

Abstract
Tapasin (Tpn) has been implicated in multiple steps of the MHC class I assembly pathway, but the mechanisms of function remain incompletely understood. Using purified proteins, we could demonstrate direct binding of Tpn to peptide-deficient forms of MHC class I molecules at physiological temperatures. Tpn also bound to M10.5, a pheromone receptor-associated MHC molecule that has an open and empty groove and that shares significant sequence identity with class I sequences. Two types of MHC class I–Tpn complexes were detectable in vitro depending on the input proteins; those depleted in β2m, and those containing β2m. Both were competent for subsequent assembly with peptides, but the latter complexes assembled more rapidly. Thus, the assembly rate of Tpn-associated class I was determined by the conditions under which Tpn–MHC class I complexes were induced. Peptide loading of class I inhibited Tpn–class I-binding interactions, and peptide-depletion enhanced binding. In combination with β2m, certain peptides induced efficient dissociation of preformed Tpn–class I complexes. Together, these studies demonstrate direct Tpn–MHC class I interactions and preferential binding of empty MHC class I by Tpn, and that the Tpn–class I interaction is regulated by both β2m and peptide. In cells, Tpn is likely to be a direct mediator of peptide-regulated binding and release of MHC class I from the TAP complex.
Keywords: antigen presentation, HLA, TAP transporter
Peptide products of foreign and self-proteins bind to MHC class I heavy chains (HC) and light chains [β2-microglubulin (β2m)] within the endoplasmic reticulum (ER) of cells and are then transported to the cell surface, where they are available for immune surveillance by cytotoxic T lymphocytes (1). The transporter associated with antigen processing (TAP) is a peptide transporter that translocates cytosolic peptides into the ER lumen, for assembly with MHC class I HC and β2m. Other ER resident proteins that assist in MHC class I (class I) assembly include the chaperones calnexin and calreticulin, the thiol-disulfide isomerase ERp57, and the MHC-encoded transmembrane protein tapasin (Tpn). Individually or in combination, these components ensure quality control of class I-peptide assembly (1).
For many human and mouse class I allotypes, Tpn increases cell surface class I expression. How Tpn mediates this increase has been a matter of considerable debate, and many functions have been proposed for Tpn. Tpn stabilizes TAP, and increases steady-state levels of TAP, thereby allowing more peptides to be translocated into the ER (2, 3). Tpn also allows a physical link between class I and the TAP (4), and by doing so, may increase the effective concentration of peptides that are available for class I binding. Tpn may also be important for preventing ER exit of peptide-deficient class I (5–7), and may thereby indirectly promote the accumulation of more stable class I at the cell surface. Soluble Tpn (sTpn) that does not stably bind TAP, enhance TAP expression levels, or mediate the TAP–class I interaction, is able to partially compensate for a defect in cellular Tpn (2), indicating that at least some of Tpn's functions are independent of its effects upon TAP. There are also indications from cell-based experiments, that Tpn edits/optimizes the class I peptide repertoire (8), or directly facilitates peptide loading of class I molecules (9–11).
The absence of a direct binding assay between Tpn and class I has hampered a better understanding of the mechanisms of Tpn's function. Although cell-based experiments have demonstrated class I-Tpn binding in the absence of other components of the class I pathway (5, 12), there are no reports thus far of direct Tpn–class I-binding interactions outside the context of a cell. Here, we reconstituted the Tpn–class I interaction by using purified proteins and investigated the effects of Tpn upon the class I–peptide interaction, and the effects of peptide upon the class I–Tpn interaction.
Results
Tpn Can Bind to Peptide-Deficient Class I Molecules at Physiological Temperature.
A FLAG-tagged sTpn was expressed in and purified from CHO cells (Fig. 6, which is published as supporting information on the PNAS web site), and soluble peptide-deficient class I heterodimers were purified from insect cells (13, 14). Fluorescent peptides and native-PAGE-based assembly assays (13) were used to ask whether sTpn binding influenced peptide loading of soluble peptide-deficient HLA-A2 (A2) or HLA-B*3501 (B35). Under the conditions that were examined, no significant differences in peptide binding were observed in the presence or absence of sTpn (Fig. 7 A–D, which is published as supporting information on the PNAS web site).
Absence of a significant effect of sTpn on peptide loading could arise because of absent or inefficient sTpn–class I-binding interactions under these in vitro conditions, or because of enhanced kinetics of class I peptide loading compared with sTpn binding. Therefore, it was important to investigate binding interactions between sTpn and A2. Equimolar amounts of A2 heterodimers and sTpn monomers were incubated for 2 h at 4°C or 37°C. Samples were then immunoprecipitated with the anti-FLAG antibody, proteins separated by SDS/PAGE, and visualized by silver-staining to observe sTpn and any A2 that bound to sTpn. A2 was visualized in the anti-FLAG immunoprecipitations (IPs) after protein incubations at 37°C (Fig. 1A, lane 6), but not 4°C (Fig. 1A, lane 3).
Fig. 1.

