

Abstract
Voltage-activated ion channels open and close in response to changes in membrane voltage, a process that is crucial for electrical signaling in the nervous system. The venom from many poisonous creatures contains a diverse array of small protein toxins that bind to voltage-activated channels and modify the gating mechanism. Hanatoxin and a growing number of related tarantula toxins have been shown to inhibit activation of voltage-activated potassium (Kv) channels by interacting with their voltage sensing domains. This review summarizes our current understanding of the mechanism by which these toxins alter gating, the location of the toxin receptor within Kv channels and the disposition of this receptor with respect to the lipid membrane. The conservation of tarantula toxin receptors among voltage-activated ion channels will also be discussed.
1. Introduction
Voltage-activated ion channels open and close in response to changes in membrane voltage, a process that underlies their fundamental roles in the generation and propagation of electrical signals within the nervous system. Not surprisingly, these crucial ion channels are popular targets for the wide array of protein toxins found in the venom of poisonous creatures. For example, venom from arachnids (spiders and scorpions), anthozoans (sea anemone), mollusks (cone snails) and reptiles (snakes) contain toxins that bind to voltage-activated potassium (Kv), sodium (Nav) and calcium (Cav) channels (Possani et al., 2000; Rash and Hodgson, 2002; Srinivasan et al., 2002; Terlau and Olivera, 2004). The most widely appreciated biological function of toxins targeting voltage-activated channels is to paralyze prey, either by inhibiting (Nav and Cav inhibitors) or excessively stimulating (Kv inhibitors and Nav activators) transmission at synapses within the nervous system.
The toxins that interact with voltage-activated ion channels can be thought of as working through two distinct targeting mechanisms. In the first mechanism the toxin binds to the outer vestibule of the ion conduction pore and inhibits the flow of ions. The best studied examples of this mechanism are the scorpion toxins charybodotoxin and agitoxin (Miller, 1995), two particularly interesting Kv channel inhibitors that were used to find the pore forming region of potassium channels (MacKinnon and Miller, 1989). In the second type of mechanism the toxin binds to a region of the channel that changes conformation during gating and influences the gating mechanism by altering the relative stability of closed, open or inactivated states. The α- and β-scorpion Nav channel toxins from Centruroides sculpturatus scorpion venom were the earliest studied protein toxins that modify gating of voltage-activated ion channels (Koppenhofer and Schmidt, 1968b, 1968a; Cahalan, 1975; Wang and Strichartz, 1983). This review primarily focuses on the more recently discovered family of toxins that modify gating of Kv channels, with occasional reference to related studies on toxins that interact with Cav and Nav channels.
1.1 Structure and gating of Kv channels
The X-ray structure of the mammalian Kv1.2 channel (Long et al., 2005) nicely illustrates the two primary regions of the channel that are targeted by toxins (Fig. 1). As the structure shows, Kv channels are tetramers, a fact that was first established from experiments with the pore blocking scorpion toxin, agitoxin (MacKinnon, 1991). Each Kv channel subunit contains 6 transmembrane segments, termed S1 to S6, arranged into two types of domains; a single pore domain formed by the S5-S6 regions from the four subunits (yellow helices), and four surrounding voltage-sensing domains, each one formed by the S1-S4 helices from a single subunit. The pore domain houses the K+ selective ion conduction pathway (demarcated by the red potassium ions in Fig 1 B,C), and the receptor for pore blocking toxins that bind to the extracellular vestibule near the selectivity filter. In the case of charybdotoxin, a Lys residue on the active surface of the toxin projects into the pore of the channel and interacts with potassium ions bound within the selectivity filter (Anderson et al., 1988; MacKinnon and Miller, 1988; Park and Miller, 1992). Many other types of K+ channels only contain the equivalent of the pore domain of Kv channels (Jan and Jan, 1997), and pore blocking scorpion and bee toxins similarly bind to the extracellular entrance of the pore in these simpler channels (Lu and MacKinnon, 1997; Jin and Lu, 1998), highlighting the conserved nature of potassium channel pores. The pore domain also contains the S6 activation gate, an intracellular region of the ion conduction pore that prevents the flow of ions in the closed state (Fig 1B) (Yellen, 2002). The S6 gate region of the protein does not appear to be directly targeted by protein toxins, presumably because poisonous creatures deposit their venom into the extracellular solution and the S6 gate is located on the opposite side of the membrane.
Fig.1.
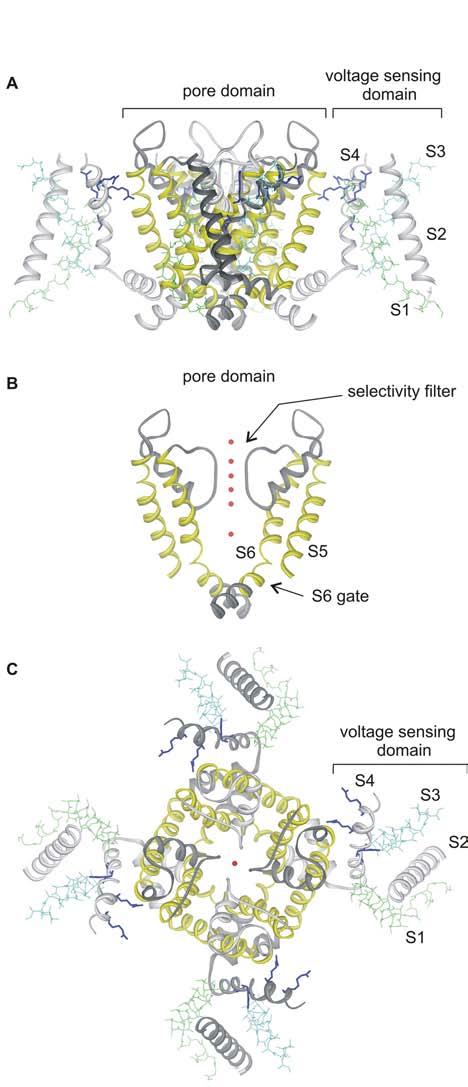
X-ray structure of the Kv1.2 channel
(A) Side view of the tetrameric Kv1.2 structure oriented so that the extracellular side of the membrane is positioned at the top. (B) Side view with front and back subunits and all four voltage sensing domains removed for clarity. Red spheres are potassium ion bound within the pore, marking the location of the ion permeation pathway. (C) View of the Kv1.2 structure from the extracellular side of the membrane. Images were created us DSViewer Pro and Protein Data Bank accession ID 2A79 (Long et al., 2005).
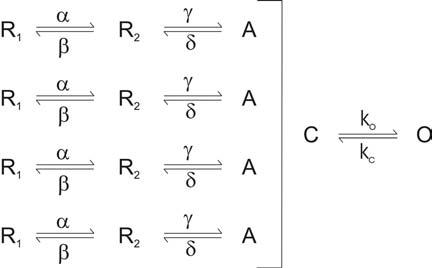
The voltage sensing domain is a second region of the channel that is widely targeted by toxins, in this case by the class of toxins that bind to and inhibit the channel by stabilizing a particular state in the gating pathway. The process of voltage-dependent gating in Kv channels is thought to involve several relatively independent motions of the four voltage sensing domains between resting (R) and activated (A) states, followed by a concerted opening transition where the S6 gate moves from a closed (C) to an open (O) state (Scheme I; (Ledwell and Aldrich, 1999)). There is growing evidence to suggest that the S4 segment within the voltage sensing domains also moves during the concerted opening transition (Smith-Maxwell et al., 1998b, 1998a; Ledwell and Aldrich, 1999; del Camino et al., 2005; Pathak et al., 2005), and therefore a toxin that binds to the voltage sensing domains could in principle inhibit channel activation by stabilizing any of the states prior to the open state.
Scheme I.
1.2 Toxins inhibiting Kv channels by targeting voltage sensors
Hanatoxin is the founding member of a family of toxins that bind to the voltage sensing domains in Kv channels and inhibit opening of these channels. This 35 residue protein toxin was originally isolated from the venom of the Chilean rose tarantula (Grammostola spatulata) during a search for new inhibitors of the Kv2.1 channel (Swartz and MacKinnon, 1995). The selectivity of hanatoxin is far from absolute, as it also inhibits Kv4.2 channels with similar affinity compared to Kv2.1 (see also Section 5.1). Although hanatoxin is one of the most extensively studied toxins targeting the voltage sensing domains in Kv channels (see below), the list of newer toxins in this class is rapidly growing (Fig 2). The closely related SGTx1 from Scodra griseipes (Marvin et al., 1999; Lee et al., 2004) also inhibits Kv2.1 and systematic mutagenesis on this toxin has identified the active face of the molecule (Wang et al., 2004). The recently discovered guangxitoxin (GxTx1E) from Plesiophrictus guangxiensis has particularly high affinity for Kv2.1 (Herrington et al., 2006), which should open up promising opportunities for better understanding the complex between this family of toxins and voltage sensing domains (see section 2.3). Stromatoxin (ScTx1) from Stromatopelma calceata and heteroscodratoxins (HmTx1,2) from Heteroscodra maculate target both Kv2 and Kv4 channels (Escoubas et al., 2002), whereas the heteropodatoxins (HpTx1-3) from Heteropoda venatoria (Sanguinetti et al., 1997; Zarayskiy et al., 2005), phrixotoxins (PaTx1,2) from Phrixotrichus auratus (Diochot et al., 1999) and TLTx1-3 from Theraphosa leblondi venom (Ebbinghaus et al., 2004) seem to primarily target Kv4 channels. One of the more recent additions to this family of toxins is VsTx1, a toxin that was isolated from Grammostola spatulata venom when screening for activity against the KvAP channel (Ruta et al., 2003; Ruta and MacKinnon, 2004), an archebacterial Kv channel for which several X-ray crystal structures are available (Jiang et al., 2003a; Lee et al., 2005). The amino acid sequences of the Kv channel toxins discussed thus far show varying similarity, ranging from 82% to 30%, and all have six cysteine residues that form 3 disulphide bonds at the core of the molecules (Fig 2; see also Section 3). It is rather interesting that with the exception of HpTx1-3, all of these Kv channel gating modifier toxins were isolated from tarantula venom. Although it is a mystery why these particular creatures are so fond of producing gating modifier toxins (and why they appear uninterested in making pore blocking protein toxins), it is clear that their venom is a particularly rich and relatively untapped repository of these toxins.
