

Abstract
We describe a Markov Chain Monte Carlo analysis of five human Y- chromosome microsatellite polymorphisms based on samples from five diverse populations. Our analysis provides strong evidence for mutational bias favoring increase in length at all loci. Estimates of population coalescent times and population size from our two largest samples, one African and one European, suggest that the African population is older but smaller and that the English East Anglian population has undergone significant expansion, being larger but younger. We conclude that Markov Chain Monte Carlo analysis of microsatellite haplotypes can uncover information not apparent when the microsatellites are considered independently. Incorporation of population size as a variable should allow us to estimate the timing and magnitude of major historical population trends.
The genetic material comprising the nonrecombining region of the Y chromosome is effectively haploid and is inherited patrilineally. Such inheritance makes Y-chromosomal polymorphisms ideal for elucidating human male genetic histories, but the scarcity of variable markers has limited progress for several years. However, the recent discovery of polymorphic Y-microsatellite repeat polymorphisms offers a range of markers suitable for studying the ancestry of human populations.
Most microsatellite mutations involve either the gain or loss of single repeat units such that alleles tend to evolve according to a one-dimensional random walk. This apparent simplicity has been exploited to formulate a number of genetic distance measures, but there remain a number of problems that need to be overcome. First is the problem we refer to as back-mutations and involves the loss of information through convergence: an allele that undergoes one increase followed by one decrease in length will appear identical to an allele of the same original length that has not mutated. Second, some mutations may involve larger changes in sizes than one repeat unit, a possibility examined by Di Rienzo et al. (1). Third, there is growing evidence that the random walk is asymmetric. At least two studies have reported that gains in length significantly outnumber losses (2, 3), though the extent of the bias has not been quantified.
For Y-chromosome microsatellites, where genetic recombination is absent, uncertainties about the mode of evolution can be overcome by using network-based approaches (4–7) to illustrate potential relationships between haplotypes. In a study of East Anglian Y-chromosome microsatellite haplotypes (4), we constructed a network in which haplotypes were ordered by total allele length summed over five loci. The resulting network is strongly structured and possesses a well-defined central core composed of several closely related, relatively common haplotypes. Because common, well-connected haplotypes are unlikely to be recent in origin, the core should be indicative of the genetic structure of this population’s ancestors. The asymmetric shape of the network, with fewer short haplotypes and greater numbers of longer haplotypes, is suggestive of the mutational asymmetry that has been documented at autosomal loci. However, without a method of assessing the probabilities associated with inferred genealogical relationships, it is impossible to know how much confidence we can have in such networks. In principle, if we knew the processes by which microsatellites evolve, we could derive a probability distribution for the ancestral state.
An approach that offers the potential both to estimate the ancestral state and the parameters associated with population history and microsatellite evolution involves the use of Markov Chain Monte Carlo (MCMC) methods. These have been used successfully in phylogenetic analyses of mitochondrial data (8, 9) and microsatellite data (10). These methods are more flexible and powerful than other methods of phylogeny analysis. Because there is no recombination between the Y-chromosome microsatellites studied, reconstruction of the ancestry is a mathematically precise problem, in that under an assumed evolutionary process, the probability distributions for the ancestor and ancestry tree are well defined. This makes MCMC attractive, because it computes the conditional probability P(ancestry, evolutionary parameters|data). The fact that back-mutations are frequent means that methods that attempt to construct unique trees will give poor results whereas MCMC techniques incorporating stochastic tree geometries can still extract evolutionary signals.
We were interested to examine the extent to which MCMC methods are able to shed light on genealogical relationships based on Y-microsatellite haplotypes and provide extra information about the underlying evolutionary processes. For comparison with our existing data set of East Anglians (n = 174), Nigerians (n = 23), and Sardinians (n = 15), we genotyped an additional 104 South African and 22 Japanese males. We then developed, tested, and applied a MCMC coalescence algorithm.
Methods
Microsatellite Typing.
Microsatellite typing was carried out as described in Cooper et al. (4), except that an additional primer was designed to allow separate amplification of locus DYS389A. The primer (DYS389R2: TGAGAGTTGGGTACAGAAGTAGG) was designed from the sequence previously described (4) (European Molecular Biology Laboratory database no. X97312). DYS389R2 was used in PCR with DYS389F to amplify a region of 118–138 bp, encompassing the single GATA repeat array corresponding to DYS389A.