Tpn can form complexes with peptide-deficient classical and nonclassical class I molecules at physiological temperatures. (A–C) sTpn–class I and sTpn–M10 complexes are detectable by coimmunoprecipitation analyses. SDS/PAGE and silver staining analyses of direct protein loads of 0.2–0.5 μg A2 (A, lane 1; B, lane 2), sTpn (B, lane 1, 0.5 μg), M10 (C, lanes 1 and 5, 0.5 μg) or HFE (B, lane 3, 1 μg) and anti-FLAG IP of indicated proteins or buffer that had been incubated at 4°C (A, lanes 2–4), 30°C (C, lanes 6 and 7) or 37°C (A, lanes 5–8; B lanes 4–9; and C, lanes 2–4). (D) A2/Tpn and M10/Tpn complexes are detectable in insect cells. SDS/PAGE phosphorimaging analyses of indicated samples. Insect cells cultured at 26°C were infected with baculoviruses encoding full length Tpn and indicated proteins, metabolically labeled, lysed, proteins immunoprecipitated with Tpn-specific antibody (PaSta-1, lane 1) or anti-His to detect His tagged proteins (M10 or A2) (lanes 2–5). (E and F) Peptide loading of class I inhibits sTpn binding. (Left) Direct protein loads of A2 (E, lane 1, 0.25 μg), LLDVPTAAV-loaded A2 (E, lane 2, 0.25 μg), or B35 (F, lane 1, 0.5 μg) and anti-FLAG IP (E, lanes 3–8; F, lanes 2–7) of indicated proteins. pA2 indicates LLDVPTAAV-loaded A2, and pB35 indicates LPSSADVEF-loaded B35. (Right) Bar graph shows class I/sTpn intensity ratio comparisons for the indicated lanes. Binding of sTpn to peptide-deficient class I was set at 100% to compare relative signals. Data are representative of numerous (A), two (B, C Right, E, and F) and four (C Left and D) independent analyses.
To examine the specificity of the A2–sTpn interaction, we tested the ability of sTpn to bind to other proteins with structures related to class I. HFE (the protein mutated in hereditary hemochromatosis) HC folds into a structure similar to class I HC, and associates with β2m, but does not bind peptides, and has a closed peptide-binding groove (15). By IP assays similar to those described for A2, sTpn did not interact with HFE (Fig. 1B, lane 6). M10.5 (M10), is a pheromone receptor-associated protein with a class I-like fold. M10 binds β2m, but the counterpart of its peptide-binding groove is open and unoccupied (16). Somewhat surprisingly, like A2, M10 bound sTpn at 37°C (Fig. 1C Left, lane 2), but not at lower temperatures (Fig. 1C Right, lane 7). Insect cells have previously been used to assess TAP/Tpn/class I complex formation and to reconstitute class I peptide loading (5, 17). Additionally, in insect cells, Tpn is the obligate mediator of the TAP–class I interaction (12). To verify that sTpn–A2 and sTpn–M10 interactions observed in vitro were not nonspecific binding interactions induced by protein incubations at 37°C, we assessed A2 and M10 binding to Tpn in insect cells. After metabolic labeling of baculovirus-infected cells, Tpn/A2 and Tpn/M10 complexes could both be visualized by IP with the anti-His antibody (both A2 and M10 are His tagged) (Fig. 1D, lanes 5 and 4, respectively). Together, these observations indicated that Tpn–A2 and Tpn–M10 interactions were not restricted to the in vitro incubation condition at 37°C, but were also observable when the proteins were coexpressed in insect cells.
The observation of M10-Tpn binding suggested preferential Tpn recognition of open and empty class I molecules. This possibility was examined further. A2 (10 μM) was incubated with 500 μM A2-specific peptide LLDVPTAAV (18) at 47°C for 2 h, to obtain a peptide-loaded version. Unbound peptide was removed from A2 by passage through a Biospin-30 column. Equal amounts of untreated or peptide-loaded A2 (pA2) were compared for tapasin binding in IP assays. In sTpn/pA2 incubations, the signal intensity in the region corresponding to A2 HC was significantly reduced compared with sTpn/untreated A2 incubations (Fig. 1E, lane 6 compared with lane 4, and quantifications, Right). We also investigated Tpn binding to B35, a class I allotype that has been described to be Tpn-dependent for its cell surface expression (19). Soluble B35 was loaded with a specific high affinity peptide LPSSADVEF (20) at 47°C for 2 h. Equal amounts of untreated or peptide-loaded B35 heterodimers (pB35) were incubated with sTpn at 37°C for 2 h, and IP assays undertaken. Peptide-deficient B35 bound to sTpn, but peptide-loaded B35 did not (Fig. 1F, lane 3 compared with lane 5). Together, these analyses indicated that Tpn preferentially recognized peptide-deficient class I molecules, as well as the empty and open class I like protein, M10.
When Added in Excess, β2m Can Be Visualized as a Component of Tpn–Class I Complexes.
After IP of sTpn-A2 mixtures with the anti-FLAG antibody, it was difficult to detect β2m by direct staining of gels (Fig. 2A Upper, lane 2). However, by immunoblotting with β2m-specific antibodies, a faint signal was observed for β2m (Fig. 2A Lower, lane 2). We compared β2m levels in the sTpn–A2 complexes against different known amounts of purified heterodimeric A2. Relative to the β2m/HC ratios in the heterodimeric A2 samples (expected to be 1:1); a significantly substoichiometric β2m/HC ratio was observed in sTpn-associated A2 HC (Fig. 2A Lower, β2m blot). To examine whether the presence of excess β2m during sTpn–class I complex formation could enhance the amount of sTpn-A2-associated β2m, we incubated A2 and sTpn in the presence of a ten-fold excess of purified β2m at 37°C for 2 h, followed by IP with the anti-FLAG antibody. Indeed, when excess β2m was present during complex formation, the amount of sTpn-associated β2m was enhanced and equal or higher β2m/HC ratios were observed than in the heterodimeric A2 samples (Fig. 2B). This increase in β2m association coincided with a decrease in the overall recovery of sTpn-associated HC.
Fig. 2.

Tpn can form complexes with β2m at physiological temperature. sTpn–A2 complexes were formed by incubating proteins (5 μM each) at 37°C either in the absence (A) or presence (B) of β2m (50 μM) and immunoprecipitated with anti-FLAG. (C) sTpn (5 μM) and β2m (50 μM), or β2m alone were incubated for 2 h at 37°C, then immunoprecipitated with anti-FLAG. (A–C Upper) Silver-stained SDS/PAGE gels showing anti-FLAG IP of indicated proteins (A, lane 2; B, lanes 1 and 2; C lanes 1 and 2) or buffer (A, lane 1; C, lane 3) and direct protein loads of indicated concentration of A2 heterodimers (A and B, lanes 3–8). (A–C Lower) Anti-β2m blot of the above gels. Data are representative of two independent analyses.