Fig.2.
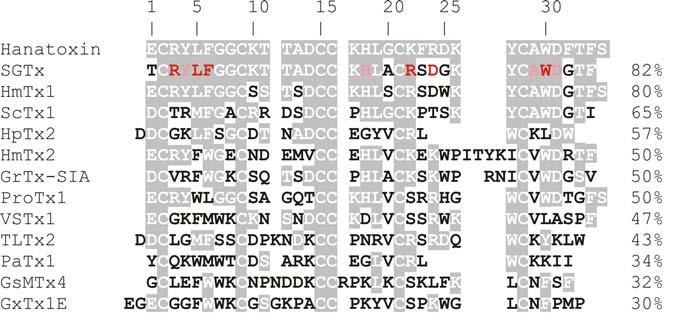
Sequence alignment of Kv channel voltage sensor toxins.
Shading indicates similarity to hanatoxin. Mutation of red and pink residues in SGTx cause dramatic (ΔΔG>1.5 kcal mol−1) and moderate (ΔΔG=1-1.5 kcal mol−1) perturbations in toxin binding to Kv2.1, respectively (Wang et al., 2004). Numbers to the right are % similarity to hanatoxin. Several toxins other than those that target Kv channels have been included for purposes of discussion, including GrTx-SIA and ProTx1 which were originally isolated as inhibitors of Cav channels, and GsMTx4, an inhibitor of stretch activated cation channels. See text for references.
It has recently become evident that sea anemone also produce gating modifier toxins that interact with Kv channels. BDS I and II from Anemonia sulcata inhibit Kv3 channels (Diochot et al., 1998) and the related APETx1 from Anthopleura elegantissima venom inhibit HERG Kv channels (Diochot et al., 2003; Restano-Cassulini et al., 2006). The sequences of these anemone toxins are quite different than other Kv channel toxins discussed here, but a growing body of work suggests that they probably inhibit through a related mechanism (Diochot et al., 2003; Yeung et al., 2005).
2. Mechanism of channel inhibition
The first indication that protein toxins inhibit Kv channels through a mechanism other than pore occlusion came from experiments on hanatoxin where transfer of the outer vestibule (S5-S6 linker) of Kv2.1 into a relatively toxin insensitive channel failed to render the recipient channel sensitive to the toxin (Swartz and MacKinnon, 1995). Binding of a Kv2.1 toxin to regions outside the pore could have been predicted from earlier studies showing that the Kv2.1 channel contains several strategically positioned basic residues within the outer vestibule that prevent tight binding of many pore blocking protein toxins (Gross et al., 1994). Although hanatoxin might have evolved a way of binding to such an unfriendly outer vestibule, targeting of a distinct region of the channel protein would seem more likely. There are now at least three lines of evidence demonstrating that hanatoxin and its close relatives inhibit Kv channels by modifying gating. First, toxin-bound channels can open and conducting ions, but the energy required to do so is considerably greater than for unbound channels, as if the toxin works through an allosteric mechanism. Second, the toxin has pronounced effects on movements of the voltage sensing domains, the region of the protein that drives opening of the channel in response to changes in membrane voltage. Third, the toxin does not interact intimately with the outer vestibule of the channel, as would be expected for a pore blocker, but instead interacts intimately with defined region within the voltage sensing domains.
2.1 Opening of toxin-bound channels
Several hallmark effects of hanatoxin are illustrated in Fig 3, where the activity of the Kv2.1 channel is assayed using voltage clamp recording techniques. Although the toxin can completely inhibit K+ channel currents evoked with a relatively weak depolarization, robust opening of the channel can be observed in response to a strong depolarization, as if the toxin shifts activation of the channel to more depolarized voltages. A key additional effect of the toxin is to produce an apparent speeding of channel closing, which can be seen by scaling the tail currents observed following repolarization of the membrane to negative voltages (Fig 3A) (Swartz and MacKinnon, 1997a). One interpretation of these phenomena is that the toxin binds to the channel and stabilizes one of the closed states in Scheme I, allowing opening of the channel with the toxin bound, but only if a sufficiently strong depolarization is applied. In principle, both of these phenomena could also be explained by a pore blocking mechanism where the blocker affinity is weaker at positive voltages; the voltage dependence here might result because the blocker enters the electric field within the pore or interacts with permeant ions within the field (MacKinnon and Miller, 1988; Lu, 2004). In this second scenario, opening of the channel in response to large depolarizations would occur because the toxin dissociates from the pore, and the tail current would be faster because it binds rapidly at negative voltages. One strong prediction of such a pore blocking mechanism is that rebinding during the tail current at negative voltages should get faster as the aqueous concentration of the toxin is increased, at least until the diffusion limit is reached. Although the tail currents show some concentration dependence, it is not strong enough to be explained by a rebinding phenomenon and the effect of the toxin on tail current kinetics saturate in the low micromolar range, far below the diffusion limit (Swartz and MacKinnon, 1997a). In addition, the kinetics of the tail currents (τ∼5ms) are more than three orders of magnitude faster than the onset of inhibition that is observed after addition of the toxin to the extracellular solution, inconsistent with rebinding of the toxin upon return to negative voltages. Collectively, these results are inconsistent with a pore blocking mechanism, but are fully compatible with the idea that Kv channels can open and close with hanatoxin bound, and that the toxin inhibits activation of the channel by stabilizing a closed state. In Section 4.2 we will revisit the question of whether the channel can gate with hanatoxin bound.
Fig.3.
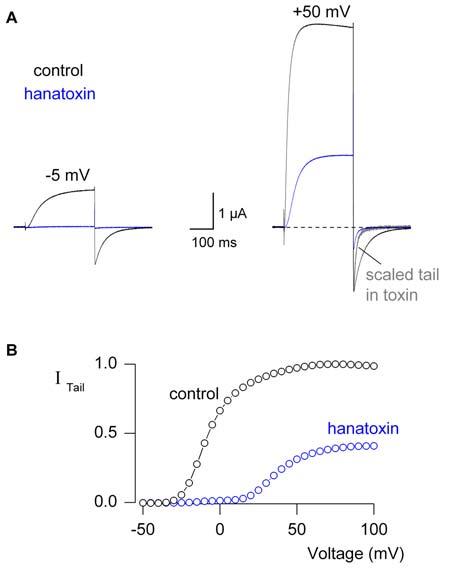
Hanatoxin modifies gating of the Kv2.1 channel.
(A) Inhibitory effects of hanatoxin (5 μM) on Kv2.1 channel ionic currents elicited by weak (left) and strong depolarizations (right). Holding voltage was −90 mV and tail voltage was −50 mV. The Kv2.1 was expressed in Xenopus oocytes and studied using a two electrode voltage clamp recording technique. (B) Voltage-activation relations obtained in the absence or presence of 5 μM hanatoxin by measuring tail currents after repolarization to −50 mV following various strength depolarizations and normalized to the maximal value in control.
In addition to shifting activation to positive voltages, hanatoxin decreases the maximal conductance at positive voltages (Fig 3), another result that is consistent with the idea that the toxin remains bound to the channel during gating. It is interesting that ω-GrTx-SIA, another gating modifier toxin from Grammostola spatulata venom (Lampe et al., 1993), inhibits Cav2.1 channels by shifting activation to depolarized voltages, much like shown in Fig 3 for hanatoxin, but in the case of ω-GrTx-SIA the maximal conductance at positive voltages actually increases (McDonough et al., 1997a). The effects of these toxins on the maximal conductance can be explained by an effect of the toxins on the concerted opening transitions (e.g. either increasing or decreasing the maximal open probability), but not by a pore blocking mechanism. This kind of differential effect on maximal conductance has recently been observed for BDSII, a sea anemone toxin that targets Kv3 channels (Yeung et al., 2005). In this case the toxin shifts activation to positive voltages and decreases the maximal conductance of the wild-type Kv3.2b channel, very similar to hanatoxin on Kv2.1 However, for a triple S4 mutant of the Kv3.2b channel, BDSII shifts activation to positive voltages and increases the maximal conductance, much like ω-GrTx-SIA on the Cav channel (Yeung et al., 2005). Although the mechanism underlying the effects of these toxins on the maximal conductance remains to be fully explained, these phenomena strongly point to an effect of the toxins on the gating mechanisms of Kv channels.
It seems likely that the other toxins discussed in the preceding section also inhibit by modifying gating. ScTx1, SGTx1 and GxTx1E shift activation of the Kv2.1 channel to positive voltages (Escoubas et al., 2002; Lee et al., 2004; Herrington et al., 2006), and similar effects are observed for PaTx1,2, HpTx1-3 and TLTx1 on Kv4 channels (Sanguinetti et al., 1997; Diochot et al., 1999; Ebbinghaus et al., 2004; Zarayskiy et al., 2005). The functional similarity of these toxins to hanatoxin, taken together with substantial sequence similarity (Fig 2), strongly suggests that they also inhibit Kv channels by modifying gating. Although the sea anemone toxins APETx1 and BDS are unrelated in sequence to hanatoxin, they similarly shift activation to positive voltages (Diochot et al., 1998; Diochot et al., 2003; Yeung et al., 2005) and other evidence discussed below suggests that they work through a mechanism similar to that of hanatoxin.
