Ca2+]i as a regulatory factor, because the oscillations were observed in excised patches and with Ba2+ (not Ca2+) on both sides of the membrane. At present, the nature of these associations remains unknown, but are clearly of tremendous interest.
What is the physiological significance of activating the Ca2+ channels by hyperpolarization? We view control by voltage as integrating the ABA signal with the prevailing transport “poise” of the membrane (17). One consequence of raising [Ca2+]i is to reduce current through inward-rectifying K+ channels while activating Cl- channels (9, 10) to drive the plasma membrane positive of EK for solute loss and stomatal closure. However, if Ca2+ channel activation functions to trigger a rise in [Ca2+]i for membrane depolarization when the voltage is well negative of EK, the same [Ca2+]i rise must be superfluous when the voltage is already positive of EK and, hence, biased for solute loss. Thus, the dual effects of voltage and ABA on the Ca2+ channel ensures that the [Ca2+]i “signature” correctly reflects cellular needs for osmotic solute loss.
In conclusion, we have identified a low-conductance Ca2+ channel at the plasma membrane of Vicia guard cells that is selective for Ca2+ (and Ba2+) over K+ and Cl−. We show that channel activation is stimulated by increasing [Ba2+] outside and suppressed by raising [Ca2+]i above physiological resting values. Ca2+ channel opening is strongly promoted by membrane hyperpolarization and by ABA, consistent with a role for the channel in triggering increases in [Ca2+]i that lead to stomatal closure. ABA evokes damped oscillations in channel activity, and the Ca2+ channel retains its sensitivity to ABA even in excised membrane patches, indicating that the essential elements for ABA perception, signal transduction, and Ca2+ channel control are retained intact on the membrane.
Acknowledgments
We thank Gerhard Thiel and Alexander Grabov for comments on the manuscript. This work was supported in part by the Biotechnology and Biological Sciences Research Council Grants P09640, C10234, and P09561, and European Community Biotech Grant CT96-0062. D.H. was a Sainsbury Ph.D. Student.
Abbreviations
- [Ca2+]i
cytosolic free Ca2+ concentration
- ABA
abscisic acid
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.080068897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.080068897
The true number of channels per cell is difficult to estimate, but is almost certainly greater than this value. Most patches showed one to three channel levels (<4% in total showed no Ca2+ channels), and as many levels again were uncovered in ABA. For this same reason, values given for Po are probably overestimated in proportion. An underestimate of 2-fold in the channel number N introduces an error of 50% in the estimate for Po (28). We have assumed an average of five channels (the mean in ABA), a patch area of 1 μm2, a cell surface area of 1,000 μm2 (17, 37), and values for Po of 0.001 before and 0.1 after adding ABA. Total membrane current I = γCa(V − ECa)NPo, where V is the membrane voltage and ECa is the Ca2+ equilibrium voltage.
References
- 1.Sanders D, Brosnan J M, Muir S R, Allen G, Crofts A, Johannes E. Biochem. Soc. Symp. 1994. 183–197. [PubMed] [Google Scholar]
- 2.Bootman M D, Berridge M J. Curr Biol. 1996;6:855–865. doi: 10.1016/s0960-9822(02)00609-7. [DOI] [PubMed] [Google Scholar]
- 3.Taylor L P, Hepler P K. Annu Rev Plant Physiol Mol Biol. 1997;48:461–491. doi: 10.1146/annurev.arplant.48.1.461. [DOI] [PubMed] [Google Scholar]
- 4.Knight H, Trewavas A J, Knight M R. Plant Cell. 1996;8:489–503. doi: 10.1105/tpc.8.3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammond-Kosack K E, Jones J D G. Plant Cell. 1996;8:1773–1791. doi: 10.1105/tpc.8.10.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehrhardt D W, Wais R, Long S R. Cell. 1996;85:673–681. doi: 10.1016/s0092-8674(00)81234-9. [DOI] [PubMed] [Google Scholar]
- 7.Ritchie S, Gilroy S. New Phytol. 1998;140:363–383. doi: 10.1111/j.1469-8137.1998.00299.x. [DOI] [PubMed] [Google Scholar]
- 8.Bush D S. Plant Physiol. 1993;103:7–13. doi: 10.1104/pp.103.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blatt M R, Grabov A. Physiol Plant. 1997;100:481–490. [Google Scholar]
- 10.Ward J M, Pei Z M, Schroeder J I. Plant Cell. 1995;7:833–844. doi: 10.1105/tpc.7.7.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnes S A, McGrath R B, Chua N H. Trends Cell Biol. 1997;7:21–26. doi: 10.1016/S0962-8924(97)10043-5. [DOI] [PubMed] [Google Scholar]