Direct incubation of sTpn with β2m alone at 37°C for 2 h (Fig. 2C Upper and Lower, lanes 1) followed by anti-FLAG IP revealed a signal for β2m that was slightly above the nonspecific control (Fig. 2C, lane 1 compared with lane 2; lane 2 was an IP of β2m alone with anti-FLAG), suggesting the possibility of a weak β2m–sTpn interaction. Together, these observations indicate that the primary site of Tpn-class I contact resides in the HC, but that β2m can be detected in complex with Tpn when it is present in stoichiometric excess.
Tpn-Associated Class I Is Subsequently Assembly Competent.
A2–sTpn complexes were preformed by incubating both proteins (5 μM each) in the presence or absence of excess β2m (50 μM) at 37°C for 2 h (in 50 mM Tris/150 mM NaCl, pH 7.5). The complexes were incubated with anti-FLAG beads overnight at 4°C. Proteins were eluted by using 100 μM FLAG peptide, and the eluates were found to contain both Tpn and A2 (Fig. 3A, lanes 1 and 2).
Fig. 3.

Class I isolated in complex with Tpn is subsequently assembly competent. (A and B Top) SDS/PAGE and silver-staining analyses of sTpn-A2 (A) or sTpn-B35 (B) complexes eluted from an anti-FLAG beads. sTpn–class I complexes formed in the absence or presence of excess β2m are shown in lanes 1 and 2, respectively. Direct protein loads of indicated amounts of class I standards are shown in lanes 3–6. (A and B Middle) Samples corresponding to proteins shown in Top were incubated with 1 μM LLDCFPTAAV (for A2, A) or LPSCFADVEF (for B35, B) and 1 μM β2m at 37°C for 1, 2, or 4 h. Class I–peptide complexes were separated from free peptide by native-PAGE, and protein–peptide complexes were visualized by fluorimaging analyses. (A and B Bottom) Peptide-binding signals from Middle were quantified by ImageQuant and are represented as bar graphs. Data are representative of three independent analyses.
The amounts of HC in the two anti-FLAG eluates were estimated by titration against known amounts of A2 heterodimers (Fig. 3A Top, lanes 3–6). sTpn-A2 eluates in which complexes were formed in the absence of excess β2m contained ≈80 ng A2 (Fig. 3A Top, compare lane 1 with lane 5 and 6), which was reduced to ≈40 ng when complexes were assembled in the presence of excess β2m (Fig. 3A Top, compare lane 2 with lane 5 and 6). To assay for relative assembly competence of the eluted A2, proteins recovered from the anti-FLAG column or the different amounts of purified A2 heterodimers were incubated with excess β2m and fluorescently tagged LLDCFPTAAV at 37°C for indicated time points (Fig. 3A Middle). Peptide binding was quantified by native-PAGE and fluorimaging analyses (13) (Fig. 3A Bottom). For eluates of the A2–sTpn complexes formed in the presence of excess β2m, the peptide-binding signals were only slightly lower than the corresponding signals obtained with the 40 ng A2 standard (Fig. 3A Middle, compare lanes 2 and 5). We concluded that majority of this eluate was assembly competent, with slightly reduced binding kinetics than that of free class I. For eluates of the A2–sTpn complexes that were formed in the absence of excess β2m (complexes that were depleted in β2m), the peptide-binding signals obtained were significantly lower than the corresponding signals obtained with the 80 ng heterodimeric A2 standard (Fig. 3A Middle, compare lanes 1 and 6). Thus, β2m binding to HC may be rate limiting for assembly of peptide-class I in these β2m-depleted sTpn–A2 complexes. β2m is a critical regulator of class I-peptide assembly rates. Peptide loading of class I heterodimers is enhanced by the presence of excess β2m even in the absence of Tpn (Fig. 8, which is published as supporting information on the PNAS web site).
A parallel set of results were obtained with B35. Under conditions where sTpn–B35 complexes were formed in the presence of excess β2m, the extent of B35-peptide assembly observed was similar to that observed with free B35 (Fig. 3B, compare lane 2 with lane 3 and 4). Under conditions where sTpn–B35 complexes were formed in the absence of excess β2m, B35-peptide assembly observed was significantly inhibited relative to that observed with free heterodimeric B35 (Fig. 3B, compare lanes 1 and 5).
Tpn Binds Preferentially to Empty Class I Molecules.
In the studies described so far, sTpn–class I complexes were observable at 37°C, but not at lower temperatures (Figs. 1A and 4A and C). It was possible that the A2 and B35 purified from insect cells were not completely empty, and that the 37°C incubations promoted dissociation of peptides endogenous to insect cells. To ask whether interactions of sTpn with completely “empty” class I could occur at lower temperatures, A2 and B35 (100–300 μg) were dialyzed against 6 M guanidine hydrochloride (GnHCL) by using Centricon (10-kDa membranes) to remove any associated peptide, and proteins refolded by gel filtration chromatography (Superose 6 column) in the presence of 100–300 μg of β2m. Fractions corresponding to heterodimers were collected and used directly in peptide-binding assays at room temperature to compare their assembly competence in the presence or absence of sTpn. Small enhancements, if any, were observed for peptide binding to empty A2 and B35 in the presence of sTpn compared with the absence of sTpn (Fig. 9, which is published as supporting information on the PNAS web site).
Fig. 4.

Tpn preferentially binds empty class I molecules. (A–C) Silver-stained SDS/PAGE showing direct protein loads of A2 (A, lane 1, 0.3 μg), B35 (C, lane 2, 0.2 μg), or sTpn (A, lane 2, 0.05 μg; B, lane 1, 0.2 μg; C, lane 1, 0.1 μg) or anti-FLAG IP (A, lanes 3 and 4; B, lanes 2 and 3; C, lanes 3–6) of indicated proteins (3 μM each) after incubations at 30°C for 6 h. Empty class I molecules are represented as eA2 and eB35, and untreated class I molecules are represented as A2 and B35. Data are representative of two (A and B) or three (C) independent analyses.
