

Abstract
Approximately 20–30% of neurons in the avian cochlear nucleus (nucleus magnocellularis) die following deafferentation (i.e. deafness produced by cochlea removal) and the remaining neurons show a decrease in soma size. Cell death is generally accepted to be a highly regulated process involving various pro-survival and pro-death molecules. One treatment that has been shown to modify the expression of these molecules is chronic administration of lithium. The present experiments examined whether lithium treatment can protect neurons from deafferentation-induced cell death. Post-hatch chicks were treated with LiCl or saline for 17 consecutive days, beginning on the day of hatching. On the 17th day, a unilateral cochlea ablation was performed. Five days following surgery, the nucleus magnocellularis neurons were counted stereologically on opposite sides of the same brains. Lithium reduced deafferentation-induced cell death by more than 50% (9.8% cell death as compared with 22.4% in saline-treated subjects). Lithium did not affect cell number on the intact side of the brain. Lithium also did not prevent the deafferentation-induced decrease in soma size, suggesting a dissociation between the mechanisms involved in the afferent control of soma size and those involved in the afferent control of cell viability. A possible mechanism for lithium’s neuroprotective influence was examined in a second set of subjects. Previous studies suggest that the pro-survival molecule, bcl-2, may play a role in regulating cell death following deafferentation. Tissues from lithium- and saline-treated subjects were examined using immunocytochemistry. Chronic administration of lithium dramatically increased the expression of bcl-2 protein in nucleus magnocellularis neurons. These data suggest that lithium may impart its neuroprotective effect by altering the expression of molecules that regulate cell death.
Keywords: apoptosis, neuroprotection, auditory brainstem
Cell death is a regulated process that is common during normal development of the nervous system, but can be initiated by a variety of events including deafferentation, disease, or injury. Neuronal cell death can also be induced during development in many sensory systems following the loss of sensory input. For example, mitral and tufted cells of the olfactory system die following unilateral naris occlusion (Frazier and Brunjes, 1988; Meisami and Safari, 1981), and in the visual system, the survival of tectal neurons depends on activity-dependent release of trophic factors from retinotectal axon terminals (Catsicas et al., 1992). In the auditory system, cell death can occur in the cochlear nucleus following deafness (Born and Rubel, 1985; Hashisaki and Rubel, 1989; Tierney et al., 1997). Although cell death following sensory deprivation has been widely studied in a variety of systems, little work has gone into efforts to protect neurons from deafferentation-induced cell death. A limited number of treatments have been shown to protect spiral ganglion cells from cell death following cochlear damage (Yamagata et al., 2004). For brain stem auditory neurons, the only promising manipulations reported to date involve either embryonic treatment with receptor antagonists (Solum et al., 1997) or genetic manipulations (Mostafapour et al., 2002).
The present experiments examine the control of cell death in the brain stem auditory system of the chick following deafness. In this system, the auditory nerve projects unilaterally to a relatively homogeneous population of neurons in the avian cochlear nucleus, nucleus magnocellularis (NM) and provides the only source of excitatory drive to those neurons (Parks and Rubel, 1978; Rubel 1978). This arrangement is ideal for studies of the role of sensory experience in development because unilateral manipulations of auditory nerve activity will affect afferent input to the ipsilateral NM but leave input to the contralateral NM intact, thus providing a within-subject control population of neurons. The most extreme manipulation of auditory input is deafness, which results in a complete cessation of activity in the ipsilateral NM (Born et al., 1991). Consequently, a common approach has been to remove one cochlea, thereby removing all excitatory afferent input to NM. Previous studies have shown that deafferentation by cochlea removal results in the death of 20–30% of the neurons in the ipsilateral NM and the remaining neurons show a decrease in soma size (Born and Rubel, 1985; Edmonds et al., 1999; Hyde and Durham, 1994).
Several events leading toward the deafferentation-induced cell death in NM have been documented. A robust decrease in both RNA and protein synthesis is observed as early as one hour after deafferentation (Garden et al., 1995; Steward and Rubel, 1985). In addition, intracellular calcium levels gradually rise in the absence of afferent activity (Zirpel et al., 1995). In vitro analyses of the early effects of deafferentation on NM neurons have shown that the maintenance of calcium homeostasis and ribosomal structure and function requires activity-dependent release from the auditory nerve and subsequent activation of metabotropic glutamate receptors (Hyson, 1997, 1998; Hyson and Rubel, 1989; Nicholas and Hyson, 2004; Zirpel and Rubel, 1996; Zirpel et al., 2000). However, these earliest changes occur across the entire population of activity-deprived neurons and it is still unclear what factors lead to the differentiation of NM neurons into living and dying populations. This segregation clearly emerges by 6–12 h after deafferentation, at which time most of the cells show a partial recovery of protein synthesis, but approximately 20–30% of the neurons show a complete cessation of protein synthesis (Steward and Rubel, 1985). Presumably the recovering neurons will go on to survive while those that had a breakdown in protein synthesis will die.