DNA Samples.
DNA samples were 174 East Anglians, 23 Nigerians, and 15 Sardinians as described (4) with the addition of 104 South African and 22 Japanese males.
MCMC Methods.
We assume a neutral evolution model in a population of constant size, with a microsatellite single-step mutation process i.e., mutations change microsatellite length by one repeat unit. Increases and decreases in length are at rates denoted μ+ and μ−, respectively, to allow estimation of the extent of any bias in mutation process from the data. All microsatellites are assumed to have the same mutation rate and extent of bias. We use MCMC techniques to compute posterior probability distributions of these parameters and the ancestry conditional on the observed data. MCMC essentially performs Monte Carlo integration (using Markov Chains) of the normalization integrals obtained through use of Bayes theorem. Our MCMC implementation is similar to that of ref. 10 except for the introduction of the mutation bias and the fact that the mutation rate and population size are not treated independently i.e., we use the variable μN instead of μ and N separately, where μ = μ+ + μ− is the mutation rate and N is the effective population size. This has the advantage that we can use uninformative priors throughout. The analysis of ref. 10 was based on the East Anglian data set previously reported and analyzed through network and classical methods (4). Full details of our MCMC methods are given in the Appendix.
Results
The evolutionary process is defined by the two variables μN, the average number of mutation events that occur in a lineage since the coalescent, and ω = (μ+ − μ−)/(μ+ + μ−), the mutation bias or balance between expansion and contraction mutations. The magnitude of bias is expected to be constant across populations, whereas μN is expected to vary between populations; the ratio between populations reflecting relative population sizes. Bayesian analysis was carried out on individual loci separately, on haplotypes constructed from all possible combinations of two, three, and four loci and on five-locus haplotypes.
Mutation Bias (ω).
The posterior probability density for the bias of the individual loci of the East Anglians and South Africans (Table 1, Fig. 1) shows weak evidence for mutation bias (single-locus best-estimates of ω are positive in nine of 10 cases) but the signal is not strong enough to yield significance in any particular instance. For five-locus haplotypes analyzed across five populations, the estimates of mutation bias are consistently positive and are significantly greater than zero for the three largest samples (East Anglians, South Africans, and Nigerians) (Table 2). Moreover, none of the five population estimates differ in magnitude from any other. These observations provide good evidence that Y microsatellites mutate asymmetrically. The average size of ω for the largest samples (East Anglians and South Africans) is 0.48, which implies that mutations resulting in the gain of a repeat unit are approximately 2.8 times more frequent than those resulting in the loss of a repeat unit. The effect of varying the number of loci used to estimate ω is shown in Fig. 2. As expected from the increasing sample size, the SD of the estimate of ω decreases as more loci are added. Adding more loci results in an asymptotic convergence of ω toward a point close to the estimate based on all five loci. This convergence suggests that the extra constraints imposed by considering multilocus haplotypes uncover information that is not resolved when individual loci are treated separately.
Table 1.
Mutation rate and bias, calculated for individual loci (based on a run of 2 × 105 steps) for East Anglians and South Africans
| DYS391 | DYS19 | DYS390 | DYS389A | DYS389B | |
|---|---|---|---|---|---|
| East Anglian | |||||
| Classical μN | 0.32 | 0.46 | 0.94 | 0.36 | 0.43 |
| Classical skewness | 0.27 | 1.45 | −0.07 | 1.18 | −0.37 |
| Range | 3 | 5 | 5 | 5 | 5 |
| MCMCμN | 1.0 (0.6) | 2.1 (1.0) | 4.9 (2.7) | 2.2 (1.0) | 3.3 (1.4) |
| MCMC bias | 0.2 (0.4) | 0.3 (0.4) | 0.2 (0.4) | 0.2 (0.4) | 0.04 (0.4) |
| South Africans | |||||
| Classical μN | 0.15 | 0.79 | 3.32 | 0.93 | 0.76 |
| Classical skewness | 1.43 | 0.08 | 0.63 | −0.71 | −0.72 |
| Range | 2 | 4 | 5 | 5 | 5 |
| MCMCμN | 0.5 (0.3) | 4.9 (2.9) | 2.2 (1.1) | 7.2 (4.2) | 4.9 (3.2) |
| MCMC bias | 0.1 (0.5) | 0.2 (0.4) | 0.5 (0.4) | −0.1 (0.4) | 0.02 (0.5) |
Standard errors are given in parentheses.