Empty class I molecules were concentrated in the presence of excess β2m (300 μg) and analyzed for binding to sTpn at lower temperature (Fig. 4). Empty A2 and B35 but not the precursor untreated proteins were observed to form complexes with sTpn at 30°C (Fig. 4B, lane 3, and Fig. 4C, lane 6). The ability of denatured/refolded class I (but not the precursor class I) to bind sTpn at 30°C also supports the possibility that sTpn preferentially binds to a conformation found in empty class I molecules. Empty A2 and B35 that were complexed to Tpn at 30°C were purified by using anti-FLAG beads, and proteins eluted with the FLAG peptide as described in Fig. 3. Eluates containing A2 and B35 were both found to be assembly competent (Fig. 10, which is published as supporting information on the PNAS web site).
Regulation of the Tpn–Class I Interaction by Peptide and β2m.
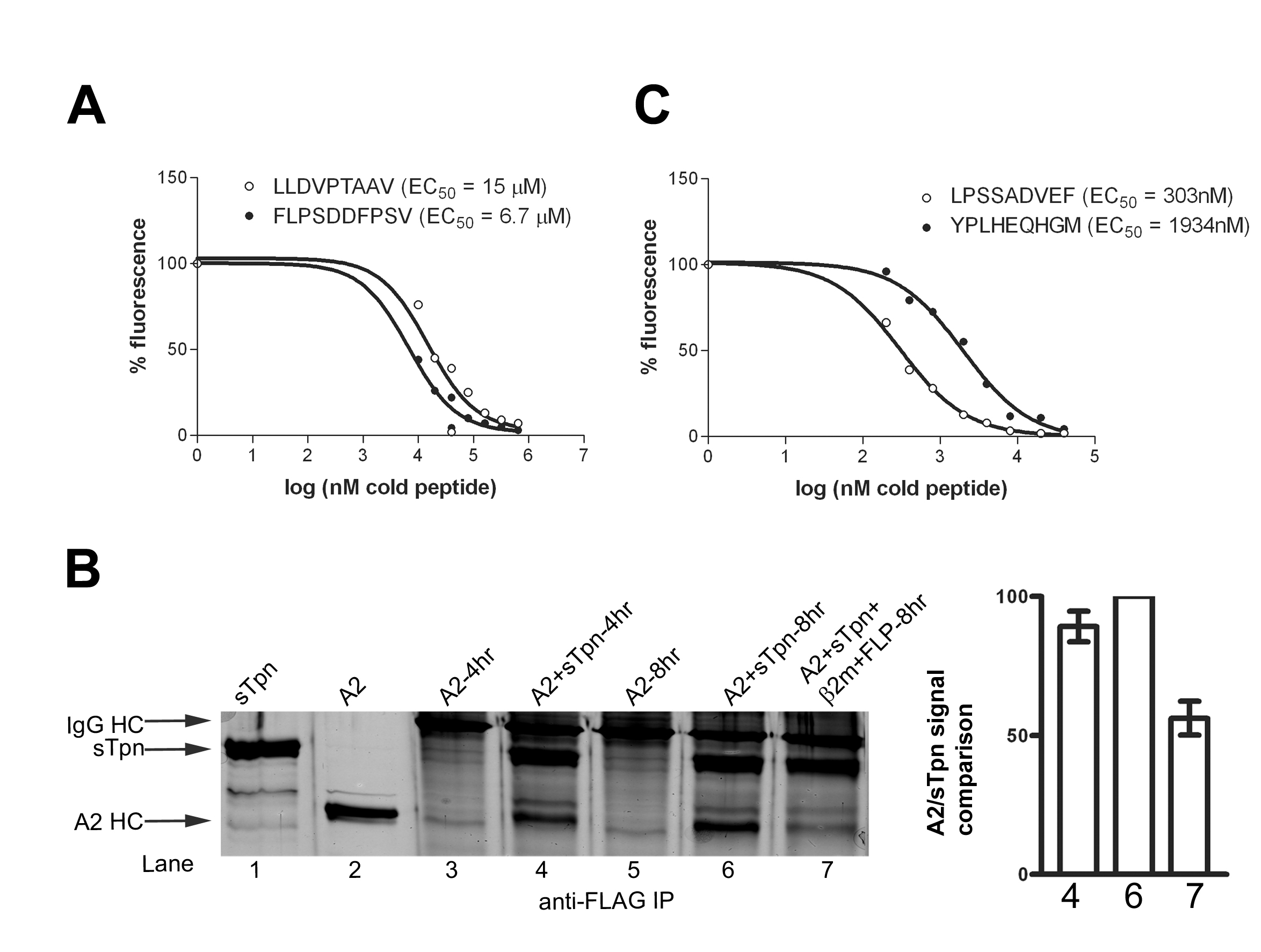
In the binding analyses of Figs. 3 and 10, peptide-loaded class I molecules migrated at the same position on a native gel regardless of whether the protein used in the assembly assays was free class I or Tpn-class I eluates from the anti-FLAG column. These results suggested that sTpn had dissociated from the peptide-loaded class I either as a result of peptide loading, or as a result of protein dilutions after protein elutions from anti-FLAG beads. To directly investigate the effect of peptide on disassembly of sTpn-class I, sTpn and A2 were incubated for 2 h in the absence of excess β2m after which, the mixture was divided into three parts. One part was stored on ice and the remaining two parts were each incubated with buffer or excess β2m and A2 specific peptides (LLDVPTAAV) at 37°C for an additional 2 h and IP analyses were undertaken (Fig. 5A). Compared with the additional incubation with buffer (Fig. 5A, lane 5), and compared with samples that were incubated for just 2 h (Fig. 5A, lane 4), additional incubation with peptide and β2m resulted in a reduction in the amount of coimmunoprecipitating A2 (Fig. 5A, lanes 6). The FLPSDDFPSV peptide that bound to A2 with 2.5-fold higher affinity than LLDVPTAAV (Fig. 11A, which is published as supporting information on the PNAS web site) also induced dissociation of A2 from Tpn to similar levels as LLDVPTAAV (Fig. 11B). However, parallel analyses revealed more efficient dissociation of Tpn-B35 induced by the high affinity peptide LPSSADVEF compared with the lower affinity YPLHEQHGM (Fig. 5 B and C, compare lanes 6). LPSSADVEF was estimated to bind B35 with ≈6-fold higher affinity than YPLHEQHGM (Fig. 11C).