Despite the massive reduction in total mRNA synthesis (Garden et al., 1995), there is a robust, but transient, upregulation of mRNA for bcl-2 following cochlea removal. This upregulation is observed 6–12 h after cochlea removal in approximately 20–30% of the neurons (Wilkinson et al., 2002). The bcl-2 protein is thought to be cytoprotective and therefore one would expect that it would be in the living population of cells and not in the dying population. However, given that there is approximately the same percentage of neurons dying as there is expressing bcl-2 mRNA, it is likely that bcl-2 mRNA is being expressed in the dying population of cells. One possible explanation for this unusual finding is that perhaps the deafferentation-induced expression of the bcl-2 mRNA is incapable of being translated in dying cells because their protein synthesis machinery has been broken down by the time that the message has been expressed. Wilkinson et al. (2002) were unable to detect an upregulation of bcl-2 protein following cochlea removal, only an upregulation of mRNA. They suggested that the initiation of cell death cascades may have signaled the nucleus to make more pro-survival molecules, but the mRNA could not be utilized to make the protein and the cell goes on to die. If this hypothesis is correct, then bcl-2 might be able to rescue the cell from deafferentation-induced cell death if it is expressed as a protein. One treatment that has been shown to increase bcl-2 protein expression in neurons is chronic treatment with lithium (Chen et al., 1999). If chronic lithium can increase bcl-2 expression, and if bcl-2 expression is capable of rescuing deafferented NM neurons, then chronic lithium treatment may have a neuroprotective effect on NM neurons following cochlea removal.
Lithium is the major treatment of choice for bipolar disorder, yet the precise mechanism for how lithium works is not clearly understood. For the present purposes, it is important to note that lithium has been found to reduce neuronal death in animal models of neurodegenerative diseases such as Huntington’s disease (Wei et al., 2001), and it reduces death induced by ischemia or stroke (Nonaka and Chuang, 1998). Lithium’s efficacy is long lasting, requires a long term pretreatment and occurs at concentrations that are therapeutic in humans. The present experiments first test the hypothesis that chronic pre-treatment with lithium protects NM neurons against deafferentation-induced cell death. Secondly, a possible mechanism of this neuroprotection, the upregulation of the pro-survival molecule bcl-2, is evaluated using immunocytochemistry.
EXPERIMENTAL PROCEDURES
All subjects were post-hatch chicks (Ross×Ross) hatched and reared at Florida State University. The surgical procedures used in these experiments were approved by the Animal Care and Use Committee at Florida State University and conform to the guidelines set forth by the National Institutes of Health. All efforts were made to minimize the number of animals used and their suffering.
Procedures
Post-hatch chicks, of either sex, received daily injections of either lithium chloride (LiCl) or saline for 17 days prior to unilateral cochlea removal. Cell size and cell death in NM were examined 5 days following surgery (n = 8 per group). A second set of subjects (n = 11 per group) was examined 7 h after the final injection of lithium and levels of the pro-survival molecule bcl-2 were measured.
Daily injections
Post-hatch chicks (P0) were started on a LiCl or saline injection regimen. Both groups of animals received a daily s.c. injection for a total of 17 days. The dose of LiCl was gradually increased across age, beginning at 1.5 mM/kg for the first four days, followed by 2.3 mM/kg for seven days, and finally, 3.0 mM/kg for the last six days. This schedule was adapted from the protocols of both Wei et al. (2001) and Nonaka and Chuang (1998). The volume of each injection was 0.01 ml/kg. Control subjects received daily 0.01 ml/kg injections of saline. Subjects received unilateral cochlea removal surgery one hour after the last daily injection on P16. For two subjects, lithium treatment was continued at the 3.0 mM/kg dose for the 5 day survival period following cochlea removal. The lithium treatment for the remaining subjects was terminated on the day of cochlea removal.
Plasma lithium levels
One group of animals (n = 6) received the chronic lithium injection regimen and serum samples were taken at the end of the 2.3, and 3.0 mM/kg treatments (P10, and P16 respectively). The blood was collected from a wing vein while the bird was under halothane anesthesia. The collected blood was then centrifuged using Becton Dickinson Compact II Centrifuge (Becton Dickinson, Franklin Lakes, NJ, USA) and the lithium and sodium concentrations of the plasma fractions were analyzed using an Electrolyte Analyzer (Beckham Coulter Synchron EL-ISE; Beckham Coulter, Fullerton, CA, USA). The plasma levels were measured 1 h postinjection on P10 (the last day of the 2.3 mM/kg treatment), and 1 and 6 h postinjection on P16 (the last day of the 3.0 mM/kg treatment), and on P17 (24 h post last injection). Fig. 1 shows the average plasma lithium concentrations at these time points. As expected, the plasma concentration of lithium increased with increasing dose, reaching a level approximately 2.5 mmol/l following the 3.0 mM/kg dose. Plasma levels of lithium were reduced by approximately 80% by 6 h postinjection, indicating that lithium is rapidly excreted. No lithium was detected in the plasma 24 h after the last injection.
Fig. 1.
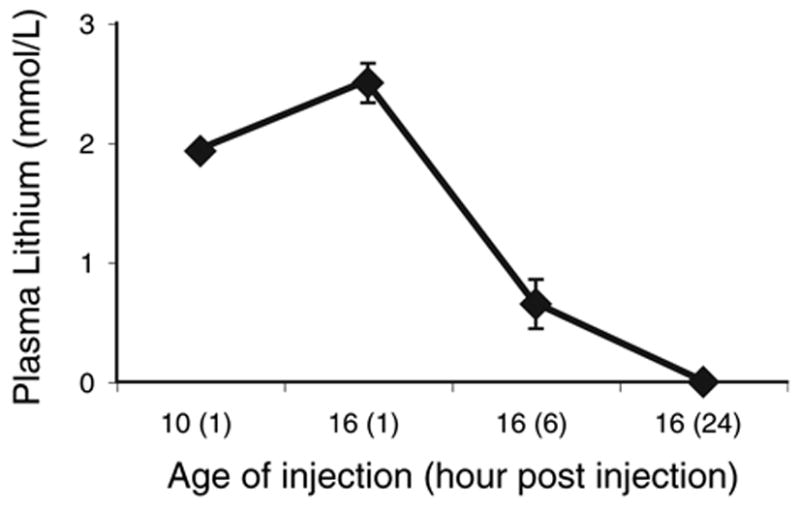
Average plasma lithium levels measured at different times during the treatment regimen. Comparison of the levels measured 1 h after injection of a 2.3 mM/kg dose on P10 with the levels measured 1 h after injection of a 3.0 mM/kg dose on P16 shows the expected increase in serum lithium with increasing dose. Lithium is rapidly excreted. By 6 h after injection of the 3.0 mM/kg dose, serum levels of lithium are reduced by approximately 80%, and no lithium is detected in the serum 24 h after the injection. Error bars represent standard error of the mean. Where bars are absent, the error was smaller than the size of the symbol.
