

Abstract
The trafficking of leukocytes from the blood to sites of inflammation is the cumulative result of receptor-ligand-mediated signaling events associated with the leukocytes themselves as well as with the underlying vascular endothelium. Our data show that Gαi signaling pathways in the vascular endothelium regulate a critical step required for leukocyte diapedesis. In vivo studies using knockout mice demonstrated that a signaling event in a non-lymphohematopoietic compartment of the lung prevented the recruitment of proinflammatory leukocytes. Intravital microscopy showed that blockade was at the capillary endothelial surface andex vivo studies of leukocyte trafficking demonstrated that a Gαi-signaling event in endothelial cells was required for transmigration. Collectively, these data suggest that specific Gαi2-mediated signaling between endothelial cells and leukocytes is required for the extravasation of leukocytes and for tissue-specific accumulation.
Keywords: G proteins, inflammation, knockout mice, leukocyte trafficking, pulmonary models
The tissue-specific recruitment of polymorphonucleated granulocytes (i.e., neutrophils, basophils, and eosinophils) and lymphocytes has been particularly well studied, including the mechanisms mediating leukocyte tethering, rolling, adhesion, and eventual transmigration from the circulation (1). These studies have suggested that receptor–ligand interactions coupled to Gαi-containing heterotrimeric G proteins in leukocytes are particularly important for the vectorial movement of leukocytes to tissues in response to chemokine gradients (2). Thus, these receptors represent potential drug targets for therapeutic approaches directed against inflammatory diseases (3).
The activation of Gαi-coupled receptors leads to the dissociation of the heterotrimeric G protein and intracellular signaling events mediated by the release of the Gαi subunit (Gαi) bound to GTP and the free Gβγ dimer (4). The Gαi family includes genes encoding the subunits Gαi1, Gαi2, Gαi3, Gαo, and Gαz (5). The activities of Gαi family members, with the exception of Gαz, are distinguishable from other Gα subunits by their susceptibility to pertussis toxin (PTX). The Gαi1, Gαi2, and Gαi3 subunits are expressed in many leukocytes and tissues involved in allergic inflammation such as granulocytes (6), lymphocytes (7), airway smooth muscle (8), airway epithelium (8), and endothelial cells (9). Leukocytes that express these Gαi family members appear to express all three Gαi subunits simultaneously. However, studies assessing expression and function have suggested that heterotrimeric complexes containing Gαi2 and Gαi3 are abundant in leucocytes (2). Moreover, the importance of Gαi-mediated signaling events in leukocyte recruitment/accumulation has been highlighted by using mouse models capitalizing on the inhibitory character of PTX (6). However, the specific mechanisms and, more importantly, the relevant cell types involved in facilitating leukocyte accumulation remain largely unknown.
Results and Discussion
Gαi2-Signaling Pathways in a Resident Cell of the Lung Are Required for the Accumulation of Eosinophils After Allergen Provocation.
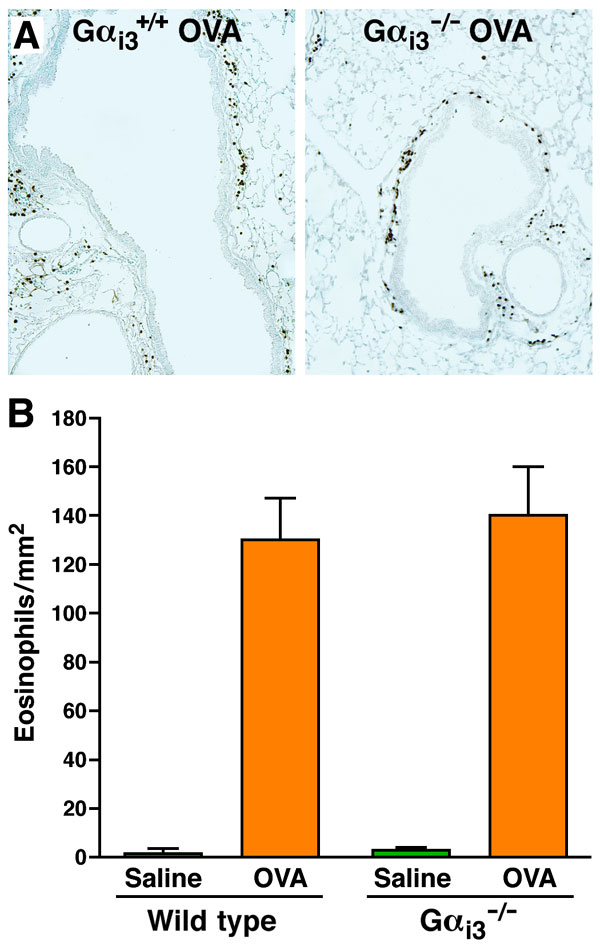
The recruitment and accumulation of eosinophils in the airway lumen and lung tissue after allergen provocation is a defining feature found in both asthma patients (10) and animal models of allergic respiratory inflammation (11). Gαi-coupled CCR3 receptor–ligand interactions promoting chemotaxis are primarily responsible for the allergen-induced accumulation of pulmonary eosinophils (12). Gαi2 and Gαi3 transcripts dominate the mRNAs encoding the PTX-sensitive Gαi subunits of mouse peripheral blood eosinophils (supporting information (SI) Fig. 5). We therefore examined mice deficient in either Gαi2 or Gαi3 for airway eosinophilia during asthma induced by sensitization and aerosol challenge with ovalbumin (OVA). The accumulation of eosinophils in the airway lumen of Gαi2−/− mice was significantly reduced relative to wild-type animals (Fig. 1A), but induced eosinophilia in Gαi3−/− was unaffected (Fig. 1B). A similar reduction in eosinophil accumulation was seen in the peribronchial areas of the lungs from Gαi2−/−mice but not Gαi3−/− mice relative to wild-type animals (SI Figs. 6 and 7). These data indicate that Gαi2-dependent signaling pathways are required for eosinophil entry into tissues from the circulation.