Fig. 5.

Dissociation of sTpn–A2 and sTpn–B35 complexes by peptides and β2m. (A–C Left) Silver-stained SDS/PAGE gels of indicated proteins (lanes 1 and 2) or anti-FLAG IP (lanes 3–7). (A and B) Indicated class I and sTpn (5 μM each) or class I alone were incubated for 2 h at 37°C (lanes 3 and 4, respectively), followed by additional 2 h incubation with buffer (lanes 5), or with 50 μM β2m and 500 μM LLDVPTAAV (LLD) or LPSSADVEF (LPS) (lanes 6). (C) B35 and sTpn were incubated for 2 h at 37°C (lane 5), followed by additional 2 h incubation with buffer (lane 3) or with 50 μM β2m and 50 μM YPLHEQHGM (YPL) (lane 6). Samples were then processed for IP as described in Experimental Procedures. (A–C Right) Bar graphs show class I/sTpn intensity ratio comparisons for the indicated lanes, with the highest ratio in each gel set at 100%. Data in Right panels are averaged over two independent analyses. (D and E) Class I binding to anti-FLAG beads depends on Tpn. Class I (5 μM) was incubated at 37°C in the presence or absence of sTpn (5 μM) and β2m (50 μM) as indicated at 37°C for 4 h. Proteins isolated with anti-FLAG beads were eluted with 100 μM FLAG peptide and analyzed by immunoblotting with anti-His (D; A2) or HC10 (E; B35). (F and G) Class I–sTpn complexes were formed as described in D and E in the presence (F) or absence (G) of excess β2m, isolated with anti-FLAG beads, and class I was eluted with combinations of specific peptide (S), nonspecific peptide (NS), and β2m as indicated, or buffer alone. Immunoblotting of A2 and B35 elutions is indicated in the Upper and Lower panels, respectively. Volumes of eluates used in the immunoblotting analyses are indicated (10, 20, or 40 μl). These data are representative of three and four independent analyses, respectively, for F and G.
To further investigate and compare conditions for dissociation of class I–Tpn, complexes were isolated by using anti-FLAG beads, and then subject to different elution conditions. The first set of elutions with the FLAG peptide verified that class I binding to the anti-FLAG beads was indeed dependent on the presence of tapasin (Fig. 5 D and E). In the next set of elutions (Fig. 5F), anti-FLAG beads containing sTpn–class I complexes formed in the presence of β2m were incubated at room temperature for 4 h with 0.1 ml buffer containing specific peptides alone, β2m and specific peptides, β2m and nonspecific peptides, or buffer alone, as indicated. Supernatants were collected and analyzed for the presence of class I by immunoblotting analyses with HC-specific antibodies. The analyses showed that specific peptides alone could elute B35 to some extent, and low levels of A2 (Fig. 5F, lanes 3 and 4 compared with lanes 7 and 8). β2m plus nonspecific peptides eluted more A2 and B35 compared with the specific peptides alone or buffer (Fig. 5F, lanes 5 and 6). The β2m plus specific peptide combination was most efficient at elution of both A2 and B35 (Fig. 5F, lanes 1 and 2); however the differences between elutions with β2m/nonspecific peptide and β2m/specific peptide were more pronounced for A2 than for B35, and indicated more efficient elution of B35 with β2m alone.
Anti-FLAG beads containing sTpn–class I complexes formed in the absence of excess β2m were incubated at room temperature for 4 h with 0.1 ml β2m and specific peptides, β2m and nonspecific peptides, β2m alone, or buffer alone, and the eluate analyzed for class I by immunoblotting (Fig. 5G). Although some elution was observed with β2m alone or β2m in combination with nonspecific peptides (Fig. 5G, lanes 1–2 and 5–6), the β2m+specific peptide combinations were more effective at dissociating class I from tapasin (Fig. 5G, lanes 3 and 4).
If Tpn associates preferentially with the peptide-free form of class I, the expectation is that different peptides would inhibit Tpn–class I complex formation to an extent that is determined by the intrinsic affinity of the particular peptide–class I interaction. Indeed, this was the observed result. When present during sTpn–class I complex formation, LLDVPTAAV was a better inhibitor of the sTpn–class I interaction than the lower affinity LLDVPTAAVQ (Fig. 7 and Fig. 12A, which is published as supporting information on the PNAS web site; see Fig. 7 for comparisons of binding of the two peptides). Likewise for B35 peptides, the higher affinity LPSCFADVEF peptide was a better inhibitor of the sTpn–class I interaction than the lower affinity YPLHEQHGM peptide (Fig. 12 B and C).