Figure 1.

Estimated probability density distribution of mutation bias for individual loci and the five-locus haplotype.
Table 2.
Estimates of number of mutations (μN), bias (ω), and coalescence time (μT) for each of our five population samples and based on a run of 2 × 105 steps
| East Anglians | South Africans | Nigerians | Japanese | Sardinians | |
|---|---|---|---|---|---|
| Sample size | 174 | 104 | 23 | 22 | 15 |
| μN | 5.03 (0.77) | 3.29 (0.61) | 3.25 (0.98) | 2.71 (0.86) | 1.87 (0.73) |
| 95% CI | 3.66–6.62 | 2.16–4.51 | 1.60–5.20 | 1.25–4.42 | 0.73–3.39 |
| ω | 0.43 (0.14) | 0.53 (0.19) | 0.70 (0.20) | 0.27 (0.41) | 0.37 (0.40) |
| 95% CI | 0.16 to 0.69 | 0.15 to 0.89 | >0.33 | −0.58 to 0.96 | >−0.39 |
| μT | 2.19 (0.68) | 3.19 (1.34) | 2.34 (1.00) | 2.00 (0.89) | 1.52 (0.68) |
| 95% CI | 1.24–3.34 | 1.77–4.25 | 1.28–4.20 | 1.07–2.99 | 0.78–2.76 |
Standard errors are given in parentheses.
Figure 2.

Effect of the number of loci used on the estimate of bias. The average across all possible combinations is shown displaced to the right, with 1 SD (average of the variances).
Rate of Mutations in Coalescent (μN).
MCMC ancestry computations were performed on the individual loci of the two largest data sets (East Anglian and South African) (Table 1), and posterior probability distributions were plotted (Fig. 3). The values of μN generated are similar to the average value of 1.65 obtained by simulation of the evolution of individual loci (4). However, the MCMC-generated values of μN are greater than those estimated by the classical method (i.e., from the variance of the allele frequency distributions; ref. 12), with the single exception of locus DYS390 in South Africans, although these differences are not significant because of the large variance in the classical estimates. The distributions for bias and μN are uncorrelated, therefore our estimates of μN are very similar to those of Wilson and Balding (10), who analyzed the East Anglian data set with zero bias.
Figure 3.

Estimated probability density distribution of μN for individual loci and the five-locus haplotype.
Our estimate for μN based on five-loci haplotypes is larger than for any of the loci individually in the East Anglian data set (significant for DYS391, DYS19, and DYS389A, all at P < 0.01). The effect of varying the number of loci used in the MCMC analysis is shown in Fig. 4. The SD of the estimates of μN decreases as more loci are added because more data are contributing to the estimate. However, as more loci are considered the behavior of μN differs between the two populations. In the East Anglians, as more loci are added μN increases, with no sign of approaching an asymptote after five loci. By contrast, in the South Africans, mean μN changes little as the number of loci considered increases.
Figure 4.

Effect of the number of loci used on the estimate of μN. The average across all possible combinations is shown displaced to the right, with 1 SD (average of the variances).
The discrepancy in the behavior of the single and multilocus estimates between the East Anglians and Africans can be attributed to differences in population history. Relative to the constant population size assumed by our Bayesian analysis, an expanding population will carry allele frequency distributions that are more leptokurtic, with longer tails and more counts in the center. With only one locus, the rare edge alleles lying in the tails of a leptokurtic distribution can be attributed to stochastic noise and the Bayesian posterior distributions will concentrate mainly on the common, central alleles. The result is an underestimate of μN. However, when additional loci are considered, generating multilocus haplotypes, the edges of the distribution will become better defined. Because the breadth of the distribution also defines its depth, this has the effect of forcing the estimate of μN upward toward its true value. Thus, the general agreement between single-locus and multilocus estimates for the African data suggest that the underlying assumption of constant population size has been met. In contrast, the disagreement seen in the East Anglian data set suggests historical population expansion, a possibility consistent with the expansion that would have followed the colonization of Europe.