2.2 Influencing voltage sensor movement
Inhibition of channel opening could be accomplished by a toxin influencing several steps in the activation mechanism of Kv channels (see Scheme I). The four voltage sensing domains are thought to move relatively independently between R and A states in response to membrane depolarization, followed by a concerted opening transition where the S6 gate opens to support ion conduction. Charged Arginine residues in the S4 helix of the voltage sensor (Fig 1; dark blue residues) move in response to a change in membrane voltage, and this movement can be detected as a non-linear capacitive current, or gating current (Bezanilla and Stefani, 1998). Although the opening transition also involves movement of the voltage sensors, which can be detected because it involves the translocation of a small amount of gating charge, the bulk of the gating charge movement occurs during the R to A transitions (Smith-Maxwell et al., 1998b, 1998a; Ledwell and Aldrich, 1999; del Camino et al., 2005; Pathak et al., 2005). Hanatoxin has been shown to produce a particularly dramatic inhibition of Kv2.1 gating currents elicited by weak depolarizations (by ∼ 80%), consistent with the idea that the toxins alters transitions between R and A states (Lee et al., 2003). When examined over a range of voltages, the toxin is observed to produce a shift of the gating charge vs voltage relation by around 40 mV to more positive voltages, which suggest that the toxin causes a relative stabilization of the R state of the voltage sensors. In addition, kinetic analysis demonstrates that toxin binding affinity becomes weaker once the voltage sensors activate (Phillips et al., 2005), providing additional support for the idea that hanatoxin stabilizes the resting conformation of the voltage sensor. The sea anemone BDSII was recently reported to have similar effects on gating currents recorded for the Kv3.2b channel (Yeung et al., 2005). Although it is clear that hanatoxin and BDSII have pronounced effects on voltage sensor movement, it will be interesting to examine the effects of these toxins in more detail, in particular to determine whether they alter forward or backward rates for specific transitions during voltage sensor activation. It will also be interesting to further investigate the effects of these toxins on the concerted opening transition. The pronounced effects of hanatoxin on deactivation and on the amplitude of the maximal conductance of the Kv2.1 channel (Section 2.1; Fig 3) suggest that the toxin also influences the concerted opening transition, a picture that fits nicely with recent evidence for voltage sensor movement during opening of Kv channels (Smith-Maxwell et al., 1998b, 1998a; Ledwell and Aldrich, 1999; del Camino et al., 2005; Pathak et al., 2005).
2.3 The toxin receptor in Kv channels
A growing body of evidence supports a direct interaction between toxins like hanatoxin and the voltage sensing domains of Kv channels. Early experiments showed that hanatoxin and the pore blocker agitoxin-2 can simultaneously bind to the Kv2.1 channel, suggesting that the hanatoxin binding site is located peripheral to the pore blocking toxin receptor within the outer vestibule of the pore (Swartz and MacKinnon, 1997a). Mutations at the peripheral edges of the external surface of the pore domain (i.e. outside the agitoxin receptor) fail to influence hanatoxin binding to Kv2.1, suggesting that the receptor for hanatoxin lies outside the pore domain entirely (Li-Smerin and Swartz, 2000). Perhaps the strongest evidence for hanatoxin binding to the voltage sensing domains comes from the data in Fig 4, which shows the results of an alanine scan of the entire S1 through S4 voltage sensing domain of the Kv2.1 channel (Li-Smerin and Swartz, 2000). Mutations at many positions within the domain have little effect on toxin binding, but those that do alter toxin binding are localized to the external ends of S3 and S4 (Figs 4,5; red highlighted residues). Further mutagenesis within the S3 and S4 helices suggests that hanatoxin interacts intimately with this region of the voltage sensing domain, with F274 and E277 in the C-terminal portion of S3 playing particularly crucial roles (Li-Smerin and Swartz, 2000) (Fig 5). At F274 systematic substitutions show that hydrophobic amino acids cause the smallest perturbation in hanatoxin binding energy. The range of ΔΔG values (ΔΔG = -RT ln (Kdwt/Kdmut)) for hydrophobic residues (Ile, Val, Trp, Tyr, Pro, Met, Ala, Cys) is 1.2 to 1.8 kcal mol−1 while the range of ΔΔG values for non-hydrophobic residues (Gly, Asp, Ser, Glu, Arg, Lys) is 2.4 to 3.7 kcal mol−1 (Li-Smerin and Swartz, 2000). Although substitution of hydrophobic residues result in the smallest perturbations, even these perturbations were quite significant, implying a very close and relatively specific interaction with hydrophobic residues on the toxin. It is also interesting that at F274, substitutions with either Lys or Arg produce the largest perturbations, with ΔΔG values of 3.7 and 3.6 kcal mol−1, respectively. One possibility is that F274 is positioned close to a basic residue on the toxin, and that substitution with basic residue at 274 weakens toxin binding by electrostatic repulsion in addition to disrupting favorable hydrophobic interactions. This is a feasible possibility given the position of key hydrophobic and basic residues in the structure of the toxin (see Sections 3.1 and 3.2). There is also an interesting relationship between the magnitude of perturbations and the nature of substituted residues at position E277 in Kv2.1 (Li-Smerin and Swartz, 2000). Substitutions with basic residues have the largest effects (ΔΔG = 2.8 kcal mol−1) while those with neutral residues have more moderate effects (ΔΔG from 1.3 to 2.2 kcal mol−1). Mutation of E277 to Asp produced no discernable change in toxin binding affinity (ΔΔG = 0.04 kcal mol−1). Although the approximate correlation between the charge of the side chain at 277 and toxin binding energy might signify a through-space electrostatic interaction between this acidic residue and a basic residue on the toxin, several observations point to a more intimate interaction. First, substitutions with various neutral amino acids produce significantly different perturbations, with ΔΔG values ranging from 1.3 to 2.2 kcal mol−1. Second, the large values of ΔΔG for all mutations (1.3 to 2.8 kcal mol−1), with the exception of Asp, are more consistent with intimate or short range interactions. If E277 interacts with a basic residue on hanatoxin through formation of a salt bridge, the apparent tranquility observed upon substitution of Asp for Glu would imply some degree of flexibility at the toxin-channel interface. Another possibility is that the unique phenotype observed for E277 mutations is the consequence of an electrostatic interaction between toxin and channel that takes place within the membrane, perhaps within the electrostatically complex interfacial regions (see Section 4). Although F274 and E277 are among the most crucial residues for toxin binding to Kv2.1, there are numerous other residues that may also interact with the toxin, including L275, T276, S278, V282, and F285 within S3, and R290 and R296 within S4 (Swartz and MacKinnon, 1997b; Li-Smerin and Swartz, 2000; Li-Smerin and Swartz, 2001).
Fig.4.
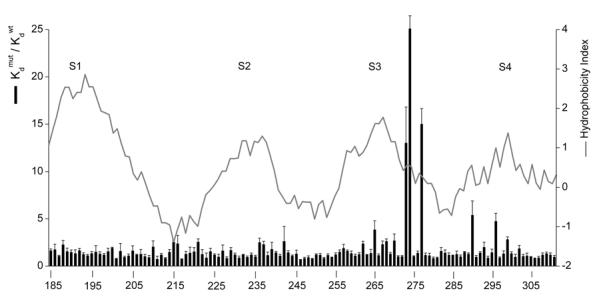
The hanatoxin receptor within Kv2.1 channels.
Bar graph plots the normalized hanatoxin Kd for each mutation spanning from K185 in S1 to T311 in S4. Data were taken from (Swartz and MacKinnon, 1997b) and (Li-Smerin and Swartz, 2000). Most residues were mutated to Ala, except for the native Ala residues which were mutated to Tyr. Solid line superimposed on the bar graph is a 17-residue window analysis of the Kyte-Doolittle hydrophobicity index (Kyte and Doolittle, 1982).
Fig.5.
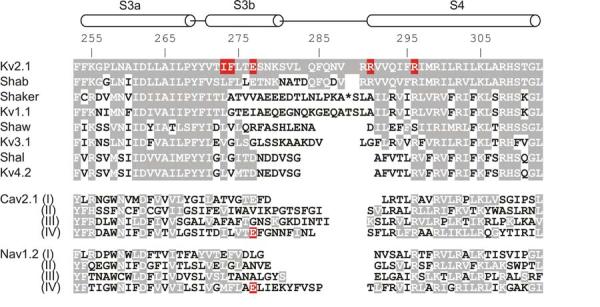
Sequence alignment for the S3 through S4 paddle region of voltage-activated cation channels.
Horizontal cylinders indicate the approximate positions of S3 (divided into S3a and S3b) and S4 helices. Numbers indicate the residue positions within the Kv2.1 channel and shading indicates similarity to Kv2.1. For the Shaker Kv channel 13 residues have been omitted for alignment purposes and their position marked by the asterisk. Red highlighting indicates residues whose mutations cause pronounced changes in the binding affinity of gating modifier toxins (see text for details).
It is particularly interesting that the region where hanatoxin binds, termed the voltage sensor paddle in structural studies on KvAP (Jiang et al., 2003a), has been proposed to be a uniquely mobile region of the voltage sensing domain and to move as a unit in response to changes in membrane voltage (Jiang et al., 2003a; Jiang et al., 2003b; Ruta et al., 2005). The paddle motif is a helix-turn-helix motif composed of the C-terminal portion of S3, (S3b) and the S4 helix (Fig 5). The mutagenesis experiments in Kv2.1 pointing to a particular crucial role for the S3b helix in forming the toxin receptor also independently revealed the helical structure of this C-terminal portion of S3 (Li-Smerin and Swartz, 2000; Li-Smerin and Swartz, 2001), further supporting a direct interaction between the toxin and the paddle motif.
The receptors for the other gating modifier toxins for Kv channels remain to be further defined, but several of them appear to interact with a similar region of the channel. In the case of VSTx1, the toxin binds equally well to the isolated S1-S4 voltage sensing domain compared to the full length channel (Ruta and MacKinnon, 2004), suggesting that its receptor is within the voltage sensor. Also, experiments with chimeric channels show that moving the S3-S4 paddle motif from KvAP into Kv2.1 transfers sensitivity to VSTx1 (Alabi and Swartz, 2006), consistent with this toxin binding to a similar region as hanatoxin. Chimera constructs between toxin sensitive and insensitive Kv channels are also consistent with HpTx2 and TLTx1 interacting with the S3-S4 region of Kv4 channels (Ebbinghaus et al., 2004; Zarayskiy et al., 2005). Finally, the effects of S3 mutants on BDSII support the interaction of this toxin with the S3 helix of Kv3.2b channels (Yeung et al., 2005).