Fig. 1.
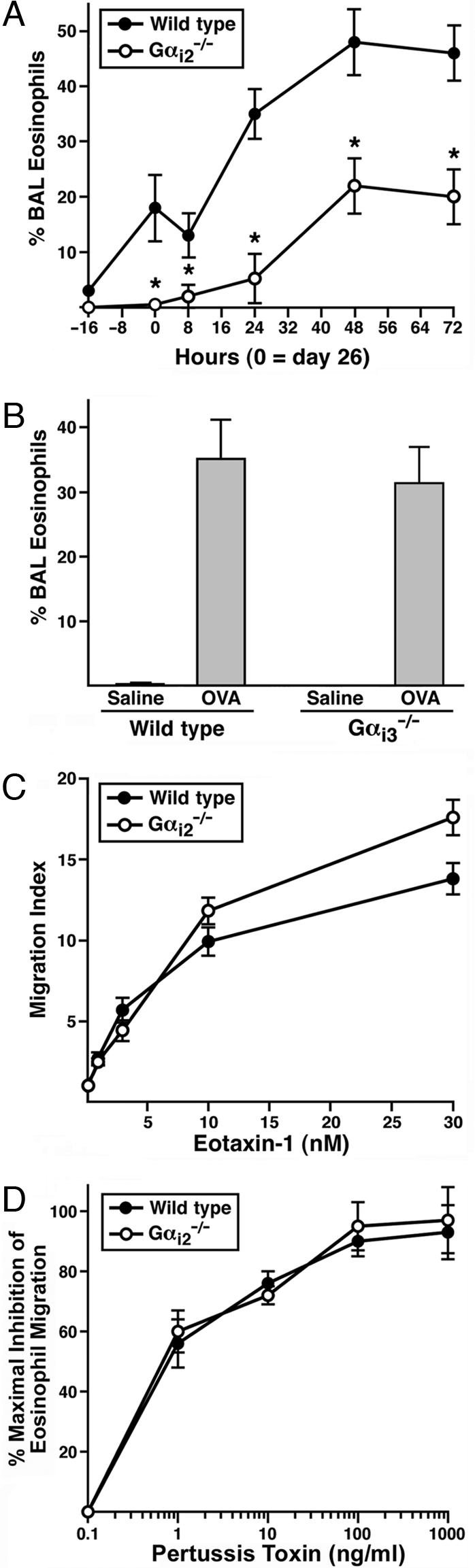
Allergen-induced eosinophil accumulation uniquely relies on Gαi2 signaling mechanisms that are independent of cell autonomous events in eosinophils. OVA-induced eosinophil accumulation in the airway lumen of Gαi2−/− (A), but not Gαi3−/− (B), mice was significantly lower (∗, P < 0.05) relative to wild-type animals (n = 8 mice per group). (C) Eosinophil transwell chemotaxis assays demonstrated that in the absence of Gαi2 signaling events, in vitro eosinophil migration to recombinant mouse eotaxin-1 was the same, if not nominally higher, relative to wild type. (D) PTX pretreatment of eosinophils before the transwell chemotaxis assay showed that the blockade of all Gαi-signaling events abolished eotaxin-1-induced chemotaxis, thus demonstrating that the CCR3 receptor-mediated eosinophil chemotaxis occurring in the absence of Gαi2 results exclusively from signaling events using the remaining PTX-sensitive Gα subunits, Gαi1 and/or Gαi3.
In vitro chemotaxis assays demonstrated that the loss of Gαi2 signaling in eosinophils did not prevent Gαi-coupled receptor-mediated chemotaxis (e.g., CCR3-mediated responses to eotaxin-1/-2; ref. 12); instead, the loss of Gαi2 may have even enhanced the ability of the granulocytes to respond to chemoattractant (Fig. 1C). Furthermore, these studies showed that the eotaxin-CCR3-mediated chemotactic response of Gαi2-deficient eosinophils was abolished by PTX (Fig. 1D), demonstrating that the signal transduction pathways mediating chemotaxis used the remaining Gαi family members. These data support the hypothesis that Gαi signaling events modulate eosinophil responses to available chemoattractants in vitro. However, the loss of allergen-induced pulmonary eosinophilia in Gαi2 knockout mice is unlikely to be a consequence of an eosinophil-specific Gαi2 deficiency but instead might depend on Gαi2 in other cells or on other signals.