Body weight
Lithium treatment had no noticeable effects on the overall health of the subjects. This is evident from the similar growth rates in the saline- and lithium-treated subjects. As can be seen in Fig. 2, there is no difference between the groups in weight gain across age (P = 0.906).
Fig. 2.
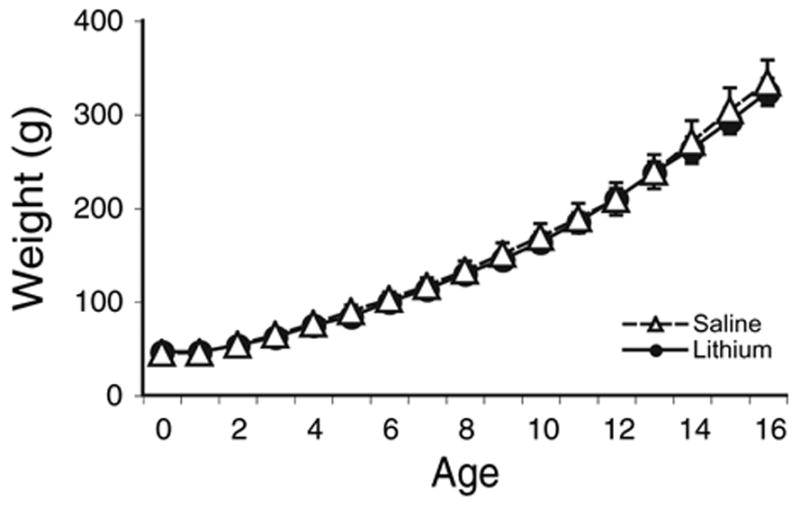
Average weights of subjects during drug treatments. No difference in weight gain was observed between the saline- and lithium-treated groups. Error bars represent standard error of the mean. Where bars are absent, the error was smaller than the size of the symbol.
Cochlea removal surgery
Subjects were anesthetized with halothane. A small incision was made to widen the ear canal and the tympanic membrane was punctured. The columella was removed, followed by the extraction of the basilar papilla (cochlea) through the oval window using forceps. The extracted tissue was placed in water and viewed under a dissection microscope to ensure complete cochlea removal. The middle ear cavity was packed with Gelfoam and the external incision sealed with surgical adhesive.
Tissue preparation
Five days following cochlea removal, subjects were deeply anesthetized with pentobarbital and perfused with 0.9% saline followed by ice cold 4% paraformaldehyde. Brainstems were removed from the cortex and post-fixed in 4% paraformaldehyde for 1–2 h followed by overnight cryoprotection at 4 °C in phosphate-buffered saline (PBS) containing 20% sucrose. The tissue was rapidly frozen in 2-methylbutane on dry ice and embedded in TBS tissue freezing medium for cryosectioning using a Leica CM 1850 cryostat (Leica Microsystems Inc., Bannockburn, IL, USA). Sections for cell counting were cut at 60 μm and were either mounted directly to slides or collected into ice cold PBS for later mounting. Every section containing NM was collected. Mounted sections were then dried overnight and stained for Nissl. To assure adequate staining throughout the relatively thick section, slides were rehydrated in dH2O for a relatively long period of time (approximately 20–30 min). The slides were then transferred to Thionin for 10 min, rinsed twice with distilled water for 2 min each, followed by two changes of 70% ethanol for five minutes each. The slides were placed into 95% ethanol for a time period that varied depending on the darkness of the stain (approximately 20 min) and then they were transferred through two 3 min changes of 100% ethanol. Finally, sections were cleared after xylene. Slides were coverslipped in DPX mounting medium and allowed to dry overnight.
Immunocytochemistry
Pairs of subjects, one treated with LiCl and one treated with saline, were processed simultaneously. Puncture marks were made in the ventral portion of the brain stem to identify treatment condition following the simultaneous processing. Perfusion and cryoprotection were identical to that described above. Cryosections (20–30 μm) containing NM were placed in a vial containing PBS. Sections were then washed 2×10 minutes in PBS and endogenous peroxidase activity was quenched by incubating in 0.03% H2O2 in methanol for 10 minutes. Following three 10 min rinses in PBS, sections were placed in a blocking solution containing 2% bovine serum albumin (BSA), 1% normal goat serum (NGS), 0.1% Triton X-100 in PBS for an hour. Sections were then transferred to a 1:500 concentration of the primary antibody Bcl-2 (n-19: sc-492, epitope mapping of amino acids 4–21 of the N-terminus of human origin of Bcl-2, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) in blocking solution and incubated on a rotator overnight at 4 °C. Control sections receiving the same treatment but with no primary antibody were processed to ensure specificity of the reaction. The following day the sections were then removed from 4 °C and allowed to incubate at room temperature for two hours. Sections were washed 3×10 min in PBS. Sections were then incubated in a 1:200 concentration of goat anti-rabbit secondary antibody in blocking solution (BSA, NGS, and Triton) for one hour. Following three 10-minute washes with PBS, sections were incubated in avidin–biotin–peroxidase complex (ABC; Vector Laboratories, Burlingame, CA, USA) for one hour. After another round of washes, with PBS, sections were reacted with diaminobenzidine (DAB) tetrahydrochloride and H2O2 for visualization. Following a final round of washes sections were mounted on slides and allowed to dry overnight. The following day, slides were cleared using graded alcohols and xylenes and coverslipped in DPX mountant.