It will be interesting to learn more about how these toxins dock onto the voltage sensor paddle motif, perhaps using mutant cycle approaches to identify interacting residues at the protein-protein interface (Hidalgo and MacKinnon, 1995; Schreiber and Fersht, 1995). GxTx1E, the newest addition to Kv channel voltage sensor toxins (Herrington et al., 2006), may be particularly useful for such studies because its low nM affinity could improve accurate determinations of the binding affinity of mutant toxins to mutant channels, a requirement of mutant cycle analysis. It may also be possible to obtain high resolution structures of a toxin-channel complex, as recently obtained for cobra and cone snail toxins and acetylcholine binding protein (Bourne et al., 2005; Celie et al., 2005; Ulens et al., 2006). The best prospects for such a complex with the toxins discussed here might be for VSTx1 and KvAP since both proteins can be produced in large quantities and either NMR or X-ray structures of the uncomplexed proteins have already been solved (Jiang et al., 2003a; Jung et al., 2005; Lee et al., 2005).
2.4 Stoichiometry between toxin and channel
The peripheral position of toxin receptors within the voltage sensing domains infers that there should be four toxin receptors per channel, one per voltage sensor. The evidence that at least two hanatoxin molecules can occupy the Kv2.1 channel is quite strong, and there is reason to believe that toxin occupancy of the channel can be as high as four. A simple experiment pointing to multiple occupancy is that the concentration dependence for inhibition of opening at negative voltages is distinct from that for shifting activation of the channel to positive voltages (Swartz and MacKinnon, 1997a). For example, 200 nM hanatoxin can completely inhibit opening of the Kv2.1 channel at −20 mV, indicating that all of the channels in the cell have at least one toxin bound. However, application of higher toxin concentrations produce further shifts of the activation curve, which must mean that at least a second toxin can bind. Another indication of multiple occupancy is seen in the kinetics of recovery following removal of the toxin from the aqueous solution bathing the cell. If only a single toxin bound to the channel the recovery (unbinding) kinetics should be well described by a single exponential function, whereas higher occupancy would result in a more complex time course involving a lag in the initial phase of recovery. In experiments using saturating concentrations of hanatoxin to achieve high occupancy, the recovery time course exhibits a dramatic lag that is most consistent with the binding of between 3 and 4 toxins per channel (Swartz and MacKinnon, 1997a; Phillips et al., 2005).
It is generally believed that all four voltage-sensors must activate before Kv channels open with significant probability (Islas and Sigworth, 1999). One might predict, therefore, that a single toxin binding to a single voltage sensor would be sufficient to inhibit opening of the channel. In this case the concentration dependence for fractional inhibition of opening at negative voltages, which approximates the fraction of channels with no toxins bound (Swartz and MacKinnon, 1997a), should be related to voltage sensor occupancy by a fourth power relation if all four voltage sensors can bind the toxin. For example, if the toxin is applied at a concentration equal to the Kd for binding to individual voltage sensors, where half of the voltage sensors have a toxin bound, the fraction of channels with no toxins bound should equal 0.54, or 0.0625. When the concentration dependence for inhibition of channel opening is assessed from the fraction of uninhibited ionic current at negative voltages it is well described by a fourth power relation where the Kd for toxin binding to each site is 104 nM (Swartz and MacKinnon, 1997a; Lee et al., 2003). Because of the fourth power relationship, the Kd here corresponds to the concentration at which only 6.25 % of channels remain uninhibited, which differs from the single site case where the Kd corresponds to the concentration at which 50% of channels remain uninhibited. How does this measure of channel occupancy compare with a measure of voltage sensor occupancy? When the concentration dependence for inhibition of voltage sensor activation is assessed from the fraction of uninhibited gating current at negative voltages it is well described by a single site relation where the Kd for toxin binding to each site is around 107 nM, which in this case corresponds to the concentration where 50% of the gating current remains uninhibited (Lee et al., 2003). In other words, there is a sizable shift in the concentration dependence for inhibition of opening compared to that for gating charge movement, and this difference corresponds to precisely what is predicted by a simple four independent site model.
3. Structures of Kv channel voltage sensor toxins
NMR solution structures have been solved for five of the Kv channel voltage sensor toxins listed in Fig 2, including hanatoxin1, SGTx1, HpTx2, PaTx1 and VSTx1 (Bernard et al., 2000; Takahashi et al., 2000; Chagot et al., 2004; Lee et al., 2004; Jung et al., 2005). Structures are also available for the related toxins GrTx-SIA (Takeuchi et al., 2002) and GsMTx4 (Oswald et al., 2002), inhibitors of Cav channels and stretch activated channels, respectively.
3.1 General structural features
The structures of the Kv channel toxins consist of a triple-stranded anti-parallel β-sheet containing 4 chain reversals, with strand-I being absent or distorted in some of the toxins. The overall similarity of the backbone folds of these toxins in illustrated in Fig 6 for hanatoxin (Takahashi et al., 2000) and VSTx1 (Jung et al., 2005). The C-termini for all of these toxins are poorly constrained and have been removed for clarity. The six cysteine residues form three disulfide bonds with connectivity of Cys2-Cys16, Cys9-Cys21 and Cys15-Cys28 (numbering here is for hanatoxin). Both the overall fold and disulfide bond connectivity are frequently observed in other inhibitory toxins, and has been termed the “inhibitory cysteine knot” (ICK) fold by Norton and colleagues (Pallaghy et al., 1994; Norton and Pallaghy, 1998). Although the ICK fold appears to be a common fold for presentation of a channel interacting surface, it doesn't appear to specify the region being targeted or the mechanism of action, as some ICK toxins act as pore blockers and others as gating modifiers. For example, the ICK toxins ω-conotoxin GVIA and Κ-conotoxin PVIIA bind to the outer vestibule of Cav and Kv channels, respectively (Boland et al., 1994; Ellinor et al., 1994; Shon et al., 1998; Garcia et al., 1999; Terlau et al., 1999; Naranjo, 2002; Boccaccio et al., 2004), whereas the Kv channel toxins discussed here bind to the voltage sensors.
Fig.6.
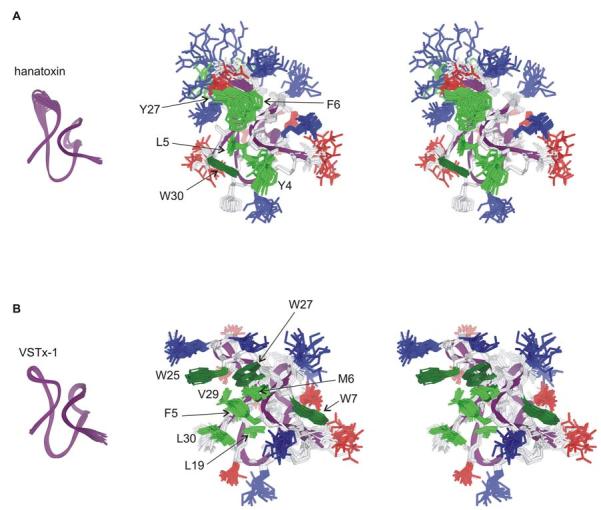
NMR solution structures of Hantoxin and VSTx-1.
(A) Stereo pairs of the hanatoxin structure represented as a bundle of 20 superimposed conforms. Residue coloring as follows: blue, basic; red, acidic; pink, Ser/Thr; green, hydrophobic (Trp is dark green). Backbone fold is shown at left in purple. (B) Stereo pairs of the VSTx1 structure represented as a bundle of 20 superimposed conforms with coloring as in A. The C-termini in both toxins (F32-S35 in hanatoxin and S32-F34 in VSTx1) are poorly constrained and have been removed for clarity. Images were created us DSViewer Pro and Protein Data Bank accession IDs 1D1H for hanatoxin (Takahashi et al., 2000) and 1S6X for VSTx1 (Jung et al., 2005).
One of the more striking structural features of the voltage sensor toxins is the amphipathic nature of the toxin (Fig 6), with one face containing a cluster of hydrophobic residues (green) that exhibit an unusually high degree of solvent exposure, surrounded by basic (blue), acidic (red) and other highly polar residues. Although the overall fold and amphipathic nature of the voltage sensor toxins are quite similar, there are detailed differences, in particular for the residues that comprise the hydrophobic surface. In hanatoxin1 and SGTx1 the hydrophobic surfaces are comprised of Y4, L5, F6, Y27 and W30, with Y4, F6 and W30 forming an aromatic sandwich around L5. Interaction among residues on the hydrophobic surface is evident in the NMR spectra of both toxins where the proton resonances for L5 are observed at unusually high field strength due to ring currents associated with surround aromatic residues (Takahashi et al., 2000; Lee et al., 2004). In VSTx1 the hydrophobic surface differs in detailed ways from the above toxins, consisting of F5, M6, W7, L19, W25, W27, V29 and L30, occupying a significantly larger surface than the equivalent region in hanatoxin and SGTx1, and containing three Trp residues as apposed to one (Fig 6)(Jung et al., 2005). Although an aromatic sandwich like that found in hanatoxin and SGTx is also present in VSTx1, in this instance consisting of F5 and W27 sandwiching M6, overall the hydrophobic surface is more distributed and less protruding than in hanatoxin and SGTx.
The hydrophobic surface of PaTx1 is more similar to VSTx1 than the other toxins, although it is somewhat smaller in that several VSTx1 hydrophobic residues (W25, V29 and L30) are absent. W5, M6, W7, L19 and W24 comprise the hydrophobic surface in PaTx1 and these residues occupy similar positions to F5, M6, W7, L19 and W27 in VSTx1. Although the structure of HpTx2 has been discussed in the context of it acting as a pore blocker (Bernard et al., 2000), the detailed structure of HpTx2 is similar to hanatoxin and SGTx, with L6, F7, W25 and L28 in HpTx2 occupying positions equivalent to L5, F6, Y27 and W30, respectively. HpTx2 is missing the equivalent of Y4 in hanatoxin and SGTx, but contains three additional hydrophobic residues (W30, Y20 and V21) that contribute to the solvent exposed hydrophobic surface. Like the other gating modifier toxins discussed here, HpTx2 shifts activation of the channel to positive voltages and chimeric channels support an interaction of the toxin with the voltage sensing domains (Sanguinetti et al., 1997; Zarayskiy et al., 2005), suggesting that HpTx2 also inhibits by binding to the voltage sensors and stabilizing resting conformations.