- 12.McAinsh M R, Brownlee C, Hetherington A M. Plant Cell. 1992;4:1113–1122. doi: 10.1105/tpc.4.9.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grabov A, Blatt M R. Plant Physiol. 1999;119:277–287. doi: 10.1104/pp.119.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leckie C P, McAinsh M R, Allen G J, Sanders D, Hetherington A M. Proc Natl Acad Sci USA. 1998;95:15837–15842. doi: 10.1073/pnas.95.26.15837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilroy S, Fricker M D, Read N D, Trewavas A J. Plant Cell. 1991;3:333–344. doi: 10.1105/tpc.3.4.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staxen I, Pical C, Montgomery L T, Gray J E, Hetherington A M, McAinsh M R. Proc Natl Acad Sci USA. 1999;96:1779–1784. doi: 10.1073/pnas.96.4.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grabov A, Blatt M R. Proc Natl Acad Sci USA. 1998;95:4778–4783. doi: 10.1073/pnas.95.8.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Webb A A R, McAinsh M R, Mansfield T A, Hetherington A M. Plant J. 1996;9:297–304. [Google Scholar]
- 19.Gelli A, Blumwald E. J Membr Biol. 1997;155:35–45. doi: 10.1007/s002329900156. [DOI] [PubMed] [Google Scholar]
- 20.Zimmermann S, Nurnberger T, Frachisse J M, Wirtz W, Guern J, Hedrich R, Scheel D. Proc Natl Acad Sci USA. 1997;94:2751–2755. doi: 10.1073/pnas.94.6.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thuleau P, Ward J M, Ranjeva R, Schroeder J I. EMBO J. 1994;13:2970–2975. doi: 10.1002/j.1460-2075.1994.tb06595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pineros M, Tester M. Planta. 1995;195:478–488. [Google Scholar]
- 23.Ding J P, Pickard B G. Plant J. 1993;3:83–110. doi: 10.1111/j.1365-313x.1993.tb00013.x. [DOI] [PubMed] [Google Scholar]
- 24.Blatt M R, Armstrong F. Planta. 1993;191:330–341. [Google Scholar]
- 25.Elzenga J T M, Keller C P, Van Volkenburgh E. Plant Physiol. 1991;97:1573–1575. doi: 10.1104/pp.97.4.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barry P H, Lynch J W. J Membr Biol. 1991;121:101–117. doi: 10.1007/BF01870526. [DOI] [PubMed] [Google Scholar]
- 27.Colquhoun D, Sigworth F J. In: Single Channel Recording. Sakmann B, Neher E, editors. New York: Plenum; 1995. pp. 397–482. [Google Scholar]
- 28.Horn R. Biophys J. 1991;60:433–439. doi: 10.1016/S0006-3495(91)82069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foehr G, Warchol W, Gratzl G. Methods Enzymol. 1993;221:149–157. doi: 10.1016/0076-6879(93)21014-y. [DOI] [PubMed] [Google Scholar]
- 30.Dietrich P, Hedrich R. Plant J. 1998;15:479–487. [Google Scholar]
- 31.Grabov A, Leung J, Giraudat J, Blatt M R. Plant J. 1997;12:203–213. doi: 10.1046/j.1365-313x.1997.12010203.x. [DOI] [PubMed] [Google Scholar]
- 32.Thiel G, Wolf A H. Trends Plant Sci. 1997;2:339–345. [Google Scholar]
- 33.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 1992. [Google Scholar]
- 34.Klusener B, Weiler E W. Plant Physiol. 1999;119:1399–1405. doi: 10.1104/pp.119.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chapman E R, An S, Edwardson J M, Jahn R. J Biol Chem. 1996;271:5844–5849. doi: 10.1074/jbc.271.10.5844. [DOI] [PubMed] [Google Scholar]
- 36.Zamponi G W, Snutch T P. Pflügers Arch Eur J Physiol. 1996;431:470–472. doi: 10.1007/BF02207290. [DOI] [PubMed] [Google Scholar]
- 37.Thiel G, MacRobbie E A C, Blatt M R. J Membr Biol. 1992;126:1–18. doi: 10.1007/BF00233456. [DOI] [PubMed] [Google Scholar]
- 38.McAinsh M R, Brownlee C, Hetherington A M. Physiol Plant. 1997;100:16–29. [Google Scholar]
- 39.McAinsh M R, Brownlee C, Hetherington A M. Proc R Soc London Ser B. 1991;243:195–202. [Google Scholar]
- 40.MacRobbie E A C. Planta. 1989;178:231–241. doi: 10.1007/BF00393199. [DOI] [PubMed] [Google Scholar]
- 41.Stoeckel H, Takeda K. J Membr Biol. 1995;146:201–209. doi: 10.1007/BF00238009. [DOI] [PubMed] [Google Scholar]
- 42.Gelli A, Higgins V J, Blumwald E. Plant Physiol. 1997;113:269–279. doi: 10.1104/pp.113.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schroeder J I, Hagiwara S. Proc Natl Acad Sci USA. 1990;87:9305–9309. doi: 10.1073/pnas.87.23.9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fairley-Grenot K A, Assmann S M. J Membr Biol. 1992;128:103–113. doi: 10.1007/BF00231883. [DOI] [PubMed] [Google Scholar]
- 45.Berridge M J. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J M, Olcese R, Qin N, Noceti F, Birnbaumer L, Stefani E. Proc Natl Acad Sci USA. 1997;94:2301–2305. doi: 10.1073/pnas.94.6.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vergani P, Hamilton D, Jarvis S, Blatt M R. EMBO J. 1998;17:7190–7198. doi: 10.1093/emboj/17.24.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blatt M R, Gradmann D. J Membr Biol. 1997;158:241–256. doi: 10.1007/s002329900261. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Z A, Neher E. J Physiol (London) 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemtiri-Chlieh F, MacRobbie E A C. J Membr Biol. 1994;137:99–107. doi: 10.1007/BF00233479. [DOI] [PubMed] [Google Scholar]
- 51.Gradmann D, Blatt M R, Thiel G. J Membr Biol. 1993;136:327–332. doi: 10.1007/BF00233671. [DOI] [PubMed] [Google Scholar]