Discussion
Our data suggest two types of Tpn–class I complexes: those containing predominantly HC and those containing both HC and β2m. β2m seems to destabilize Tpn–HC interactions, as indicated by the observations of inhibited HC-Tpn binding in the presence of excess β2m (Fig. 3), as well as β2m-induced elution of class I HC from Tpn (Fig. 5 F and G). Excess β2m may simply reduce the exposure of Tpn-binding residues by stabilization of heterodimeric HC–β2m interactions (in the same manner that peptides do), rendering HC–Tpn interaction weaker, but also more dynamic and assembly competent (compared with the HC–Tpn interaction formed in the absence of excess β2m; Fig. 3). The observations that B35 was more easily dissociated by β2m/peptide combinations and β2m alone compared with A2 (Fig. 5) is reminiscent of previous findings that HLA-B*3501/TAP interactions were not detectable in cell lysates under conditions where A2/TAP interactions were readily detectable (21). The stabilities of peptide-deficient forms of class I allotypes may be variable, which in turn could be reflected in the extent of steady-state Tpn/TAP binding. Additionally, for some class I molecules such as A2, assembly of HC/β2m/peptide trimers may be a cooperative event, whereas the heterodimeric intermediates may be more stable for other class I allotypes.
That β2m destabilizes HC–Tpn interactions may seem surprising in light of reports that Tpn–HC interactions were significantly reduced in β2m-deficient cells compared with β2m-sufficient cells (22). However, in cells, the absence of β2m markedly decreases levels of HLA class I HC, making it difficult to directly quantify relative propensities of free HC and β2m-associated HC for Tpn binding (as was the case in insect cells; C. Perria and M.R., unpublished observations). Alternatively/additionally, in cells, in the absence of β2m, free HC may be sequestered from Tpn binding by other chaperones such as calnexin.
Cell surface expression of many class I allotypes is markedly increased by the presence of Tpn, and Tpn increases MHC class I export from the ER (11), class I thermostability (8), and cell surface stability (11). How might the data described here explain these assembly-promoting effects of Tpn? Our studies indicate preferential recognition of empty class I by Tpn. Furthermore, sampling of the peptide environment by the MHC class I seems to be permissive under conditions where Tpn binding was observed (Fig. 5), either because MHC class I is in dynamic equilibrium between Tpn-free and Tpn-associated conformations, or because a Tpn-bound conformation of class I is also directly permissive for peptide loading. Higher affinity peptides were better inducers of Tpn-class I dissociation (Fig. 5 B compared with C) and stronger inhibitors of the Tpn class I binding (Fig. 12). It seems likely based on these data that the presence of Tpn could allow for a more stringent class I-peptide affinity checkpoint, consistent with models of Tpn-mediated editing/optimization (8). Only peptides that are able to efficiently dissociate class I from tapasin will be found in association with class I on the surface of Tpn-sufficient cells. Zarling et al. (11) have suggested a role for Tpn as a peptide loading facilitator rather than editor. This type of activity might have been reflected by an increase in peptide loading by class I in Tpn-associated complexes, but our studies revealed very small effects if any (Fig. 10). Additional conditions may need to be explored, varying protein concentrations and including Tpn-associated factors. Compared with the in vitro experiments shown here, within the ER, assembly and degradation of peptide-deficient class I are likely to be strongly competing processes. It has been shown recently that Tpn efficiently recruits ERp57 into the class I assembly complex (23), and it is well known that ERp57 is found in physical association with the lectin chaperones calreticulin/calnexin. Thus, the presence of Tpn may serve to efficiently recruit a chaperone network around the assembling empty class I, which is supported by previous observations that interactions within the assembly complex are all cooperative to some extent (reviewed in ref. 1). Assembly within this protective chaperone network environment may be more highly favored than outside this environment, where proteases and ER degradation pathways could compete more effectively with assembly of class I with peptide.
By preferential recognition of the empty conformation of class I, Tpn could also actively enhance dissociation of low affinity peptides and promote peptide exchange, as previously observed for HLA-DM in the MHC class II antigen presentation pathway (reviewed in ref. 24). A detailed analysis of this possibility will be important by using real time measurements of class I-peptide dissociation and exchange in the presence and absence of Tpn.
Although our data indicate preferential recognition of empty class I by Tpn (Fig. 4), Tpn may not be essential for ER retention of empty MHC class I in all cases (9, 10, 25). For example, human MHC class I molecules seem to be retained efficiently in the ER even in the absence of Tpn (25). However, Tpn seems to be quite important for ER retention of murine MHC class I (5–7). In the absence of Tpn, increased ER escape and cell surface expression of empty and suboptimally loaded class I could also, at least in part explain the reduction in MHC class I surface expression in some Tpn-deficient cells.
The interaction of Tpn with M10 was initially surprising in light of the report that TAP is not expressed in cells that express M10 (26), but subsequent comparison of the sequences of M10 and class I molecules revealed 40%, 40%, and 74% sequence identity in the α1, α2, and α3 domains, respectively. The extent of sequence identity between HFE and class I was considerably less significant (31%, 25%, and 41% sequence identity in the α1, α2, and α3 domains, respectively). Thus, whereas Tpn may not be a bona fide assembly factor for M10 in cells, the significant sequence identity between class I and M10 HC, taken together with its “open and empty groove” structure may promote Tpn-M10 cross-reactivity in vitro (Fig. 1C), or under conditions of coexpression (Fig. 1D). Residues that have been implicated in the Tpn–class I interaction include residues 132 (27) and 134 (28, 29) in the α2 domain of human class I. These residues are conserved between A2 and M10, but not between HFE and A2, supporting the possibility that this region forms a contact site for Tpn binding. Also conserved in M10 are D227 and E229 in the α3 domain, additional residues implicated in Tpn binding by human MHC class I (27). It will be important to investigate whether mutations within both the α2 and α3 domains disrupt or destabilize Tpn-MHC class I binding interactions by the assay described here.
A significant finding of these studies is that conditions that promoted heterodimer assembly induced dissociation of MHC class I-Tpn. Other investigators have reported difficulties in demonstrating peptide-induced dissociation of class I from the loading complex (30). Our data indicate that both β2m (Figs. 3 and 5) and the type of peptide used (Fig. 5) are likely to be important determinants of the extent of dissociation of class I from a Tpn-associated complex. Furthermore, as illustrated in Fig. 3, the specific conditions under which peptide loading complexes were isolated could determine the assembly kinetics of Tpn-associated MHC class I molecules.