Stereology
Sections containing NM neurons were analyzed using a stereological program (Stereo Investigator, Microbrightfield, Inc., Williston, VT, USA) to compare the number of neurons on the deafferented side versus the intact side. The optical fractionator, an unbiased and efficient stereological method, was used. Cell counts using this method are unaffected by tissue shrinkage and the objects are counted in an objective, systematic way. Each cell has an equal chance of being counted and the optical fractionator reduces the chance of double counting cells. The end result is an estimate of cell number in the nucleus. The Gundersen coefficient of error for these estimates was <0.05 in each case. The parameters of the program were set such that a grid of 40×40 μm boxes separated by 100 μm were randomly placed over user-generated outlines of NM and the investigator counted the number of cells in each box. A cell was counted if its right-most portion was located inside the box or was touching the right side or top line of the box. Typical section thickness after processing was approximately 18–21 μm. To avoid double counting, only cells whose right-most portion was located between 3 μm and 18 μm from the top of the section were counted. A 1:2 series of the entire extent of NM was included in the analysis. The investigator performing the counting was blind to treatment condition. Once all sections were counted, the data were sorted into their respective LiCl or saline treatment groups and the effects of deafferentation and lithium were analyzed using analysis of variance (ANOVA).
Cell size of NM neurons was also measured on opposite sides of the same brains. These measurements were made using a video image analysis program (Neurolucida, Microbrightfield, Inc.). All measurements of cell size were from NM neurons located midway in the anterior-to-posterior extent of the nucleus. Cell size was determined by outlining the stained portion of the cell body, its the largest diameter visualized under a 100× objective. Only neurons with a clearly distinguishable nucleolus were included in the analysis. At least 50 neurons were measured on both sides of the brain.
Analysis of immunocytochemistry
Tissues from both the lithium-treated and control groups were processed in the same vials. The level of immunolabeling was objectively measured using a video image analysis program (NIH Image J). The light levels and camera settings remained constant for each pair of subjects. The mean gray-scale density over approximately 20 NM neurons in a given tissue section was measured beginning with cells at the medial edge of the nucleus and proceeding laterally. Differences in the density of labeling between saline- and lithium-treated subjects were compared using a t-test. To compare labeling differences across different pairs of subjects, the labeling density of each cell was transformed to a z-score based on the mean and standard deviation of the gray-scale density of cells in the saline-treated member of the pair. This transform provides a measure of the difference in labeling in terms of the number of standard deviations of labeling density, rather than simply a percentage change in mean level of the somewhat arbitrary gray scale. The investigator performing the measurements was blind to treatment condition.
RESULTS
Lithium reduces deafferentation-induced cell death
Unilateral cochlea removal produced cell death in the deafferented NM. An example of this effect can be seen in the upper panel of Fig. 3. As seen in the lower panel of Fig. 3, chronic treatment with lithium reduced the degree of deafferentation-induced cell death. Stereological analysis of Nissl-stained tissue revealed that cochlea removal produced 22.4% cell death in saline-treated subjects (Fig. 4). This is in line with previous reports of 20–30% cell death in NM neurons following cochlea removal (Born and Rubel, 1985). However, only 9.8% cell death was observed when LiCl was administered for 17 days prior to cochlea removal. Statistical analyses on these percent difference scores revealed that there was a reliable difference between the saline and lithium treated groups (t(14) = 3.2, P<0.01). When lithium was continued throughout the survival period there was a trend toward less cell death (5.1±2.91%, n = 2) than when it was terminated on the day of cochlea removal (11.43±2.62%, n = 6), but with this small sample size, the trend was not statistically reliable (P = 0.25).
Fig. 3.
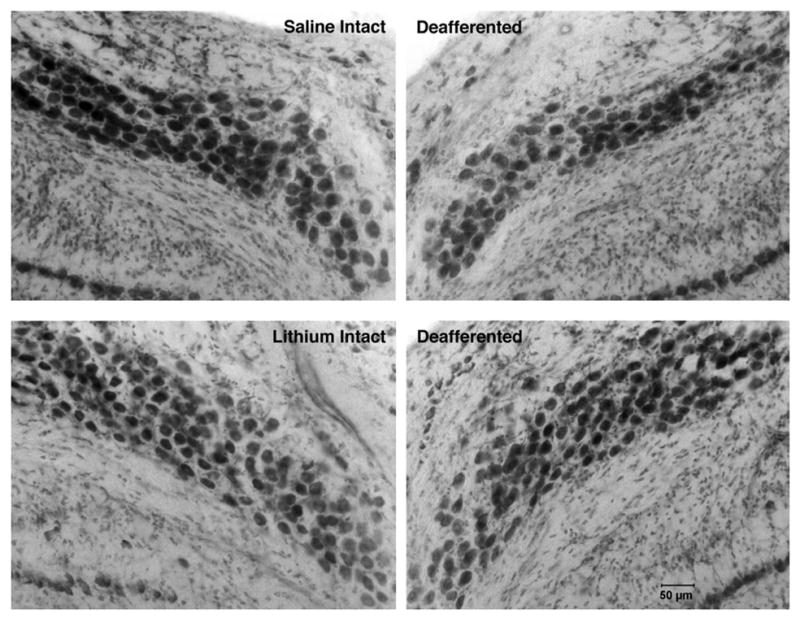
Photomicrographs of intact and deafferented sides of saline- (top) and lithium-treated (bottom) subjects 5 days after cochlea removal. Fewer cells are observed in NM on the deafferented side of the brain. Fewer cells are lost following cochlea removal in the lithium-treated subjects.