Adoptive transfer of wild-type vs. Gαi2−/− eosinophils into OVA-treated wild-type recipient mice was performed as an in vivo approach to confirm our in vitro observation that the loss of Gαi2 in eosinophils appeared to enhance chemotactic responses and therefore was not directly responsible for the lack of eosinophil accumulation observed in the lungs of OVA-treated Gαi2−/− mice. IL-5 transgenic mice (NJ.1638; ref. 13) or compound Gαi2-deficient IL-5 transgenic animals (Gαi2−/−/NJ.1638) were used to isolate pure (>98.5%) populations of wild-type and Gαi2−/− peripheral blood eosinophils, respectively. Adoptive transfer was achieved by the repeated i.p. instillation of eosinophils into sensitized wild-type mice during the challenge phase of the OVA protocol. In some studies, the eosinophils were labeled ex vivo with the fluorescent tag carboxyfluorescein diacetate-succinimidyl ester (CFDA-SE; Molecular Probes Inc., Eugene, OR) to confirm the identity of the eosinophils in the lung as being derived from those adoptively transferred (SI Fig. 8). Assessment of airway cell populations after adoptive transfer of wild-type eosinophils demonstrated that their increased availability in circulation resulted in a greater number of airway eosinophils (Fig. 2A). However, the adoptive transfer of the same number of Gαi2−/− eosinophils into OVA-treated wild-type animals resulted in an elevated OVA-induced airway eosinophilia that was even greater than the increase observed after transfer of wild-type eosinophils (Fig. 2A). This pattern also extended to lung tissue where transfer of Gαi2−/− eosinophils resulted in higher levels of accumulation compared with transfer of wild-type eosinophils (Fig. 2A and SI Fig. 9). These transfer studies collectively confirmed our in vitro transwell chemotaxis results and showed that Gαi2−/− eosinophils are capable of responding to chemotactic gradients and, if anything, may have a greater inherent ability for migration relative to wild-type eosinophils. Interestingly, the reciprocal transfer of wild-type eosinophils into OVA-treated Gαi2−/− mice did not result in a pulmonary eosinophilia. Thus, allergen-induced recruitment of eosinophils in this model was absolutely dependent on the presence of a Gαi2 signaling pathway in the recipient mice, regardless of the Gαi2−/− status of the eosinophils (Fig. 2A).
Fig. 2.

Gαi2 signaling in a non-lymphohematopoietic compartment(s) in the lung is necessary for eosinophil accumulation after allergen provocation. (A) Adoptive transfer of eosinophils into OVA-treated wild-type recipients (n = 8–10 mice per group) demonstrated that Gαi2−/− eosinophils have an increased ability to traffic to both the airway lumen and the peribronchial areas of the lung. In contrast, the reciprocal transfer of wild-type eosinophils into OVA-treated Gαi2−/− recipients demonstrated that eosinophil accumulation depended on lung-associated Gαi2 signaling. Ø, transfer of PBS vehicle alone. ∗, P < 0.05; †, significantly different (P < 0.05) from all other OVA groups examined. (B) Bone marrow engraftment of wild type recipients (Donor Marrow) demonstrated that the lack of eosinophil recruitment to the lung observed in Gαi2−/− mice (Non-Irradiated) was a consequence of a Gαi2 signaling event(s) in a lung structural cell type(s); n = 10–12 mice per group, ∗, P < 0.05. (C) OVA-induced pulmonary Th2 cytokines and IFN-γ levels in Gαi2−/− mice are unaffected relative to OVA-treated wild type (Non-Irradiated). Moreover, OVA-induced Th2 cytokine and IFN-γ levels in OVA-treated mice after engraftment of Gαi2 marrow into wild-type recipients was also unaffected relative to OVA-treated wild-type controls (Donor Marrow). All values presented are means ± SEM (n = 8 mice per group).