In summary, our studies demonstrate that Tpn binds to peptide-deficient class I, that physiological temperatures promote binding, and that conditions favorable for class I assembly dissociate Tpn–class I complexes. In cells, Tpn interaction with empty class I may serve to recruit nascent class I into a chaperone network comprising Tpn, ERp57, and the lectin chaperones, which together, may create an environment that favors peptide loading and assembly over degradation.
Experimental Procedures
Proteins and Peptides.
Soluble A2/β2m, B35/β2m, and full-length Tpn expression in insect cells has been previously described (13, 14, 31). Expression and purification of soluble tapasin and β2m and detection of class I/Tpn complexes in insect cells are described in Supporting Materials and Methods, which is published as supporting information on the PNAS web site. Fluorescently labeled versions of class I-binding peptides were obtained by cysteine substitutions at positions 4 or 5 of the parent sequences, and labeling with iodoacetamidofluorescein, as previously described (13, 14). Purified human HFE (15), mouse M10.5 (16), and a baculovirus encoding mouse M10.5/human β2m (16) were obtained from the laboratory of P. J. Bjorkman (California Institute of Technology, Pasadena, CA).
Detection of Tpn–Class I Complexes Using Purified Proteins.
Soluble class I, HFE, or M10 (2.5–5 μM each) was incubated with sTpn (2.5–5 μM) in 50 mM Tris, 150 mM NaCl, pH 7.5, at indicated temperatures for indicated time. After incubations, the samples were centrifuged, diluted in the same buffer containing 1% Triton-X-100 to 1 ml, and incubated with anti-FLAG antibody (3–4 μg per immunoprecipitation) overnight at 4°C. The supernatants were further centrifuged and incubated for 2–4 h at 4°C with protein G beads. The beads were washed three times with buffer containing 0.25% gelatin (10 mM Tris/10 mM phosphate buffer/130 mM NaCl/1% Triton X-100, pH 7.5) and once with buffer without gelatin. The beads were boiled in SDS/PAGE buffer, samples were resolved by SDS/PAGE, and immunoprecipitated proteins were visualized by silver staining. For β2m detection, the anti-FLAG immunoprecipitated samples or direct proteins loads were transferred to PVDF membrane, immunoblotted with anti-β2m antisera (Roche, Nutley, NJ), HRP-conjugated secondary antibody, and developed by ECL plus kit (Amersham Biosciences, Piscataway, NJ).
Isolation of Tpn–Class I Complexes with Anti-FLAG Beads.
sTpn–class I complexes were formed by incubation of proteins (5 μM) in the presence or absence of 50 μM β2m in a total volume of 40 μl at 37°C for 2–4 h. Proteins were then diluted to 1 ml buffer (50 mM Tris/150 mM NaCl/1% Triton X-100), incubated overnight at 4°C with 100-μl FLAG-beads, loaded onto an empty column, and washed with 5–15 ml buffer to remove free class I. For some analyses, beads were eluted with FLAG peptide (100 μM in 100 μl). For other analyses, beads were then incubated with 100 μl of buffer (50 mM Tris/150 mM NaCl) containing 50 μM β2m, 50 μM β2m plus 500 μM specific or nonspecific peptides, or buffer alone at room temperature for 4 h. Specific peptide used for A2 was FLPSDDFPSV and for B35 was LPSSADVEF and nonspecific peptide for both was RRYQKSTEL. After incubations, supernatants were collected and analyzed by SDS/PAGE and immunoblotting analyses with anti-His (for A2) or HC10 (for B35) antibodies.
Supplementary Material
Acknowledgments
We thank Chris Perria for conducting the experiment shown in Fig. 1D; Dr. Peter Cresswell (Yale University School of Medicine, New Haven, CT) for the PaSta-1 antibody; Dr. Pamela Bjorkman (California Institute of Technology, Pasadena, CA) for M10 and HFE constructs; the University of Michigan Biomedical Research Core Facilities for DNA sequencing and peptide syntheses and purification; the Hybridoma Core for ascites production; and the Rheumatic Diseases Core Center and the Michigan Diabetes Research and Training Centers for financial support. This work was supported by National Institutes of Health Grant AI-44115 and a Cancer Research Institute Investigator Award (to M.R.).