Fig. 4.
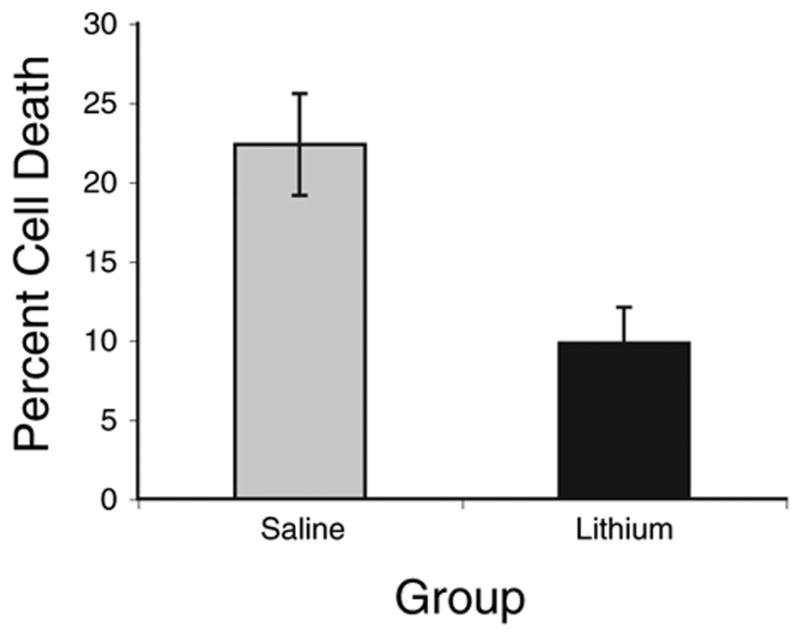
Average percent neuronal loss in NM on the cochlea removal side of the brain compared with the intact side of the same brain ((intact–deafferented)/intact×100). Fewer cells are lost following cochlea removal in subjects treated with lithium for 17 days prior to cochlea removal (n = 8, P<0.01). Error bars represent standard error of the mean.
Fig. 5 displays the raw cell count data as the mean number of cells on each side of the brain. As can be seen, there is no difference between the lithium and saline-treated groups in the number of neurons on the intact side of the brain. However, there are more neurons on the cochlea removal side of the brain in the lithium-treated group than in the saline-treated group. Statistical analyses confirmed these conclusions. A two-way mixed ANOVA, using drug treatment as the between variable and side of the brain as the within variable, revealed a main effect of Side (F(1, 14) = 46.5, P<0.0001), no main effect of Drug Treatment (F(1, 14)<1.0), but, importantly, a reliable Side× Drug Treatment interaction (F(1, 14) = 6.40, P<0.05). Post hoc pairwise comparisons (Newman-Keuls, P<0.05) revealed that there were fewer cells on the cochlea removal side of the brain in the saline-treated group than in the lithium-treated group, yet there was no reliable difference between saline- and lithium-treated groups in the number of cells on the intact side of the brain. For both groups of subjects, there were reliably fewer cells on the cochlea removal side of the brain than on the intact side of the brain.
Fig. 5.
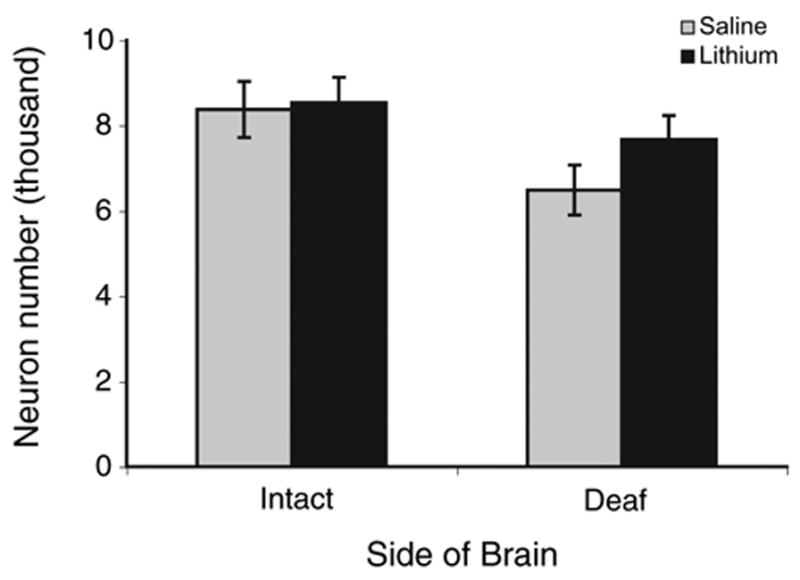
Total number of neurons in NM on each side of the brain for subjects treated for 17 days with either saline or lithium. There were fewer neurons on the deafferented side of the brain in the saline-treated group compared with all other groups, including the deafferented side of lithium-treated animals (P<0.05). There is no difference between saline- and lithium-treated subjects in the number of neurons on the intact side of the brain. Error bars represent standard error of the mean.