The widespread expression of Gαi2 among all leukocytes, including cells necessary for acquired immune responses (e.g., dendritic cells, B cells, and T lymphocytes; ref. 4), suggested the possibility that the observed decrease in eosinophil accumulation in the lungs of OVA-treated Gαi2−/− mice was simply a consequence of signaling defects limiting immune responses with a consequent loss of eosinophilia. To test this hypothesis, we examined asthma-induced eosinophilia in bone marrow reconstituted mice in which all hematopoietic, but not the other host cell, lacked Gαi2. OVA sensitization and challenge of mice reconstituted with wild-type cells revealed bronchoalveolar lavage (BAL) eosinophilia comparable to normal mice. In contrast the eosinophilic response in mice reconstituted with Gαi2−/− bone marrow was nearly 3-fold greater. Thus the requirement for Gαi2 in asthma-induced eosinophilia is not dependent on lymphohematopoietic cells. In accord with this finding, the immune response itself during OVA-induced asthma was not reduced in Gαi2−/− mice, as assessed by production of IL-4, IL-5, or IFN-γ (Fig. 2C). Moreover, OVA treatment of irradiated wild-type recipients after engraftment of wild-type vs. Gαi2−/− bone marrow also displayed equivalent BAL cytokine levels, demonstrating that the Gαi2-dependent differences in pulmonary eosinophil accumulation were not the result of compromised OVA-induced immune responses (Fig. 2C). Attempts to do the reciprocal engraftment of wild-type marrow into Gαi2−/− recipient mice have been unsuccessful (i.e., the mice die of an apparent septicemia within 5–7 days of engraftment), suggesting that Gαi2 signaling events in the non-lymphohematopoietic tissues of these knockout recipients may be necessary for the recruitment and incorporation of stem cells in the marrow compartments of these mice.
Our studies involving the transfer of eosinophils indicated that the lack of pulmonary eosinophils during asthma in Gαi2−/− mice was not due to a lack of production of eosinophils. Furthermore, assessments of eosinophilopoiesis in the bone marrow of Gαi2−/− animals showed no effect on the production of circulating eosinophils in allergen naïve and allergen sensitized/aerosol-challenged mice (data not shown). However, leukocyte counts and cell differentials did show that eosinophils “backed-up” in the peripheral blood of Gαi2−/− mice, accumulating to higher levels in the peripheral blood of both allergen naïve and OVA-treated animals (Fig. 3A). Moreover, and in agreement with published observations (14), this increased accumulation extended equally to all white blood cells, each differentially accumulating to higher levels in Gαi2−/− mice relative to wild-type control groups (Fig. 3A).
Fig. 3.

The loss of Gαi2 signaling in knockout mice leads to nonspecific increases in all circulating white blood cell types and severely limits LPS-induced airway neutrophil accumulation. (A) Increase in all white blood cell types is observed in both allergen-naïve and OVA-treated Gαi2−/− mice. The data presented represent means ± SEM (n = 5 mice per group). ∗, significantly different (P < 0.05) from wild-type saline control mice. †, significantly different (P < 0.05) from OVA-treated wild-type mice. (B) LPS administered to Gαi2−/− or Gαi3−/− mice (n = 7–10 mice per group; wild-type animals served as negative controls) showed that the induced BAL neutrophil levels 16 h after administration were significantly decreased in Gαi2−/− mice but unaffected in Gαi3−/− animals. ∗, P < 0.05. (C) Transwell chemotaxis assays demonstrated that in the absence of Gαi2 signaling events, in vitro neutrophil migration to MIP-2 was not lower but instead nominally higher relative to wild type.
Endotoxin-Mediated Pulmonary Accumulation of Neutrophils also Occurs as a Function of a Gαi2-Signaling Pathways in a Resident Cell of the Lung.
To extend our studies on the role of Gαi2 in leukocyte migration to the lungs, we next assessed the recruitment and tissue accumulation of neutrophils as a function of an innate response to endotoxin; a model system that has been exploited in pulmonary studies by intranasal administration of LPS (see, for example, ref. 15). As with eosinophils, neutrophils have been previously characterized as expressing both Gαi2 and Gαi3 subunits (16), but the accumulation of these leukocytes in the lung after LPS administration was different in Gαi2 vs. Gαi3 knockout mice. The loss of Gαi2 nearly abolished the LPS-induced pulmonary neutrophilia (Fig. 3B), whereas the neutrophilia induced in Gαi3-deficient mice was unaffected relative to wild-type animals (Fig. 3B). Despite the loss of LPS-induced neutrophil accumulation in the lungs of Gαi2−/− mice, this phenomenon did not appear to be a consequence of reduced chemotaxis resulting from the Gαi2 deficiency in the neutrophil itself. In vitro assessments of chemotaxis by using transwell assays (Fig. 3C) demonstrated that Gαi2-deficient neutrophils were capable of migrating in response to a ligand for the Gαi-coupled receptor CXCR2 (MIP-2; ref. 17). Interestingly, in vitro MIP-2-mediated chemotaxis of Gαi2-deficient neutrophils appeared to be enhanced relative to wild-type controls, consistent with our observations of eosinophils and previous studies by others investigating leukocytes deficient in genes mediating G protein-coupled receptor signal transduction (18). The similarity of the data examining both neutrophil and eosinophil recruitment again supports the leukocyte-independent character of the Gαi2 deficiency in knockout mice. Further, the accumulation of leukocytes in the blood suggests a deficiency at an early stage in cell exodus from the circulation. Once out of the vasculature, the leukocytes appear to have the ability to traffic along chemoattractant gradients and accumulate within specific tissue compartments.
Leukocyte Diapedesis and, in Turn, Tissue Accumulation Occurs as a Function of a Gαi2-Signaling Event(s) in Endothelial Cells.