Comparisons Across Loci.
Although our analysis does not detect any differences in the bias between the loci, there is evidence for heterogeneity between the mutation rates of the individual loci. On the basis of the estimates of μN, the mutation rate of locus DYS391 differs significantly from locus DYS390 (P < 0.05, both East Anglian and South African data sets), from loci DYS19, DYS389A and DYS389B (P < 0.01, South African data set) and from the joint five-locus haplotype estimate (P < 0.001 for both populations). However, removing locus DYS391 only weakly affects our estimates of the bias and of μN. The remaining four loci are consistent with each other, although as discussed above the five-locus haplotype estimate for μN is significantly larger than loci DYS19 (P = 0.008) and DYS389A (P = 0.01) on the East Anglian data set, possibly as a result of population expansion. In contrast, for the South Africans no such differences between the five-locus haplotypes and the individual loci was observed (P > 0.3), except for locus DYS391.
Ancestral Haplotypes/Coalescence.
The fact that microsatellite data do not contain a strong signal of the ancestry tree is clear from the very low frequency with which specific trees are observed. For example, in the East Anglian data (174 people), in 40,000 samples one tree was observed three times, another 100 trees were observed twice, and the rest were unique trees. This low repeatability is a reflection of the large number of possible trees (approximately n! with n people) and poor signal. The ancestral states of our two largest population samples predicted by MCMC are shown in Table 3, with current states for comparison. For each locus-population combination, the mean allele size in the sample is greater than for the ancestral state calculated from the MCMC coalescence algorithm, which is consistent with the biased mutation process indicated by ω. The estimated coalescence time of the East Anglian population (2.19 mutations since coalescent, SD = 0.98) is approximately two-thirds that of the Africans (3.19 mutations since coalescent, SD = 1.34), but the difference between these estimates is not significant. Interestingly, this relationship is reversed for μN, where the value for East Anglians is larger than for South Africans. By implication, the South African population is smaller but older than the East Anglian population, which appears recently expanded.
Table 3.
Mean and modal allele sizes in samples and for ancestral states arising from MCMC for East Anglians and South Africans
| DYS391 | DYS19 | DYS390 | DYS389A | DYS389B | |
|---|---|---|---|---|---|
| East Anglians | |||||
| Sample mean | 2.529 | 3.310 | 3.402 | 3.190 | 3.891 |
| (SD) | (0.566) | (0.677) | (0.967) | (0.602) | (0.658) |
| Sample mode | 2 | 3 | 4 | 3 | 4 |
| Ancestral mean | 1.701 | 2.772 | 2.766 | 2.766 | 2.985 |
| (SD) | (0.833) | (0.874) | (0.961) | (0.832) | (1.097) |
| Ancestral mode | 2 | 3 | 3 | 3 | |
| South Africans | |||||
| Sample mean | 2.154 | 4.173 | 2.471 | 4.154 | 4.048 |
| (SD) | (0.388) | (0.886) | (1.822) | (0.963) | (0.874) |
| Sample mode | 2 | 4 | 1 | 5 | 4 |
| Ancestral mean | 0.886 | 2.319 | 1.743 | 2.159 | 1.835 |
| (SD) | (1.303) | (1.365) | (1.441) | (1.389) | (1.444) |
| Ancestral mode | 1 | 3 | 2 | 3 | 2 |
This pattern of older African and recently expanded European populations fits well with the “out of Africa” hypothesis of human evolution (13–15) and other Y-chromosome studies and microsatellite studies (16, 17) and suggests that our MCMC algorithm has successfully recognized important complexities in the histories of these two populations. Such an ability could play a vital role in distinguishing between alternative models for the origins of modern human populations (18). In particular, our data suggest that African and non-African populations retain radically different historical size signatures, and this would seem incompatible with models that require continuous gene flow in and out of Africa (18). With larger sample sizes and the incorporation of new MCMC methods for treating population expansion and migration (11, 19), we are hopeful that estimates can soon be made for the magnitudes and timing of historical gene flow and population size fluctuation.