Abbreviations
- HC
heavy chain
- ER
endoplasmic reticulum
- TAP
transporter associated with antigen processing
- β2m
β2-microglubulin
- class I
MHC class I
- Tpn
tapasin
- sTpn
soluble Tpn
- A2
HLA-A2
- B35
HLA-B*3501
- IP
immunoprecipitation
- M10
M10.5.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
References
- 1.Cresswell P. Traffic. 2000;1:301–305. doi: 10.1034/j.1600-0854.2000.010402.x. [DOI] [PubMed] [Google Scholar]
- 2.Lehner PJ, Surman MJ, Cresswell P. Immunity. 1998;8:221–231. doi: 10.1016/s1074-7613(00)80474-4. [DOI] [PubMed] [Google Scholar]
- 3.Garbi N, Tiwari N, Momburg F, Hammerling GJ. Eur J Immunol. 2003;33:264–273. doi: 10.1002/immu.200390029. [DOI] [PubMed] [Google Scholar]
- 4.Ortmann B, Copeman J, Lehner PJ, Sadasivan B, Herberg JA, Grandea AG, Riddell SR, Tampe R, Spies T, Trowsdale J, Cresswell P. Science. 1997;277:1306–1309. doi: 10.1126/science.277.5330.1306. [DOI] [PubMed] [Google Scholar]
- 5.Schoenhals GJ, Krishna RM, Grandea AG III, Spies T, Peterson PA, Yang Y, Fruh K. EMBO J. 1999;18:743–753. doi: 10.1093/emboj/18.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnden MJ, Purcell AW, Gorman JJ, McCluskey J. J Immunol. 2000;165:322–330. doi: 10.4049/jimmunol.165.1.322. [DOI] [PubMed] [Google Scholar]
- 7.Grandea AG III, Golovina TN, Hamilton SE, Sriram V, Spies T, Brutkiewicz RR, Harty JT, Eisenlohr LC, Van Kaer L. Immunity. 2000;13:213–222. doi: 10.1016/s1074-7613(00)00021-2. [DOI] [PubMed] [Google Scholar]
- 8.Williams AP, Peh CA, Purcell AW, McCluskey J, Elliott T. Immunity. 2002;16:509–520. doi: 10.1016/s1074-7613(02)00304-7. [DOI] [PubMed] [Google Scholar]
- 9.Chun T, Grandea AG III, Lybarger L, Forman J, Van Kaer L, Wang CR. J Immunol. 2001;167:1507–1514. doi: 10.4049/jimmunol.167.3.1507. [DOI] [PubMed] [Google Scholar]
- 10.Lybarger L, Yu YY, Chun T, Wang CR, Grandea AG III, Van Kaer L, Hansen TH. J Immunol. 2001;167:2097–2105. doi: 10.4049/jimmunol.167.4.2097. [DOI] [PubMed] [Google Scholar]
- 11.Zarling AL, Luckey CJ, Marto JA, White FM, Brame CJ, Evans AM, Lehner PJ, Cresswell P, Shabanowitz J, Hunt DF, Engelhard VH. J Immunol. 2003;171:5287–5295. doi: 10.4049/jimmunol.171.10.5287. [DOI] [PubMed] [Google Scholar]
- 12.Thammavongsa V, Raghuraman G, Filzen TM, Collins KL, Raghavan M. J Immunol. 2006;177:3150–3161. doi: 10.4049/jimmunol.177.5.3150. [DOI] [PubMed] [Google Scholar]
- 13.Mancino L, Rizvi SM, Lapinski PE, Raghavan M. Proc Natl Acad Sci USA. 2002;99:5931–5936. doi: 10.1073/pnas.092031799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rizvi SM, Mancino L, Thammavongsa V, Cantley RL, Raghavan M. Mol Cell. 2004;15:913–923. doi: 10.1016/j.molcel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Lebron JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, Feder JN, Bjorkman PJ. Cell. 1998;93:111–123. doi: 10.1016/s0092-8674(00)81151-4. [DOI] [PubMed] [Google Scholar]
- 16.Olson R, Huey-Tubman KE, Dulac C, Bjorkman PJ. PLoS Biol. 2005;3:e257. doi: 10.1371/journal.pbio.0030257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lauvau G, Gubler B, Cohen H, Daniel S, Caillat-Zucman S, van Endert PM. J Biol Chem. 1999;274:31349–31358. doi: 10.1074/jbc.274.44.31349. [DOI] [PubMed] [Google Scholar]
- 18.Wei ML, Cresswell P. Nature. 1992;356:443–446. doi: 10.1038/356443a0. [DOI] [PubMed] [Google Scholar]
- 19.Park B, Lee S, Kim E, Ahn K. J Immunol. 2003;170:961–968. doi: 10.4049/jimmunol.170.2.961. [DOI] [PubMed] [Google Scholar]
- 20.Morel S, Ooms A, Van Pel A, Wolfel T, Brichard VG, van der Bruggen P, Van den Eynde BJ, Degiovanni G. Int J Cancer. 1999;83:755–759. doi: 10.1002/(sici)1097-0215(19991210)83:6<755::aid-ijc10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 21.Neisig A, Wubbolts R, Zang X, Melief C, Neefjes J. J Immunol. 1996;156:3196–3206. [PubMed] [Google Scholar]
- 22.Bangia N, Lehner PJ, Hughes EA, Surman M, Cresswell P. Eur J Immunol. 1999;29:1858–1870. doi: 10.1002/(SICI)1521-4141(199906)29:06<1858::AID-IMMU1858>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 23.Peaper DR, Wearsch PA, Cresswell P. EMBO J. 2005;24:3613–3623. doi: 10.1038/sj.emboj.7600814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, Hornell TM, Mellins ED. Immunol Rev. 2005;207:242–260. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 25.Grandea AG III, Lehner PJ, Cresswell P, Spies T. Immunogenetics. 1997;46:477–483. doi: 10.1007/s002510050308. [DOI] [PubMed] [Google Scholar]
- 26.Loconto J, Papes F, Chang E, Stowers L, Jones EP, Takada T, Kumanovics A, Fischer Lindahl K, Dulac C. Cell. 2003;112:607–618. doi: 10.1016/s0092-8674(03)00153-3. [DOI] [PubMed] [Google Scholar]
- 27.Harris MR, Lybarger L, Myers NB, Hilbert C, Solheim JC, Hansen TH, Yu YY. Int Immunol. 2001;13:1275–1282. doi: 10.1093/intimm/13.10.1275. [DOI] [PubMed] [Google Scholar]
- 28.Lewis JW, Neisig A, Neefjes J, Elliott T. Curr Biol. 1996;6:873–883. doi: 10.1016/s0960-9822(02)00611-5. [DOI] [PubMed] [Google Scholar]
- 29.Peace-Brewer AL, Tussey LG, Matsui M, Li G, Quinn DG, Frelinger JA. Immunity. 1996;4:505–514. doi: 10.1016/s1074-7613(00)80416-1. [DOI] [PubMed] [Google Scholar]
- 30.Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. Immunol Rev. 2005;207:145–157. doi: 10.1111/j.0105-2896.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 31.Raghuraman G, Lapinski PE, Raghavan M. J Biol Chem. 2002;277:41786–41794. doi: 10.1074/jbc.M207128200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}