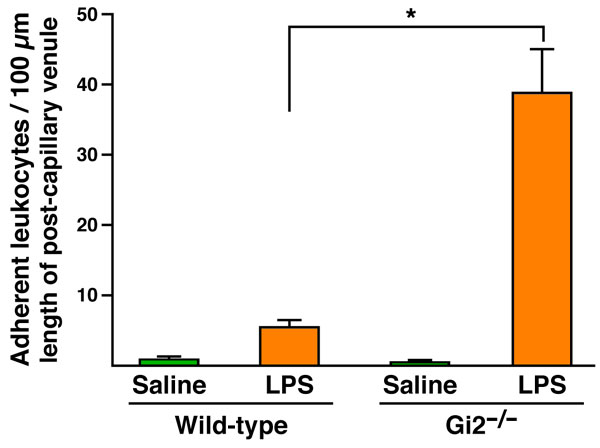
Because our results suggested a leukocyte-independent impairment early in leukocyte trafficking from the blood, we next used intravital microscopy to examine endothelial cell–leukocyte interactions to determine whether a Gαi2 deficiency in endothelial cells alters interactions with leukocytes in such a way as to limit extravasation. Visualization of abdominal mesenteric postcapillary venules from wild-type and Gαi2−/− mice (Fig. 4A and SI Movie 1) after systemic i.p. administration of LPS [10 μg in 100 μl of phosphate-buffered saline (PBS)] demonstrated enhanced accumulation of stationary leukocytes on the endothelial surfaces in Gαi2−/− animals relative to wild-type mice. Specifically, quantitative assessments of the leukocytes adhering to the venules demonstrated that whereas LPS exposure of wild-type mesentery induced only a nominal increase in static adherence to the underlying endothelium, the loss of Gαi2 led to a >7-fold increase in the number of immobilized leukocytes (Fig. 4A and SI Fig. 10). This greater number of stationary leukocytes in knockout mice could be a consequence of enhanced cell adhesion mediated by the absence of a Gαi2-signaling event(s) in the endothelium. Alternatively, the loss of Gαi2 in the endothelium of knockout mice may slow down or block diapedesis, increasing the steady-state number of static endothelial-bound leukocytes which “back-up” on the endothelial surface. Our observation that all leukocyte subtypes accumulate in the blood of Gαi2−/− mice, together with data demonstrating that different leukocyte subtypes use diverse repertoires of receptor–ligand interactions to mediate adhesion, suggest that a Gαi2-dependent effect on cell adhesion is unlikely. This conclusion was confirmed by using ex vivo laminar flow assays to assess lymphocyte adherence and migration through an endothelial cell monolayer (Fig. 4 B and C). In these studies, Gαi-signaling events were abolished in the endothelial cell monolayer by pretreatment with PTX. This pretreatment of the endothelial cells had no effect on cytotoxicity/viability or surface expression levels of the vascular cell adhesion molecule 1 (VCAM-1) (SI Figs. 11 and 12, respectively). The laminar flow study showed that α4-integrin/VCAM-1-dependent firm adhesion of lymphocytes was unaffected in the absence of endothelial cell Gαi signaling (Fig. 4B), suggesting that effects on adhesion mediated by endothelial Gαi2 signaling were not responsible for the blockade of leukocyte recruitment in knockout mice. However, the subsequent migration of firmly adherent lymphocytes through the endothelial cell monolayer (i.e., transmigration) in the laminar flow assay showed that this process was significantly inhibited after PTX treatment (i.e., a Gαi-dependent event) of the monolayer (Fig. 4C). This result suggests that a signaling event(s) in the venule endothelium of Gαi2−/− mice specifically is required for efficient transmigration/diapedesis.
Fig. 4.

The loss of Gαi2 signaling in the vascular endothelium leads to the accumulation of immobilized cells in postcapillary venules through a blockade of diapedesis. (A) Intravital photomicroscopy of LPS-exposed mesentery postcapillary venules showed that the loss of Gαi2 resulted in a significant increase of stationary leukocytes adherent to the vascular endothelium. Numbered white arrows indicate individual rolling leukocytes and black arrows identify stationary leukocytes. The Gαi-dependence of lymphocyte binding and migration through an endothelial cell monolayer was assessed by using an ex vivo parallel plate flow chamber under conditions of laminar flow comparable to pressures observed in postcapillary venules [i.e., 2 dynes (1 dyne = 10 μN)/cm2]. (B) VCAM-1-dependent firm adhesion was blocked by antibodies against either endothelial VCAM-1 or lymphocyte associated α4-integrin but was unaffected by PTX pretreatment of the endothelial cell monolayer. ∗, significantly different (P < 0.05) from no-antibody control group. (C) Lymphocyte migration through an endothelial cell monolayer is blocked in a concentration-dependent fashion by pretreatment of the endothelial cells with PTX. “No treatment,” endothelial cells treated with PBS. ∗, significantly different (P < 0.05) from PBS-treated control group.