3.2 The active surface of SGTx
Alanine scanning mutagenesis of SGTx1 (Wang et al., 2004) has provided an essentially complete map of the functionally important surfaces of the toxin (Fig 7A,B). The active face of the toxin contains the solvent exposed hydrophobic surface in addition to several key surrounding basic residues. Of the hydrophobic residues, mutation of L5, F6, and W30 result in dramatic weakening of apparent toxin affinity (Fig 7B, red residues), while mutation of Y4 results in a more moderate perturbations (Fig 7B, pink residues). All of these mutants are concentrated on the side of the molecule that contains the hydrophobic patch, whereas the many Ala mutants that have little effect on toxin affinity (gray residues) tend to be on the opposite side of the toxin. Mutagenesis of the Kv2.1 channel suggests that several hydrophobic residues, F274A within the S3b helix in particular, participate in intimate hydrophobic interactions with the toxin (Li-Smerin and Swartz, 2000). Although it is likely that L5, F6 or W30 interact with F274A, some of the effects of these mutants could be the result of local changes in the structure of the hydrophobic surface of the toxin, in particular given the intimate interactions of L5, F6 and the surrounding aromatic residues that is evident in the NMR structures of these toxins (Takahashi et al., 2000; Lee et al., 2004). Of the polar residues, R3 and R22 dramatically weaken apparent toxin affinity, while H18 and D31 result in moderate weakening. As discussed above, E277 stands out as being the only acidic residue within the voltage sensor of Kv2.1 where mutations dramatically weaken toxin binding (Li-Smerin and Swartz, 2000), making it a strong candidate for interaction with either R3 or R22. An intimate interaction between F274 in Kv2.1 and residues in the toxin's hydrophobic protrusion is compatible with an interaction between E277 in Kv2.1 and either SGTx R3 or R22, since both of these basic residues are located an equal distance (∼10-12 Å) from the hydrophobic protrusion. The D24A mutant is interesting in this regard because truncation to Ala actually increases affinity 20-fold (Wang et al., 2004). D24 is adjacent to R22 in the SGTx structure and thus there could be electrostatic repulsion between D24 and E277 as this later residue interacts with R22. In addition to perturbing the protein-protein interface between toxin and channel, some of these SGTx1 mutants could also disrupt interactions of voltage sensor toxins with the lipid membrane and it will be interesting to examine this possibility further (see Section 4).
Fig.7.
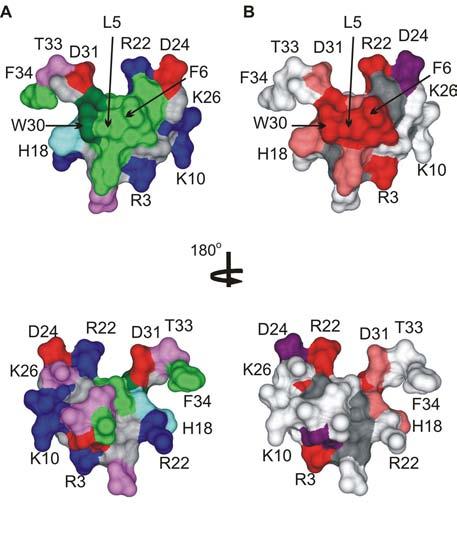
The active surface of SGTx.
(A) Surface rendering of the SGTx structure illustrating the amphipathic nature of the toxin. Residue coloring as follows: blue, basic; red, acidic; violet, Ser/Thr; green, hydrophobic (Trp is dark green). (B) Surface rendering of SGTx colored according to mutagenesis results. Backbone atoms and unstudied residues (L19, Y27 and 6 Cys) are colored dark gray. Mutants that do not significantly alter Kd (|ΔΔG| < 1 kcal mol−1) are colored light gray. Mutants that display weaker binding affinity are colored pink if |ΔΔG| = 1-1.5 kcal mol−1 and red if |ΔΔG| > 1.5 kcal mol−1. Two mutants displaying stronger binding affinity are colored purple. See (Wang et al., 2004) for details. Bottom images were obtained by rotating around the axis shown. Images were created us DSViewer Pro and Protein Data Bank accession IDs 1LA4 for SGTx (Lee et al., 2004).
Although mutagenesis data are not yet available for the other toxins that interact with voltage sensors in Kv channels, inspection of their sequences provides a good indication of those that are likely to be quite similar to SGTx1, and those which might have significantly different interaction surfaces with voltage sensors. For example, of the ten most important residues in SGTx1, eight are identical in hanatoxin and one represents a conservative substitution (R22 in SGTx to K22 in hanatoxin) (Fig 2). This comparison strongly suggests that SGTx and hanatoxin are likely to present very similar surfaces to the Kv2.1 channel. However, a very different picture emerges when SGTx1 and VSTx1 are compared. Of the ten most crucial residues in SGTx1, none are strongly conserved in VSTx1 (Fig 2), suggesting that there may be key differences in the surfaces that toxins like hanatoxin and VSTx1 present to Kv channels.
4. Membrane partitioning of voltage sensor toxins
Based on the X-ray structures of the KvAP channel and biotin accessibility studies (Jiang et al., 2003a; Jiang et al., 2003b; Ruta et al., 2005), MacKinnon and colleagues proposed that the voltage sensor paddle moves relatively large distances through the lipid bilayer in response to changes in membrane voltage. The voltage sensor paddle, a helix-turn-helix motif composed of both S3b and S4 helices, is positioned largely within the inner leaflet of the membrane at negative membrane voltages in these models and it then translocates 15 to 25Å to reside within the outer leaflet or extracellular solution with membrane depolarization. These new models raise the intriguing possibility that the target of voltage sensor toxins like hanatoxin is actually submerged within the membrane, and that these toxins might need to partition into the membrane in order to bind to the channel (Lee et al., 2003).
4.1 Toxin interactions with membranes
VSTx1, GsMTx4 and hanatoxin have all been shown to partition into model membranes (Lee and MacKinnon, 2004; Suchyna et al., 2004; Jung et al., 2005; Phillips et al., 2005), supporting the possibility that partitioning is involved in the mechanism of inhibition. Membrane partitioning of hanatoxin is illustrated in Fig 8 (A,B), where the fluorescence emission of Trp30 on the toxin exhibits a blue shift (towards lower wavelengths) upon binding to phospholipid vesicles. The amphipathic character evident in the toxin structures (Bernard et al., 2000; Takahashi et al., 2000; Chagot et al., 2004; Lee et al., 2004; Jung et al., 2005), as well as the importance of the hydrophobic face of SGTx1 (Wang et al., 2004), are consistent with the notion that membrane partitioning may be required for the toxin to reach its target. In thinking about the position of the voltage sensor paddle within the membrane it is interesting to consider how far these toxins might partition into the membrane. This question has been examined in the case of hanatoxin, as illustrated in Fig 8 (C,D), and the initial answer is that the toxin takes on a relatively superficial position within the membrane (Phillips et al., 2005). Bromide atoms attached to the lipid hydrocarbon chain quench the fluorescence of Trp30 on hanatoxin, and the efficiency of quenching is highest when the quencher is attached near the middle of the chain, suggesting that Trp30 is positioned between 8 to 9 Å from the center of the bilayer. A relatively interfacial position of the toxin would allow the hydrophobic face of the toxin to interact with the hydrophobic core of the membrane and the polar toxin residues to interact with the polar head groups of the lipid (Fig 9).
Fig.8.
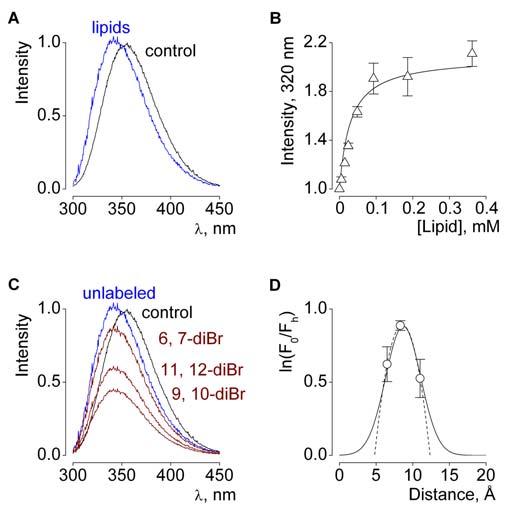
Membrane partitioning of hanatoxin.
(A) Hanatoxin fluorescence in the absence (black) or presence of vesicles composed of a 1:1 mix of POPC:POPG (blue). Lipid concentration is 1.2 mM and toxin concentration is 2 μM. (B) Fluorescence intensity plotted as a function of available lipid concentration (60% of total lipids) for vesicles containing both POPC:POPG. Smooth curve corresponds to a partition function with a mole fraction partition coefficient (Kx) of 1.8 × 106. (Milescu, M. and Swartz, K.J., unpublished data). (C) Fluorescence emission spectra for hanatoxin in the absence (black) and presence of 1.3 mM lipid vesicles containing unlabeled (blue) or brominated (diBr; maroon) lipids. The 6,7 diBr lipid has Br attached closest to the polar head group. (D) Analysis of the depth-dependent quenching profiles for hanatoxin. Fractional quenching (F0/Fh) by brominated lipids plotted vs average distance of bromine atoms from the center of the bilayer. See (Phillips et al., 2005) for details.
Fig.9.
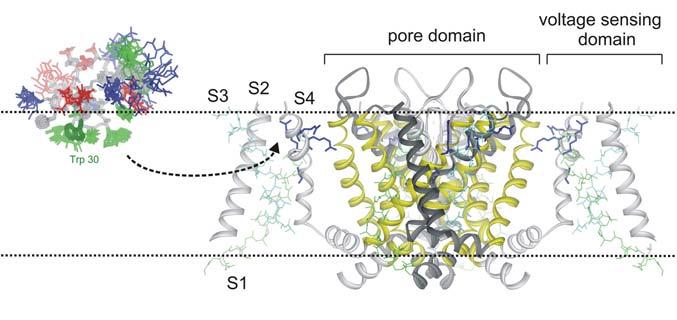
Illustration of hanatoxin partitioning into the membrane before binding to a voltage sensing domain.