Unilateral cochlea removal also resulted in a decrease in the soma size of NM neurons on the deafferented side of the brain (P<0.01 for both saline- and lithium-treated groups). There was no difference between lithium- and saline-treated groups in the percent difference in cell size (19.3% versus 19.8% respectively, P>0.85). Analyses of the absolute numbers also revealed that there was no overall effect of lithium on cell size. Means size of cells on the intact side of the brain for saline-treated subjects was 302.8±15.3 μm2 compared with 296.1±12.1 μm2 for lithium-treated subjects (P>0.69). Group means for cells on the deafferented sides were 246.9±14.3 μm2 and 238.4±14.77 μm2 for saline and lithium, respectively (P>0.56). There was no significant correlation between the percent change in cell size and the percent change in cell number in either group (r = −0.33 and −0.06 for saline- and lithium-treated groups, respectively).
Lithium increases Bcl-2 expression
Chronic lithium administration resulted in darker immunolabeling for bcl-2 in NM. An example of this effect can be seen in Fig. 6. Objective analysis of the tissue sections using densitometry confirmed these visual impressions. Fig. 6 displays the average gray scale density measurements of NM neurons in lithium- and saline-treated subjects. This difference was statistically reliable (t(10) = 4.15, P<0.01). On average, the lithium-treated subject of the pair of simultaneously processed tissues was 56±16% greater in gray scale density than the saline-treated subject. Transforming the data to z-scores revealed that, on average, the gray scale density of NM neurons in the lithium-treated subjects was 3.1±0.9 standard deviations greater than the level of labeling observed in the saline-treated subjects. The pair shown in Fig. 7 is representative of this difference and had a mean z-score difference of 3.2.
Fig. 6.
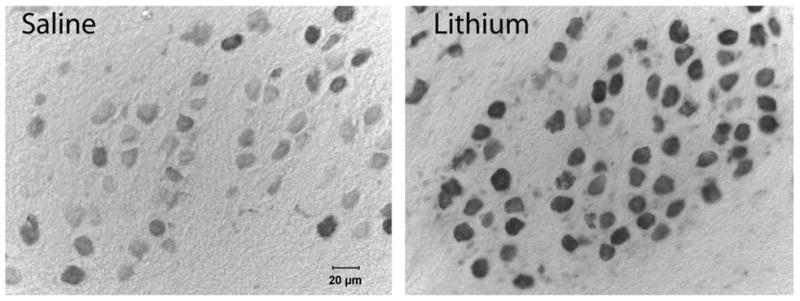
Representative photomicrographs of neurons in NM in saline- (left) and lithium-treated (right) subjects. Tissue sections were immunolabeled for bcl-2 and tissue from the two different subjects was processed simultaneously. Lithium-treated subjects consistently showed greater labeling for bcl-2.
Fig. 7.
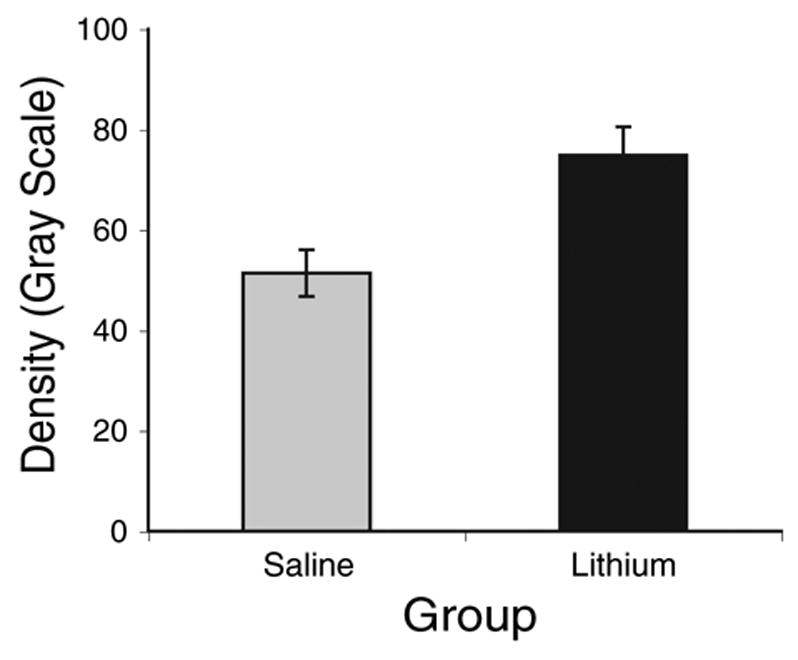
Average gray scale density measured over individual NM neurons immunolabeled for bcl-2. Lithium-treated subjects showed reliably greater labeling than saline-treated controls (P<0.05). Error bars represent standard error of the mean.
DISCUSSION
Stereological analysis
Stereological analysis of Nissl-stained tissue sections showed that there were approximately 22% fewer cells in NM on the deafferented side of the brain five days after unilateral cochlea removal. This is in agreement with previous studies, which have shown that cochlea removal produces 20–30% cell death in NM (Born and Rubel, 1985). However, chronic administration of lithium prior to cochlea removal reduced cell death. When lithium was administered for 17 days prior to cochlea removal, only 9.8% of the deafferented neurons died. If lithium treatment was continued throughout the 5 day survival period, a trend toward even less cell death (5.1%, n = 2) was observed. The reduction in the percentage of cell death following lithium treatment was due to less cell death in the deafferented NM and not a loss of cells on the intact side of the brainstem. There was no difference between lithium-and saline-treated subjects in the number of NM neurons on the intact side of the brain (see Fig. 5). Rather, there were more NM neurons still present on the deafferented side of the brain in the lithium-treated subjects. Thus, lithium administration protects the NM neurons from deafferentation-induced cell death.