Activated leukocyte trafficking and, in particular, the specificity involved in the extravasation cascade, requires a complex exchange of signals (“handshakes”) between the mobile activated leukocytes and underlying vascular endothelial cells. The general responses of leukocytes and endothelial cells to chemokines have been studied (19) as has the role of adhesion molecules and their receptors in the trafficking response (20). The involvement of Gαi-coupled receptors in leukocyte activation, rolling, and subsequent arrest at the endothelial surface before diapedesis has been shown to involve a PTX-sensitive step at the level of the leukocyte (e.g., Gαi-coupled chemokine-mediated recruitment). However, our data also suggest that the complex interaction between endothelial cells and leukocytes responding to inflammatory signals includes a signal transduction event involving the Gαi2 isoform in endothelial cells, which leads to the extravasation of circulating leukocytes. The activation of this pathway may occur in several different ways. For example, the interaction of endothelial cells with leukocytes activates a variety of ectoenzymes. Thus, transmigration may require a Gαi2-mediated response in the endothelial cells to signals or ligands generated by these ectoenzymes (reviewed in ref. 21). Alternatively, local changes in nitric oxide have been shown to modify the levels of RGS (Regulators of G protein Signaling) proteins that are required for regulating Gαi2-signaling events in endothelial cells (22). Moreover, “outside-in” Gαi2-mediated signaling events in endothelial cells leading to the release and movement of previously stationary leukocytes may occur through the mobilization and activation of intercellular adhesion molecule/VCAM-tetraspanin adhesion complexes (23) or the regulation of endothelial integrity through Pky2-mediated affects on vascular endothelial-cadherin adhesion (24). Regardless of the specific mechanism(s) mediating tissue accumulation of leukocytes, our data have surprisingly shown that a Gαi2−/−-dependent endothelial cell signaling event(s) is required for efficient diapedesis. Equally surprising was the fact that the overlapping functions of other Gαi family members that are generally capable of compensating for the loss of Gαi2 in leukocytes are not able to do so in the endothelial cells.
These observations have clinical implications and may offer new possibilities for the treatment of inflammatory diseases. In particular, the observation that an endothelial cell-specific signal transduction event(s) is required for the extravasation of multiple proinflammatory leukocyte subtypes suggests that this could be a rate-limiting step regulating their tissue accumulation in a myriad of diseases. Moreover, the events leading to diapedesis may be the result of a number of endothelial cell receptors that are coupled only to Gαi2-containing G proteins. Therefore, these receptors may potentially represent targets for drug intervention that are potentially applicable to a wide variety of both chronic and acute inflammatory diseases.
Materials and Methods
Mice.
Gαi2 and Gαi3-null mice were generated by homologous recombination in embryonic stem cells (background strain: 129) as described (25). Subsequent generations of G protein-deficient animals were the result of backcrosses (>F5) onto the inbred strain C57BL/6J, a strain that does not display the baseline inflammatory responses observed in the original 129 knockout animals (25). Eosinophils were isolated from Gαi2−/− or Gαi3−/− mice constitutively expressing mouse IL-5 that resulted from crosses of the respective G protein-deficient animals with the IL-5 transgenic line NJ.1638 [C57BL/6J (>20 backcross generations)] (13).
Induction of Allergic Airway Inflammation.
The OVA model of allergic pulmonary inflammation has been described (26). For details see SI Materials and Methods.
LPS-Induced Airway Inflammation.
Mice were lightly anesthetized with isoflurane before intranasal administration of 10 μg (20 μl total volume) of LPS (Sigma, St. Louis, MO). The number of neutrophils recruited to the airways 16 h post-LPS administration were determined by BAL, modifying a described protocol (27). BAL fluid was recovered following instillation of saline supplemented with 2% FCS (1 ml). The BAL fluid was centrifuged at low speed (10 min at 400 × g; 4°C). The recovered cells were counted by using a hemocytometer, and cell differentials were performed on stained cytospin preparations (Diff-Quik; Dade Behring, Newark, DE) counting >300 cells.
Lung Histology and Immunohistochemistry.
Lung tissue for histological analysis was obtained by instilling ≈1 ml of 10% neutral-buffered formalin (30 cm H2O constant pressure) through a cannula inserted into the trachea. The excised lung was immersed in formalin for 24 h (at 4°C). Parasagittal sections (4 μm) were obtained from paraffin-embedded tissue, stained with hematoxylin and eosin, and analyzed by bright field microscopy. Eosinophil recruitment to lung tissue compartments was determined by immunohistochemistry using a rabbit polyclonal anti-mouse major basic protein (MBP) antisera (28). Eosinophils surrounding the airways were quantified by counting the number of MBP-positive cells per square millimeter of submucosal tissue surrounding the bronchioles.
Isolation of Mouse Eosinophils, Isolation of Splenocytes, andIn Vitro Assessment of Leukocyte Chemotaxis.
For details, see SI Materials and Methods.
Eosinophil Adoptive Transfer andEx Vivo Labeling of Eosinophils and Monitoring the Recruitment/Accumulation of Labeled Eosinophils Following Adoptive Transfer to the Peritoneal Cavity.