Conclusions
We show that MCMC implementation of Bayesian posterior probability density analysis offers a powerful method for estimating population genetic parameters from Y-microsatellite haplotypes, extracting evolutionary signals that are not apparent through classical analysis of individual microsatellite allele frequency distributions. Most importantly, we provide strong evidence that Y-microsatellites are prone to mutation bias favoring expansion, in just the same way that autosomal loci appear to be. Although estimates of the key parameter μN, the product of the effective population size and the mutation rate, appear confounded by population history, this problem is partly resolved by considering the way in which the estimate of μN varies with the number of loci considered. In our African sample, little variation is noted, implying a population at or near equilibrium. Among East Anglians, multilocus haplotypes yield significantly higher estimates of μN, implying a recent population expansion. This pattern is supported by the contrasting μN and μT estimates, which imply phylogenies that are relatively deep and narrow for South Africans and shallow but wide for East Anglian. Obtaining a more precise description of these population histories should be the focus of future work, which would incorporate variation in population size as a variable in the MCMC algorithm, potentially allowing an estimation of both the extent and the timing of this expansion.
Acknowledgments
We thank Drs. S. Orru, J. Old, J. Tokunaga, M. Kotze, and R. Ramesar and Professor T. Jenkins for DNA samples, and Keith Yates for help with testing the MCMC algorithm. This work was supported by the Leverhulme Trust (Grant F/752/A). N.J.B. is an Engineering and Physical Sciences Research Council Advanced Fellow. D.C.R. is a Glaxo Wellcome Research Fellow.
Abbreviation
- MCMC
Markov Chain Monte Carlo
Appendix
Our MCMC program impliments the neutral evolution (single-step) model allowing for biased mutations. The underlying Markov Chain is based on the coalescence of the observed microsatellites back to the common ancestor. The likelihood of the observed data D and ancestry tree S for N individuals is given by
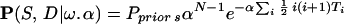 |
1 |
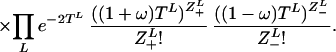 |
This consists of the changes in microsatellite length (Poisson distributed increasing and decreasing single-step events Z±) that occur between coalescent events on the tree S. Linked coalescent events are separated by time TL (link L), while times Ti denote the times between consecutive coalescence events when these are time ordered, i.e. Ti is the time to next coalescence event when there are i + 1 people. Coalescence is an exponential process eith rate α, the term ½i(i + 1) arising from the ½i(i + 1) possible coalescent events that can occur with i + 1 people. The mutation rate μ is set to 2 by time rescaling. Therefore with bias ω the mutation rate of positive/negative steps are 1 ± ω.
The Markov Chain consists of the tree S, the number of mutation steps between coalescence events Z±L (on link L), the times Ti and the bias ω. The coalescence rate α is removed from the Markov Chain through integration, updating Ti with a single-component Metropolis-Hasting algorithm based on the posterior distribution
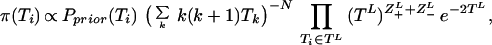 |
2 |
using the proposal distribution
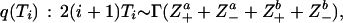 |
which is the leading order approximation to the posterior (Eq. 2) since Ti > Ti+1 on average, and therefore only the coalescing links (descendants a and b) give significant contributions. The RV α is sampled by α∑i ½i(i + 1)Ti ∼ Γ(N), i.e. a gamma variate.
Internal genotypes and mutations are updated by use of the following distribution
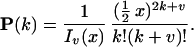 |
3 |
The normalization factor Iv(x) is the modified Bessel function. Given genotypes at either end of a link (and therefore the difference Z+ − Z−), the number of positive changes P(Z+|Z+ − Z− = v) is given by Eq. 3. A genotype g (microsatellite length) is determined by the conditional
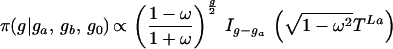 |
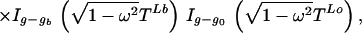 |
where g0 is the genotype of the immediate ancestor, and ga, gb are the genotypes of the two descendants, with times TL0, TLa and TLb, respectively. This follows from the likelihood (Eq. 1). We update g by normalizing this expression numerically. The corresponding changes Z+L then are updated under a Gibbs sampler.