4.2 Kinetic implications of a partitioning step
The possibility of toxin partitioning into the membrane has important implications for the kinetics and affinity of toxin binding to voltage sensors because it adds an additional step that requires consideration. Scheme II describes a scenario wherein a voltage sensor toxin partitions from the extracellular aqueous phase (Txaq) into the membrane (Txmem) and then binds the channel (Ch) to form a toxin-channel complex (Tx-Ch). It has been proposed that partitioning serves to concentrate the toxin near its target so that a low affinity (e.g. mM) protein-protein interaction would be sufficient to achieve high apparent affinity (Lee and MacKinnon, 2004). If the protein-protein interaction between toxin and channel has low affinity, toxin unbinding (koff) should be as fast as channel gating (e.g. ms) and the conformational changes that underlie voltage-dependent gating in the presence of toxin might only occur when the toxin is not bound. In the case of hanatoxin, the recovery of channel activity that is observed following removal of the toxin from the aqueous solution is quite slow (∼1000 s) (Swartz and MacKinnon, 1997a; Phillips et al., 2005), which in the case of a low affinity protein-protein interaction would necessarily arise from slow departitioning (low kout) rather than slow unbinding (low koff). This possibility was recently examined in the case of hanatoxin by determining the effects of paddle mutations on the slow recovery kinetics (Phillips et al., 2005). The F274A mutation within S3b increases the apparent equilibrium dissociation constant (Kd) for hanatoxin by 28-fold in experiments where [Taq] is varied and the fractional occupancy of the channel determined electrophysiologically. According to Scheme II, a change of this magnitude would have minimal effects on recovery in the case of slow departitioning and fast unbinding, but dramatic effects on recovery in the case of fast departitioning and slow unbinding. It was found that the F274A mutation dramatically speeds the kinetics of recovery, and to an extent similar to that predicted if unbinding of the toxin largely determines the slow recovery kinetics observed for wild-type channels (Phillips et al., 2005). These results support the idea that the koff for hanatoxin is low and that the binding affinity of the toxin is high. The long dwell time of the toxin on the resting voltage-sensor, approaching 250 s for the Wt Kv2.1 channel (Swartz and MacKinnon, 1997a; Phillips et al., 2005), is orders of magnitude slower than channel gating (ms), strongly suggesting that the voltage-sensors activate with the toxin continuously bound. At least in the case of hanatoxin, these results rule out the possibility that membrane partitioning plays an energetically dominant for the interaction of the toxin with Kv2.1 channels, but leave open the possibility that partitioning is necessary for the toxin to reach the voltage-sensor. The picture of voltage sensors activating with toxin on board implies that the magnitude of voltage sensor motion is rather limited with respect to the dimensions of the lipid bilayer. It is hard to imagine how voltage sensors could move >25 Å through the bilayer, as in the original paddle model (Jiang et al., 2003b), dragging along multiple hanatoxin molecules. In addition, even in the event that membrane partitioning is necessary, the localization of the toxin within interfacial regions of the bilayer (Section 4.1), together with the preference of the toxin for the resting voltage-sensor (Sections 2.1 and 2.2), imply that the voltage-sensor paddle motif is positioned within the outer half of the bilayer in its resting conformation (Phillips et al., 2005). A more recent version of the paddle model (Ruta et al., 2005) posits smaller movements of the paddle (∼15 Å) with a shift of the resting paddle in the direction of the extracellular side of the membrane, which is compatible with the constraints imposed by hanatoxin, particularly given the uncertainties in how either the toxin or voltage sensor perturb the structure of the membrane.
Scheme II.
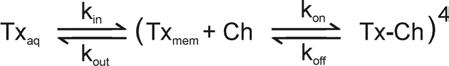
The implications of membrane partitioning in the case of toxins binding to voltage sensors in Nav channels has also been considered. Several of these toxins can partition into model membranes (Smith et al., 2005), but the affinity of the protein-protein interaction between toxin and channel was nonetheless found to be high (Rogers et al., 1996; Cohen et al., 2006), consistent with the results discussed above for hanatoxin and Kv channels. It is important to emphasize that none of the results discussed here preclude the possibility that partitioning of gating modifier toxins into the membrane is a necessary step in binding to voltage sensors. The results on both Kv and Nav channel toxins simply rule out the possibility that membrane partitioning dominates the energetics of the toxin-channel interaction by dramatically concentrating the toxins near their receptors and in doing so enabling a low affinity protein-protein interaction to achieve high apparent affinity. At present, the possible involvement of membrane partitioning is an intriguing prospect (Fig 9), but one that remains to be further explored. There is clearly much to learn about how these toxins interact with lipid membranes, how the structure of the membrane may be influenced by toxin partitioning and whether partitioning is actually necessary for voltage sensor toxins to reach their receptors.
5. Conservation and adaptation of voltage sensing domains
One of the more interesting aspects of gating modifier toxins is that many structurally diverse toxins interact with the equivalent region of different voltage-activated cation channels. For example, the mutagenesis results identifying the hanatoxin receptors on Kv channels are remarkably consistent with similar efforts to identify the receptors for gating modifier toxins on Nav and Cav channels. Mutations at E1613 in the fourth repeat of the Nav 1.2 channel alters the binding affinity of α-scorpion toxin and sea anemone toxin, two types of toxins that slow inactivation of Nav channels (Rogers et al., 1996). Also, mutation of E1658 in repeat four of the Cav 2.1 channel disrupts the binding of ω–Aga-IVA (Winterfield and Swartz, 2000), a gating modifier toxin that inhibits activation of the Ca2+ channel (Mintz et al., 1992; McDonough et al., 1997b). In sequence alignments both E1613 in the Nav channel and E1658 in the Cav channel are at positions equivalent to E277 in the Kv2.1 channel (Fig 5). It is also interesting that Kv4.2, another channel that is highly sensitive to hanatoxin, contains an Asp residue at the position equivalent to E277 in Kv2.1, and it's tempting to speculate that this acidic residue is crucial for the binding of voltage sensor toxins to the Kv4 family of channels. Although the effects of these different gating modifier toxins are rather diverse, with the Nav channel toxins slowing inactivation and the Kv and Cav channel toxins inhibiting those channels by stabilizing a closed state, it is remarkable that they have all evolved to interact with a common region of the channel. This common targeting of the voltage sensor paddle motif is consistent with recent evidence suggesting that the paddle motif is a uniquely mobile region within the voltage sensing domains (Ruta et al., 2005).
5.1 Promiscuity of voltage sensor toxins
The connection between gating modifier toxins for different types of voltage-activated channels is paralleled by several reported examples where gating modifier toxins interact rather promiscuously with Kv, Cav and Nav channels. For example, GrTx-SIA, a gating modifier of the Cav2.1 channel (McDonough et al., 1997a), also inhibits the Kv2.1 channel and the binding affinity of GrTx-SIA to this Kv channel is altered by mutations at I273, F274 or E277 within the S3b helix (Li-Smerin and Swartz, 1998). In addition, hanatoxin can bind to and inhibit the Cav2.1 channel. Kurtoxin is an α-scorpion toxin that slows inactivation of the Nav1.2 channel, but also binds to and inhibits Cav3 T-type channels by shifting activation to more depolarized voltages (Chuang et al., 1998). Perhaps the richest example of promiscuity for gating modifier toxins is ProTxI (Fig 2), a toxin isolated from the venom of the Thrixopelma pruriens tarantula. ProTxI inhibits activation of various Nav channels, the Kv2.1 channel, and the Cav2.1 channel (Middleton et al., 2002). Taken together, these results suggest that gating modifier toxins interact with a common region of the voltage-sensing domains in different voltage-gated ion channels and that this region probably adopts a conserved three-dimensional structure. Although the work on hanatoxin and several Nav channel toxins highlight the importance of protein-protein interactions in toxin binding to voltage-activated channels (see Section 4.2), it is also quite possible that lipids constitute an important part of the toxin receptor and that this contributes to the striking promiscuity of voltage sensor toxins.
5.2 Voltage sensor relatives and isolated voltage sensors
There are many other channels that are related to voltage-activated channels, but where the primary activating stimulus does not appear to be membrane voltage. Cyclic nucleotide gated channels (Craven and Zagotta, 2006) and the large family of TRP channels (Ramsey et al., 2006a) are examples of cation channels that have S1-S4 domains that are similar to those found in voltage-activated channels, but whose function is relatively unexplored. TRPV1 channels, also known as capsaicin receptors, have recently been found to exhibit significant voltage-dependence (Voets et al., 2004) and several key ligands for these channels appear to interact with the S1-S4 domain (Chou et al., 2004), supporting the possibility that this region serves a related function to its counterpart in classical voltage-activated channels. One of the more remarkable recent advances in the area of voltage sensing has been the discovery of proteins that contain a voltage sensing domain without a pore domain. These new voltage sensing proteins couple instead to a soluble phosphatase domain (Murata et al., 2005) or act as voltage-activated proton channels themselves (Ramsey et al., 2006b; Sasaki et al., 2006). It will be interesting to explore whether toxins have evolved to interact with the S3-S4 regions of cyclic nucleotide gated channels, TRP channels or the new voltage sensor proteins. Tarantula venom would be an interesting place to begin looking because they seem particularly fond of making toxins that target this region. Voltage sensor toxins could be very useful pharmacological probes for the function of all of these protein families, and they might provide a valuable link to relate the voltage sensors found in these proteins to those in classical voltage-activated channels.