For details, see SI Materials and Methods.
Hematopoietic Engraftment by Bone Marrow Transfer.
Exposing female wild-type mice to 1,100-cGy whole body lethal irradiation generated complete bone marrow chimeras. Within 3 h of irradiation, 1 × 107 bone marrow cells from wild-type or Gαi2−/− male donors were transferred by tail vein injection. Engrafted mice were used in experiments after a >45-day recovery period. Recovered mice were sensitized and aerosol challenged with OVA (saline for controls) by using the protocol noted above, and BAL eosinophils were enumerated 24 h after the last challenge. Eosinophils comprise <1% of leukocytes in the airways of saline-challenged mice of any group. Donor cell engraftment of >90% was achieved in all recipients as determined by a PCR assay designed to quantify X vs. Y chromosome-specific sequences (29).
Cytokine Assays.
Cytokine levels in BAL fluid were determined by ELISA. Mouse IL-4, IL-5, IFN-γ, and IL-12 ELISA kits (R&D Systems, Minneapolis, MN) were used according to the manufacturer's protocol. The limits of detection for each assay were as follows: IFN-γ ≈ 30 pg/ml, IL-4 ≈ 10 pg/ml, IL-5 ≈ 10 pg/ml, and IL-12 ≈ 10 pg/ml.
In vivo Assessment of Leukocyte Intracapillary Rolling and/or Adhesion in Postcapillary Venules by Intravital Microscopy andEx Vivo Assessments of Lymphocyte Adhesion and Migration.
For details, see SI Materials and Methods.
Determination of VCAM-1 Endothelial Cell Expression Using Flow Cytometry.
mHEVa endothelial cell monolayers (before or after exposure to PTX) were disassociated into single-cell suspensions by using 0.3% EDTA, and the recovered cells were washed in RPMI-1640 containing 20% FCS. Cells were stained in PBS/0.5% BSA/0.15% NaN3 with rat anti-mouse VCAM-1 antibody (normal rat IgG was used as a negative control for primary antibody staining) and visualized with biotin-conjugated goat anti-rat antibodies and strepavidin-phycoerythrin. Analysis was performed on a FACScan flow cytometer (BD Biosciences, Palo Alto, CA) with CellQuest Pro software (BD Biosciences).
Statistical Analysis.
All data presented are the means ± standard errors (SEM). Statistical analysis was performed on parametric data by using t tests with differences between means considered significant when P < 0.05.
Supplementary Material
Acknowledgments
We thank numerous individuals, including Dana Colbert, Tracy L. Ansay, Edith M. Hines, Travis Biechele, and Alfred D. Doyle. We also thank the Mayo Clinic Arizona Core facilities (Histology, Lisa Barbarisi; Immunology, Tammy Brehm-Gibson; Mouse Special Animal Services, Suresh Savarirayan; Medical Graphic Arts, Marv Ruona; Video Production and Services, Randall J. Raish; Research Library Services, Joseph Esposito). In addition, we thank our administrative staff, Linda Mardel and Margaret (Peg) McGarry. This work was supported in part by the Mayo Foundation and grants from the National Institutes of Health to J.J.L. (HL065228 and K26-RR019709), N.A.L. (HL058723), J.M.C.-M. (HL68171), P.S. (AI35796 and HL079304), M.I.S. (GM34236), and L.B. (DK-19318).
Abbreviations
- PTX
pertussis toxin
- OVA
ovalbumin
- VCAM
vascular cell adhesion molecule
- BAL
bronchoalveolar lavage
- PBS
phosphate-buffered saline.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0700185104/DC1.