The tree geometry S is updated by using a Metropolis-Hastings algorithm at each coalescent event, similar to ref. 10. The proposal distribution consists of removing the branch of a tree and placing it on any of the other links existing at that time, retaining its genotype. This move affects only a few of the links and hence the acceptance probability is efficient to calculate.
Finally the bias ω is updated with a Gibbs method, the conditional being a beta variate π(ω) ∝ (1 + ω)∑LZ+L (1 + ω)∑LZ−L.
All our priors are uninformative.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Di Rienzo A, Peterson A C, Garza J C, Valdes A M, Slatkin M. Proc Natl Acad Sci USA. 1994;91:3166–3170. doi: 10.1073/pnas.91.8.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amos W, Sawcer S J, Feakes R, Rubinsztein D C. Nat Genet. 1996;13:390–391. doi: 10.1038/ng0896-390. [DOI] [PubMed] [Google Scholar]
- 3.Primmer C, Ellegren H, Saino N, Møller A P. Nat Genet. 1996;13:391–393. doi: 10.1038/ng0896-391. [DOI] [PubMed] [Google Scholar]
- 4.Cooper G, Amos W, Hoffman D, Rubisztein D C. Hum Mol Genet. 1996;5:1759–1766. doi: 10.1093/hmg/5.11.1759. [DOI] [PubMed] [Google Scholar]
- 5.Deka R, Jin L, Shriver M D, Yu M, Saha N, Barrantes R, Chakraborty R, Ferrell E. Genome Res. 1996;6:1177–1184. doi: 10.1101/gr.6.12.1177. [DOI] [PubMed] [Google Scholar]
- 6.Roewer L, Kayser M, Dieltjes P, Nagy M, Bakker E, Krawczak M, de Knijff P. Hum Mol Genet. 1996;5:1029–1033. doi: 10.1093/hmg/5.7.1029. [DOI] [PubMed] [Google Scholar]
- 7.Malaspina P, Cruciani F, Ciminelli B, Terrenato L, Santolamazza P, Alonso A, Banyko J, Brdicka R, Garcia O, Gaudiano C, et al. Am J Hum Genet. 1998;63:847–860. doi: 10.1086/301999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffiths R C, Tavaré S. Stat Sci. 1994;9:307–319. [Google Scholar]
- 9.Yang Z, Rannala B. Mol Biol Evol. 1997;14:717–724. doi: 10.1093/oxfordjournals.molbev.a025811. [DOI] [PubMed] [Google Scholar]
- 10.Wilson I J, Balding D J. Genetics. 1998;150:499–510. doi: 10.1093/genetics/150.1.499. . P. & Felsenstein, J. (1999) Genetics152, 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beerli P, Felsenstein J. Genetics. 1999;152:763–773. doi: 10.1093/genetics/152.2.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slatkin M. Genetics. 1995;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lahr M M, Foley R A. Yearbook Phys Anthropol. 1998;41:137–176. doi: 10.1002/(sici)1096-8644(1998)107:27+<137::aid-ajpa6>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 14.Harpending H C, Batzer M A, Gurven M, Jorde L B, Rogers A R, Sherry S T. Proc Natl Acad Sci USA. 1998;95:1961–1967. doi: 10.1073/pnas.95.4.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harpending H C, Relethford J, Sherry S T. In: Molecular Biology and Human Diversity. Boyce A J, Mascie-Taylor C, editors. Cambridge: Cambridge Univ. Press; 1996. pp. 283–299. [Google Scholar]
- 16.Hammer M F, Karafet T, Rasanayagam A, Wood E T, Altheide T K, Jenkins T, Griffiths R C, Templeton A R, Zegura S L. Mol Biol Evol. 1998;15:427–441. doi: 10.1093/oxfordjournals.molbev.a025939. [DOI] [PubMed] [Google Scholar]
- 17.Kimmel M, Chakraborty R, King J P, Bamshad M, Watkins W S, Jorde L B. Genetics. 1998;148:1921–1930. doi: 10.1093/genetics/148.4.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Templeton A R. Curr Opin Genet Dev. 1997;7:841–847. doi: 10.1016/s0959-437x(97)80049-4. [DOI] [PubMed] [Google Scholar]
- 19.Tavaré S, Balding D J, Griffiths R C, Donelly P. Genetics. 1997;145:505–518. doi: 10.1093/genetics/145.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]