Acknowledgments
I thank AbdulRasheed Alabi, Frank Bosmans, Dmitriy Krepkiy, Mirela Milescu and L. Revell Phillips for helpful discussions. I also thank Mirela Milescu for making Figures 7 and 8. This work was supported by the Intramural Research Program of the NINDS, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alabi AA, Swartz KJ. Compatibility of the paddle region revealed by chimeras between archebacterial and eukaryotic Kv channels. Biophys J. 2006;90:B287. [Google Scholar]
- Anderson CS, MacKinnon R, Smith C, Miller C. Charybdotoxin block of single Ca2+-activated K+ channels. Effects of channel gating, voltage, and ionic strength. J Gen Physiol. 1988;91:317–333. doi: 10.1085/jgp.91.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Legros C, Ferrat G, Bischoff U, Marquardt A, Pongs O, Darbon H. Solution structure of hpTX2, a toxin from Heteropoda venatoria spider that blocks Kv4.2 potassium channel. Protein Sci. 2000;9:2059–2067. [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Stefani E. Gating currents. Methods Enzymol. 1998;293:331–352. doi: 10.1016/s0076-6879(98)93022-1. [DOI] [PubMed] [Google Scholar]
- Boccaccio A, Conti F, Olivera BM, Terlau H. Binding of kappa-conotoxin PVIIA to Shaker K+ channels reveals different K+ and Rb+ occupancies within the ion channel pore. J Gen Physiol. 2004;124:71–81. doi: 10.1085/jgp.200409048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland LM, Morrill JA, Bean BP. omega-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J Neurosci. 1994;14:5011–5027. doi: 10.1523/JNEUROSCI.14-08-05011.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y, Talley TT, Hansen SB, Taylor P, Marchot P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake alpha-neurotoxins and nicotinic receptors. Embo J. 2005;24:1512–1522. doi: 10.1038/sj.emboj.7600620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD. Modification of sodium channel gating in frog myelinated nerve fibres by Centruroides sculpturatus scorpion venom. J Physiol (Lond) 1975;244:511–534. doi: 10.1113/jphysiol.1975.sp010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celie PH, Kasheverov IE, Mordvintsev DY, Hogg RC, van Nierop P, van Elk R, van Rossum-Fikkert SE, Zhmak MN, Bertrand D, Tsetlin V, Sixma TK, Smit AB. Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an alpha-conotoxin PnIA variant. Nat Struct Mol Biol. 2005;12:582–588. doi: 10.1038/nsmb951. [DOI] [PubMed] [Google Scholar]
- Chagot B, Escoubas P, Villegas E, Bernard C, Ferrat G, Corzo G, Lazdunski M, Darbon H. Solution structure of Phrixotoxin 1, a specific peptide inhibitor of Kv4 potassium channels from the venom of the theraphosid spider Phrixotrichus auratus. Protein Sci. 2004;13:1197–1208. doi: 10.1110/ps.03584304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MZ, Mtui T, Gao YD, Kohler M, Middleton RE. Resiniferatoxin binds to the capsaicin receptor (TRPV1) near the extracellular side of the S4 transmembrane domain. Biochemistry. 2004;43:2501–2511. doi: 10.1021/bi035981h. [DOI] [PubMed] [Google Scholar]
- Chuang RS, Jaffe H, Cribbs L, Perez-Reyes E, Swartz KJ. Inhibition of T-type voltage-gated calcium channels by a new scorpion toxin. Nat Neurosci. 1998;1:668–674. doi: 10.1038/3669. [DOI] [PubMed] [Google Scholar]
- Cohen L, Gilles N, Karbat I, Ilan N, Gordon D, Gurevitz M. Direct evidence that receptor site-4 of sodium channel gating modifiers is not dipped in the phospholipid bilayer of neuronal membranes. J Biol Chem. 2006 doi: 10.1074/jbc.M603212200. [DOI] [PubMed] [Google Scholar]
- Craven KB, Zagotta WN. CNG and HCN channels: two peas, one pod. Annu Rev Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. [DOI] [PubMed] [Google Scholar]
- del Camino D, Kanevsky M, Yellen G. Status of the intracellular gate in the activated-not-open state of shaker K+ channels. J Gen Physiol. 2005;126:419–428. doi: 10.1085/jgp.200509385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diochot S, Drici MD, Moinier D, Fink M, Lazdunski M. Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br J Pharmacol. 1999;126:251–263. doi: 10.1038/sj.bjp.0702283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diochot S, Loret E, Bruhn T, Beress L, Lazdunski M. APETx1, a new toxin from the sea anemone Anthopleura elegantissima, blocks voltage-gated human ether-a-go-go-related gene potassium channels. Mol Pharmacol. 2003;64:59–69. doi: 10.1124/mol.64.1.59. [DOI] [PubMed] [Google Scholar]
- Diochot S, Schweitz H, Beress L, Lazdunski M. Sea anemone peptides with a specific blocking activity against the fast inactivating potassium channel Kv3.4. J Biol Chem. 1998;273:6744–6749. doi: 10.1074/jbc.273.12.6744. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus J, Legros C, Nolting A, Guette C, Celerier ML, Pongs O, Bahring R. Modulation of Kv4.2 channels by a peptide isolated from the venom of the giant bird-eating tarantula Theraphosa leblondi. Toxicon. 2004;43:923–932. doi: 10.1016/j.toxicon.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Zhang JF, Horne WA, Tsien RW. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature. 1994;372:272–275. doi: 10.1038/372272a0. [DOI] [PubMed] [Google Scholar]
- Escoubas P, Diochot S, Celerier ML, Nakajima T, Lazdunski M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol Pharmacol. 2002;62:48–57. doi: 10.1124/mol.62.1.48. [DOI] [PubMed] [Google Scholar]
- Garcia E, Scanlon M, Naranjo D. A marine snail neurotoxin shares with scorpion toxins a convergent mechanism of blockade on the pore of voltage-gated K channels. J Gen Physiol. 1999;114:141–157. doi: 10.1085/jgp.114.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, Abramson T, MacKinnon R. Transfer of the scorpion toxin receptor to an insensitive potassium channel. Neuron. 1994;13:961–966. doi: 10.1016/0896-6273(94)90261-5. [DOI] [PubMed] [Google Scholar]
- Herrington J, Zhou YP, Bugianesi RM, Dulski PM, Feng Y, Warren VA, Smith MM, Kohler MG, Garsky VM, Sanchez M, Wagner M, Raphaelli K, Banerjee P, Ahaghotu C, Wunderler D, Priest BT, Mehl JT, Garcia ML, McManus OB, Kaczorowski GJ, Slaughter RS. Blockers of the delayed-rectifier potassium current in pancreatic beta-cells enhance glucose-dependent insulin secretion. Diabetes. 2006;55:1034–1042. doi: 10.2337/diabetes.55.04.06.db05-0788. [DOI] [PubMed] [Google Scholar]
- Hidalgo P, MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- Islas LD, Sigworth FJ. Voltage sensitivity and gating charge in Shaker and Shab family potassium channels. J. Gen. Physiol. 1999;114:723–741. doi: 10.1085/jgp.114.5.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Voltage-gated and inwardly rectifying potassium channels. J Physiol. 1997;505(Pt 2):267–282. doi: 10.1111/j.1469-7793.1997.267bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003a;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003b;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- Jin W, Lu Z. A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1998;37:13291–13299. doi: 10.1021/bi981178p. [DOI] [PubMed] [Google Scholar]
- Jung HJ, Lee JY, Kim S, Eu YJ, Shin SY, Milescu M, Swartz KJ, Kim JI. Solution Structure and Lipid Membrane Partitioning of VSTx1, an Inhibitor of the KvAP Potassium Channel. Biochemistry. 2005;44:6015–6023. doi: 10.1021/bi0477034. [DOI] [PubMed] [Google Scholar]
- Koppenhofer E, Schmidt H. Effect of scorpion venom on ionic currents of the node of Ranvier. I. The permeabilities PNa and PK. Pflugers Arch. 1968a;303:133–149. doi: 10.1007/BF00592631. [DOI] [PubMed] [Google Scholar]
- Koppenhofer E, Schmidt H. Effect of scorpion venom on ionic currents of the node of Ranvier. II. Incomplete sodium inactivation. Pflugers Arch. 1968b;303:150–161. doi: 10.1007/BF00592632. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lampe RA, Defeo PA, Davison MD, Young J, Herman JL, Spreen RC, Horn MB, Mangano TJ, Keith RA. Isolation and pharmacological characterization of omega-grammotoxin SIA, a novel peptide inhibitor of neuronal voltage-sensitive calcium channel responses. Mol Pharmacol. 1993;44:451–460. [PubMed] [Google Scholar]
- Ledwell JL, Aldrich RW. Mutations in the S4 Region Isolate the Final Voltage-dependent Cooperative Step in Potassium Channel Activation. J Gen Physiol. 1999;113:389–414. doi: 10.1085/jgp.113.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Kim S, Roh SH, Endoh H, Kodera Y, Maeda T, Kohno T, Wang JM, Swartz KJ, Kim JI. Solution Structure and Functional Characterization of SGTx1, a Modifier of Kv2.1 Channel Gating. Biochemistry. 2004;43:890–897. doi: 10.1021/bi0353373. [DOI] [PubMed] [Google Scholar]
- Lee HC, Wang JM, Swartz KJ. Interaction between extracellular Hanatoxin and the resting conformation of the voltage-sensor paddle in Kv channels. Neuron. 2003;40:527–536. doi: 10.1016/s0896-6273(03)00636-6. [DOI] [PubMed] [Google Scholar]
- Lee SY, Lee A, Chen J, Mackinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci U S A. 2005 doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, MacKinnon R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature. 2004;430:232–235. doi: 10.1038/nature02632. [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y, Swartz KJ. Gating modifier toxins reveal a conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc Natl Acad Sci U S A. 1998;95:8585–8589. doi: 10.1073/pnas.95.15.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y, Swartz KJ. Localization and molecular determinants of the hanatoxin receptors on the voltage-sensing domain of a K+ channel. J Gen Physiol. 2000;115:673–684. doi: 10.1085/jgp.115.6.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y, Swartz KJ. Helical structure of the COOH terminus of S3 and its contribution to the gating modifier toxin receptor in voltage-gated ion channels. J Gen Physiol. 2001;117:205–218. doi: 10.1085/jgp.117.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- Lu Z. Mechanism of rectification in inward-rectifier K+ channels. Annu Rev Physiol. 2004;66:103–129. doi: 10.1146/annurev.physiol.66.032102.150822. [DOI] [PubMed] [Google Scholar]