References
- 1.Worthylake RA, Burridge K. Curr Opin Cell Biol. 2001;13:569–577. doi: 10.1016/s0955-0674(00)00253-2. [DOI] [PubMed] [Google Scholar]
- 2.Jiang M, Spicher K, Boulay G, Martin-Requero A, Dye CA, Rudolph U, Birnbaumer L. Methods Enzymol. 2002;344:277–298. doi: 10.1016/s0076-6879(02)44721-0. [DOI] [PubMed] [Google Scholar]
- 3.Wise A, Gearing K, Rees S. Drug Discov Today. 2002;7:235–246. doi: 10.1016/s1359-6446(01)02131-6. [DOI] [PubMed] [Google Scholar]
- 4.Wettschureck N, Offermanns S. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 5.Wilkie TM, Gilbert DJ, Olsen AS, Chen X-N, Amatruda TT, Korenberg JR, Trask BJ, de Jong P, Reed RR, Simon MI, et al. Nat Genet. 1992;1:85–91. doi: 10.1038/ng0592-85. [DOI] [PubMed] [Google Scholar]
- 6.Goldman DW, Chang FH, Gifford LA, Goetzl EJ, Bourne HR. J Exp Med. 1985;162:145–156. doi: 10.1084/jem.162.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudolph U, Spicher K, Birnbaumer L. Proc Natl Acad Sci USA. 1996;93:3209–3214. doi: 10.1073/pnas.93.8.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emala CW, Yang J, Hirshman CA, Levine MA. Life Sci. 1994;55:593–602. doi: 10.1016/0024-3205(94)00485-4. [DOI] [PubMed] [Google Scholar]
- 9.Fabian G, Szabo CA, Bozo B, Greenwood J, Adamson P, Deli MA, Joo F, Krizbai IA, Szucs M. Neurochem Int. 1998;33:179–185. doi: 10.1016/s0197-0186(98)00008-4. [DOI] [PubMed] [Google Scholar]
- 10.Bousquet J, Chanez P, Lacoste JY, Barneon G, Ghavanian N, Enander I, Venge P, Ahlstedt S, Simony-Lafontaine J, Godard P, Francois-Bernard M. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 11.Kips JC, Anderson GP, Fredberg JJ, Herz U, Inman MD, Jordana M, Kemeny DM, Lotvall J, Pauwels RA, Plopper CG, et al. Eur Respir J. 2003;22:374–382. doi: 10.1183/09031936.03.00026403. [DOI] [PubMed] [Google Scholar]
- 12.Pope SM, Zimmermann N, Stringer KF, Karow ML, Rothenberg ME. J Immunol. 2005;175:5341–5350. doi: 10.4049/jimmunol.175.8.5341. [DOI] [PubMed] [Google Scholar]
- 13.Lee NA, McGarry MP, Larson KA, Horton MA, Kristensen AB, Lee JJ. J Immunol. 1997;158:1332–1344. [PubMed] [Google Scholar]
- 14.Han SB, Moratz C, Huang NN, Kelsall B, Cho H, Shi CS, Schwartz O, Kehrl JH. Immunity. 2005;22:343–354. doi: 10.1016/j.immuni.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 15.Finsnes F, Skjonsberg OH, Lyberg T, Christensen G. Eur Respir J. 2000;15:743–750. doi: 10.1034/j.1399-3003.2000.15d19.x. [DOI] [PubMed] [Google Scholar]
- 16.Keil ML, Solomon NL, Lodhi IJ, Stone KC, Jesaitis AJ, Chang PS, Linderman JJ, Omann GM. Biochem Biophys Res Commun. 2003;301:862–872. doi: 10.1016/s0006-291x(03)00057-3. [DOI] [PubMed] [Google Scholar]
- 17.Ley K. Am J Physiol Lung Cell Mol Physiol. 2004;286:L463–L464. doi: 10.1152/ajplung.00386.2003. [DOI] [PubMed] [Google Scholar]
- 18.Jiang H, Kuang Y, Wu Y, Xie W, Simon MI, Wu D. Proc Natl Acad Sci USA. 1997;94:7971–7975. doi: 10.1073/pnas.94.15.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moser B, Wolf M, Walz A, Loetscher P. Trends Immunol. 2004;25:75–84. doi: 10.1016/j.it.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Springer TA. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 21.Salmi M, Jalkanen S. Nat Rev Immunol. 2005;5:760–771. doi: 10.1038/nri1705. [DOI] [PubMed] [Google Scholar]
- 22.Hu RG, Sheng J, Qi X, Xu Z, Takahashi TT, Varshavsky A. Nature. 2005;437:981–986. doi: 10.1038/nature04027. [DOI] [PubMed] [Google Scholar]
- 23.Barreiro O, Yanez-Mo M, Sala-Valdes M, Gutierrez-Lopez MD, Ovalle S, Higginbottom A, Monk PN, Cabanas C, Sanchez-Madrid F. Blood. 2005;105:2852–2861. doi: 10.1182/blood-2004-09-3606. [DOI] [PubMed] [Google Scholar]
- 24.van Buul JD, Anthony EC, Fernandez-Borja M, Burridge K, Hordijk PL. J Biol Chem. 2005;280:21129–21136. doi: 10.1074/jbc.M500898200. [DOI] [PubMed] [Google Scholar]
- 25.Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, Boulay G, Bradley A, Birnbaumer L. Nat Genet. 1995;10:143–150. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- 26.Borchers MT, Crosby JR, Farmer SC, Sypek J, Ansay TL, Lee NA, Lee JJ. Am J Physiol. 2001;280:L813–L821. doi: 10.1152/ajplung.2001.280.4.L813. [DOI] [PubMed] [Google Scholar]
- 27.Lundy SK, Berlin AA, Lukacs NW. Am J Pathol. 2003;163:1961–1968. doi: 10.1016/S0002-9440(10)63554-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denzler KL, Farmer SC, Crosby JR, Borchers MT, Cieslewicz G, Larson KA, Cormier-Regard S, Lee NA, Lee JJ. J Immunol. 2000;165:5509–5517. doi: 10.4049/jimmunol.165.10.5509. [DOI] [PubMed] [Google Scholar]
- 29.Novak EK, Reddington M, Zhen L, Stenberg PE, Jackson CW, McGarry MP, Swank RT. Blood. 1995;85:1781–1789. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}