The amount of cell death detected in the saline-treated animals was within the range of that has been observed in most of the previous reports on this system. For example, Born and Rubel (1985) and Edmonds et al. (1999) reported that approximately 25% of the NM neurons die two to five days after deafferentation in 1–2 week old chickens. In the present study, 22% of the neurons died 5 days after cochlea removal, which is identical to that reported by Hyde and Durham (1994). Although the percentage of cell death did match those of previous reports, our stereological measurements estimated a somewhat higher absolute number of neurons than previously reported. This may simply be due to variation in the precise strain and age of bird examined. In most studies previously reported, White Leghorn chicks were analyzed at an age of 7–10 days. The present study used Ross×Ross chicks at 16 days of age at the time of cochlea removal. Another possible contributor to the larger number of cells observed in NM is that the present analysis included much of the posterior region of NM. It appears that this so-called “hook” region was not included in the previous reports because the amount of cochlear innervation to this region is uncertain. The addition of this posterior region may have diluted the level of cell death observed and increased the number of neurons on both sides of the brain. Finally, the greater number of NM neurons estimated in the present study could be partially attributable to a difference in the method of counting neurons. The optical-fractionator stereological method used in the present report has not been used in the past. Despite these methodological differences, however, the percentage of cell death observed following cochlea removal was similar to that reported in previous studies. Importantly, the level of cell death observed in subjects pre-treated with lithium is well below the level of cell death observed in any published report on this system.
Lithium appears to protect NM neurons from deafferentation-induced cell death, but has no effect on the deafferentation-induced decrease in soma size. Cell size decreased by approximately 19% in both saline- and lithium-treated subjects following cochlea removal. There was no difference between lithium and saline-treated subjects in the size of NM neurons on the intact side of the brain, indicating that lithium had no overall effect on cell size. The magnitude of the deafferentation-induced decrease in cell size was comparable to the previously reported effects of cochlea removal in untreated subjects (Born and Rubel, 1985). The fact that lithium affects deafferentation-induced death, but not deafferentation-induced changes in cell size, is not entirely unexpected. Independence of cell size and cell death effects has also been observed in developmental analyses of the mammalian cochlear nucleus following cochlear damage. In the gerbil, for example, cell death following cochlear damage is only observed in subjects up to 7 days old, but cell size decreases following cochlear damage are observed at all ages (Tierney et al., 1997). Together, these findings suggest that the mechanisms involved in the afferent control of soma size are different than those involved in the afferent control of cell viability.
Plasma levels and general health
Because of the differences in species and route of administration, it is difficult to compare the effective level of lithium that produced neuroprotection with the level typically used for the treatment of humans with bipolar disorder or used previously for neurodegeneration studies in rats. The maximum dose used in the present study in chicks (3.0 mM/kg or 0.12 g/kg) was higher than that typically given to human patients (0.014–0.028 g/kg) (Silverstone and Romans, 1996), but the protocol for lithium treatment was similar to that used in studies of neurodegeneration in rats (Nonaka and Chuang, 1998; Wei et al., 2001). Subjects in the present study did not show any signs of toxicity during the course of this treatment. This conclusion was further supported by the fact that there was no difference in growth rate between lithium and saline-treated subjects. The lithium was rapidly excreted in birds. By 6 h postinjection of the 3.0 mM/kg dose, over 75% of the drug had been excreted from the body and by 24 h there was no detection of lithium in the serum. The rapid clearance of lithium might account for why there was a trend toward greater neuroprotection when the treatment was continued throughout the 5 day survival period compared with when the treatment was terminated on the day of cochlea ablation. Long term effects of lithium treatment following cochlea removal were not tested in the present study since previous work has shown that cell death is complete by 2–5 days following surgery. However, it remains possible that lithium merely delays deafferentation-induced death, and once the lithium is fully cleared from the system, the cells will begin to die. This hypothesis was not tested in the present study.
Lithium increases bcl-2 expression
Lithium rescued greater than 50% of the dying neurons, but exactly how lithium is having this neuroprotective effect is not yet clear. One of the possible ways that lithium may be protecting neurons is by its influence on the levels of bcl-2 protein (Manji and Chen, 2002). In the present study, lithium substantially increased bcl-2 expression in NM neurons. Bcl-2 has been implicated in the regulation of deafferentation-induced cell death in the cochlear nucleus. Wilkinson et al. (2002) reported that bcl-2 mRNA is robustly increased 6–12 h following deafferentation in approximately 20–30% of the NM neurons on the deafferented side of the brain. They suggested that the message for this pro-survival molecule was being upregulated in the dying population of NM neurons following cochlea removal. This led to the obvious question of why a pro-survival molecule would be upregulated in dying neurons. They hypothesized that the initiation of some cell death cascade resulted in a cellular signal to produce molecules that could protect the cell. However, by the time the bcl-2 mRNA is upregulated, dying neurons appear to have stopped making protein (Steward and Rubel, 1985). Consequently, the bcl-2 mRNA is not translated into protein so it cannot have its neuroprotective effect. Presumably, if bcl-2 protein was expressed, it could have a neuroprotective influence. Indeed, bcl-2 has been implicated as having a neuroprotective effect in mammalian cochlear nucleus neurons following deafferentation (Mostafapour et al., 2002). Based on these studies, one would expect that if bcl-2 protein were upregulated prior to cochlea removal, then fewer cells would die. This logic motivated the present study. Since chronic lithium has been shown to upregulate bcl-2 expression (Chen et al., 1999) then perhaps lithium could protect neurons from deafferentation-induced death.
In the present study, lithium did upregulate bcl-2 protein in NM and this upregulation was apparent at the time that cells would typically divide into living and dying populations following cochlea removal (6 h). To control for processing variables that could dramatically influence the darkness of immunolabeling, tissue from pairs of lithium-and saline-treated subjects were reacted simultaneously. Objective analysis showed that the level of labeling in NM neurons of lithium-treated subjects was more that 3 standard deviations greater than the level of labeling in the matched saline-treated subjects. The increase in bcl-2 protein suggests that upregulation of this molecule prior to cochlea removal can protect neurons from deafferentation-induced death and provides a possible mechanism for the neuroprotective action of lithium in this system.