- Lu Z, MacKinnon R. Purification, characterization, and synthesis of an inward-rectifier K+ channel inhibitor from scorpion venom. Biochemistry. 1997;36:6936–6940. doi: 10.1021/bi9702849. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- MacKinnon R, Miller C. Mechanism of charybdotoxin block of the high-conductance, Ca2+- activated K+ channel. J Gen Physiol. 1988;91:335–349. doi: 10.1085/jgp.91.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R, Miller C. Mutant potassium channels with altered binding of charybdotoxin, a pore- blocking peptide inhibitor. Science. 1989;245:1382–1385. doi: 10.1126/science.2476850. [DOI] [PubMed] [Google Scholar]
- Marvin L, De E, Cosette P, Gagnon J, Molle G, Lange C. Isolation, amino acid sequence and functional assays of SGTx1. The first toxin purified from the venom of the spider scodra griseipes. Eur J Biochem. 1999;265:572–579. doi: 10.1046/j.1432-1327.1999.00726.x. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Lampe RA, Keith RA, Bean BP. Voltage-dependent inhibition of N- and P-type calcium channels by the peptide toxin omega-grammotoxin-SIA. Mol Pharmacol. 1997a;52:1095–1104. doi: 10.1124/mol.52.6.1095. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Mintz IM, Bean BP. Alteration of P-type calcium channel gating by the spider toxin omega- Aga-IVA. Biophys J. 1997b;72:2117–2128. doi: 10.1016/S0006-3495(97)78854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton RE, Warren VA, Kraus RL, Hwang JC, Liu CJ, Dai G, Brochu RM, Kohler MG, Gao YD, Garsky VM, Bogusky MJ, Mehl JT, Cohen CJ, Smith MM. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry. 2002;41:14734–14747. doi: 10.1021/bi026546a. [DOI] [PubMed] [Google Scholar]
- Miller C. The charybdotoxin family of K+ channel-blocking peptides. Neuron. 1995;15:5–10. doi: 10.1016/0896-6273(95)90057-8. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature. 1992;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239–1243. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- Naranjo D. Inhibition of single Shaker K channels by kappa-conotoxin-PVIIA. Biophys J. 2002;82:3003–3011. doi: 10.1016/S0006-3495(02)75641-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton RS, Pallaghy PK. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon. 1998;36:1573–1583. doi: 10.1016/s0041-0101(98)00149-4. [DOI] [PubMed] [Google Scholar]
- Oswald RE, Suchyna TM, McFeeters R, Gottlieb P, Sachs F. Solution structure of peptide toxins that block mechanosensitive ion channels. J Biol Chem. 2002;277:34443–34450. doi: 10.1074/jbc.M202715200. [DOI] [PubMed] [Google Scholar]
- Pallaghy PK, Nielsen KJ, Craik DJ, Norton RS. A common structural motif incorporating a cystine knot and a triple-stranded beta-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994;3:1833–1839. doi: 10.1002/pro.5560031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CS, Miller C. Interaction of charybdotoxin with permeant ions inside the pore of a K+ channel. Neuron. 1992;9:307–313. doi: 10.1016/0896-6273(92)90169-e. [DOI] [PubMed] [Google Scholar]
- Pathak M, Kurtz L, Tombola F, Isacoff E. The cooperative voltage sensor motion that gates a potassium channel. J Gen Physiol. 2005;125:57–69. doi: 10.1085/jgp.200409197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips LR, Milescu M, Li-Smerin Y, Mindell JA, Kim JI, Swartz KJ. Voltage-sensor activation with a tarantula toxin as cargo. Nature. 2005;436:857–860. doi: 10.1038/nature03873. [DOI] [PubMed] [Google Scholar]
- Possani LD, Merino E, Corona M, Bolivar F, Becerril B. Peptides and genes coding for scorpion toxins that affect ion-channels. Biochimie. 2000;82:861–868. doi: 10.1016/s0300-9084(00)01167-6. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006a;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Moran MM, Chong JA, Clapham DE. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006b;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash LD, Hodgson WC. Pharmacology and biochemistry of spider venoms. Toxicon. 2002;40:225–254. doi: 10.1016/s0041-0101(01)00199-4. [DOI] [PubMed] [Google Scholar]
- Restano-Cassulini R, Korolkova YV, Diochot S, Gurrola G, Guasti L, Possani LD, Lazdunski M, Grishin EV, Arcangeli A, Wanke E. Species diversity and peptide toxins blocking selectivity of ether-a-go-go-related gene subfamily K+ channels in the central nervous system. Mol Pharmacol. 2006;69:1673–1683. doi: 10.1124/mol.105.019729. [DOI] [PubMed] [Google Scholar]
- Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- Ruta V, Chen J, MacKinnon R. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell. 2005;123:463–475. doi: 10.1016/j.cell.2005.08.041. [DOI] [PubMed] [Google Scholar]
- Ruta V, Jiang Y, Lee A, Chen J, MacKinnon R. Functional analysis of an archaebacterial voltage-dependent K+ channel. Nature. 2003;422:180–185. doi: 10.1038/nature01473. [DOI] [PubMed] [Google Scholar]
- Ruta V, MacKinnon R. Localization of the voltage-sensor toxin receptor on KvAP. Biochemistry. 2004;43:10071–10079. doi: 10.1021/bi049463y. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Johnson JH, Hammerland LG, Kelbaugh PR, Volkmann RA, Saccomano NA, Mueller AL. Heteropodatoxins: peptides isolated from spider venom that block Kv4.2 potassium channels. Mol Pharmacol. 1997;51:491–498. [PubMed] [Google Scholar]
- Sasaki M, Takagi M, Okamura Y. A Voltage Sensor-Domain Protein is a Voltage-Gated Proton Channel. Science. 2006;312:589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Fersht AR. Energetics of protein-protein interactions: analysis of the barnase- barstar interface by single mutations and double mutant cycles. J Mol Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Shon KJ, Stocker M, Terlau H, Stuhmer W, Jacobsen R, Walker C, Grilley M, Watkins M, Hillyard DR, Gray WR, Olivera BM. kappa-Conotoxin PVIIA is a peptide inhibiting the shaker K+ channel. J Biol Chem. 1998;273:33–38. doi: 10.1074/jbc.273.1.33. [DOI] [PubMed] [Google Scholar]
- Smith-Maxwell CJ, Ledwell JL, Aldrich RW. Role of the S4 in cooperativity of voltage-dependent potassium channel activation. J Gen Physiol. 1998a;111:399–420. doi: 10.1085/jgp.111.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Maxwell CJ, Ledwell JL, Aldrich RW. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation. J Gen Physiol. 1998b;111:421–439. doi: 10.1085/jgp.111.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Alphy S, Seibert AL, Blumenthal KM. Differential phospholipid binding by site 3 and site 4 toxins: Implications for structural variability between voltage-sensitive sodium channel domains. J Biol Chem. 2005 doi: 10.1074/jbc.M412552200. [DOI] [PubMed] [Google Scholar]
- Srinivasan KN, Gopalakrishnakone P, Tan PT, Chew KC, Cheng B, Kini RM, Koh JL, Seah SH, Brusic V. SCORPION, a molecular database of scorpion toxins. Toxicon. 2002;40:23–31. doi: 10.1016/s0041-0101(01)00182-9. [DOI] [PubMed] [Google Scholar]
- Suchyna TM, Tape SE, Koeppe RE, 2nd, Andersen OS, Sachs F, Gottlieb PA. Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature. 2004;430:235–240. doi: 10.1038/nature02743. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, MacKinnon R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 1995;15:941–949. doi: 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, MacKinnon R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 1997a;18:665–673. doi: 10.1016/s0896-6273(00)80306-2. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, MacKinnon R. Mapping the receptor site for hanatoxin, a gating modifier of voltage- dependent K+ channels. Neuron. 1997b;18:675–682. doi: 10.1016/s0896-6273(00)80307-4. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kim JI, Min HJ, Sato K, Swartz KJ, Shimada I. Solution structure of hanatoxin1, a gating modifier of voltage-dependent K+ channels: common surface features of gating modifier toxins. J Mol Biol. 2000;297:771–780. doi: 10.1006/jmbi.2000.3609. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Kim JI, Takahashi H, Swartz K, Shimada I. Solution structure of ω-grammotoxin SIA, a gating modifier of P/Q and N type calcium channels. J Mol Biol. 2002;321:517–526. doi: 10.1016/s0022-2836(02)00595-8. [DOI] [PubMed] [Google Scholar]
- Terlau H, Boccaccio A, Olivera BM, Conti F. The block of Shaker K+ channels by kappa-conotoxin PVIIA is state dependent. J Gen Physiol. 1999;114:125–140. doi: 10.1085/jgp.114.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- Ulens C, Hogg RC, Celie PH, Bertrand D, Tsetlin V, Smit AB, Sixma TK. Structural determinants of selective alpha-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc Natl Acad Sci U S A. 2006;103:3615–3620. doi: 10.1073/pnas.0507889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Droogmans G, Wissenbach U, Janssens A, Flockerzi V, Nilius B. The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature. 2004;430:748–754. doi: 10.1038/nature02732. [DOI] [PubMed] [Google Scholar]
- Wang GK, Strichartz GR. Purification and physiological characterization of neurotoxins from venoms of the scorpions centruroides sculpturatus and leiurus quinquestriatus. Mol Pharmacol. 1983;23:519–533. [PubMed] [Google Scholar]
- Wang JM, Roh SH, Kim S, Lee CW, Kim JI, Swartz KJ. Molecular surface of tarantula toxins interacting with voltage sensors in Kv channels. J Gen Physiol. 2004;123:455–467. doi: 10.1085/jgp.200309005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterfield JR, Swartz KJ. A hot spot for the interaction of gating modifier toxins with voltage- dependent ion channels. J Gen Physiol. 2000;116:637–644. doi: 10.1085/jgp.116.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The voltage-gated potassium channels and their relatives. Nature. 2002;419:35–42. doi: 10.1038/nature00978. [DOI] [PubMed] [Google Scholar]
- Yeung SY, Thompson D, Wang Z, Fedida D, Robertson B. Modulation of Kv3 subfamily potassium currents by the sea anemone toxin BDS: significance for CNS and biophysical studies. J Neurosci. 2005;25:8735–8745. doi: 10.1523/JNEUROSCI.2119-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarayskiy VV, Balasubramanian G, Bondarenko VE, Morales MJ. Heteropoda toxin 2 is a gating modifier toxin specific for voltage-gated K+ channels of the Kv4 family. Toxicon. 2005;45:431–442. doi: 10.1016/j.toxicon.2004.11.015. [DOI] [PubMed] [Google Scholar]