There are several possible cascades by which lithium could lead to the upregulation of bcl-2. A commonly proposed pathway begins with lithium’s inhibition of glycogen synthase kinase-3 (GSK-3) (Gould et al., 2003; Grimes and Jope, 2001), GSK-3 regulates the activation of a variety of transcription factors, such as nuclear factor kappa B (Demarchi et al., 2003; Pahl, 1999; Tamatani et al., 1999) and AKT (Chalecka-Franaszek and Chuang, 1999; De Sarno et al., 2002), which could go on to promote bcl-2 transcription. Lithium also influences the activation of transcription factors, polyomavirus enhancer binding protein (PEBP-2β), cAMP response element binding protein (CREB), and mitogen/extracellular regulated kinase (Chen et al., 1999; Kopinsky et al., 2003; Pardo et al., 2003), and decreases levels of p53 (Chen and Chuang, 1999). The decrease in p53 and increase in PEBP-2β, and CREB results in an increase in the expression of bcl-2 protein, ultimately leading to neural protection (Chen and Chuang, 1999; Chuang et al., 2002).
While the results of the present experiment are in agreement with the hypothesis that lithium may be neuroprotective through the upregulation of bcl-2 protein, lithium’s neuroprotective action could result from its effect on any of several cellular or molecular events that have been implicated in the control of cell death. For example, lithium has been shown to decrease the levels of the pro-apoptotic protein bax thereby increasing the bcl-2/bax ratio in favor of the anti-apoptotic protein (Chen and Chuang, 1999). Einat et al. (2003) showed that the phosphorylated transcription factors such as CREB are increased and genes such as Bad are decreased when given long term treatments of lithium and valproate. Chronic lithium treatment also protects cultured rat cerebellar, cortical, and hippocampal neurons against glutamate-induced excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx (Nonaka et al., 1998). Given this broad range of effects on molecules known to regulate cell death, it is not surprising that lithium has a neuroprotective influence in a variety of model systems, including deafferentation-induced cell death.
Acknowledgments
Supported by NIDCD grant RO1 DC 00858 and NIH jointly-sponsored grant T32 NSO7437.
The authors would like to thank Dr. Frank Johnson, Dr. Barbara Licht, Todd Stincic and Alexander Nicholas for their comments on an earlier version of this manuscript. We would also like to thank Dr. Kathleen Curtis for her assistance with the serum analysis, and Katie Carzoli for her assistance in tissue processing.
Abbreviations
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- CREB
cAMP response element binding protein
- GSK-3
glycogen synthase kinase-3
- LiCl
lithium chloride
- NGS
normal goat serum
- NM
nucleus magnocellularis
- PBS
phosphate-buffered saline
- PEBP-2
polyomavirus enhancer binding protein
References
- Born DE, Rubel EW. Afferent influences on brainstem auditory nuclei of the chicken, neuron number and size following cochlea removal. J Comp Neurol. 1985;231:435–445. doi: 10.1002/cne.902310403. [DOI] [PubMed] [Google Scholar]
- Born DE, Durham D, Rubel EW. Afferent influences on brainstem auditory nuclei of the chick: nucleus magnocellularis neuronal activity following cochlea removal. Brain Res. 1991;557:37–47. doi: 10.1016/0006-8993(91)90113-a. [DOI] [PubMed] [Google Scholar]
- Catsicas M, Pequignot Y, Clarke PG. Rapid onset of neuronal death induced by blockade of either axoplasmic transport or action potentials in afferent fibers during brain development. J Neurosci. 1992;12:4642–4650. doi: 10.1523/JNEUROSCI.12-12-04642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalecka-Franaszek E, Chuang D-M. Lithium activates the serine/threonine kinase AKT-1 and suppresses glutamate-induced inhibition of AKT activity in neurons. Proc Natl Acad Sci U S A. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Chuang D. Long term lithium treatment suppresses p53 and bax expression but increases bcl-2 expression. J Biol Chem. 1999;274:6039–6042. doi: 10.1074/jbc.274.10.6039. [DOI] [PubMed] [Google Scholar]
- Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH, Manji HK. The mood stabilizing agents lithium and valproate robustly increases the levels of the neuroprotective protein Bcl-2 in the CNS. J Neurochem. 1999;72:879–882. doi: 10.1046/j.1471-4159.1999.720879.x. [DOI] [PubMed] [Google Scholar]
- Chuang D, Chen R, Chalecka-Franaszek E, Ren M, Hashimoto R, Senatorov V, Kanai H, Hough C, Hiroi T, Leeds P. Neuroprotective effects of lithium in cultured cells and animal models of diseases. Bipolar Disord. 2002;4:129–136. doi: 10.1034/j.1399-5618.2002.01179.x. [DOI] [PubMed] [Google Scholar]
- Demarchi F, Bertoli C, Sandy P, Schneider C. Glycogen synthase kinase-3β regulates NF-κB1/p105 stability. J Biol Chem. 2003;278:39583–39590. doi: 10.1074/jbc.M305676200. [DOI] [PubMed] [Google Scholar]
- De Sarno P, Li X, Jope RS. Regulation of AKT and glycogen synthase kinase-3beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- Edmonds JL, Hoover LA, Durham D. Breed differences in deafferentation-induced neuronal cell death and shrinkage in chick cochlear nucleus. Hear Res. 1999;127:62–76. doi: 10.1016/s0378-5955(98)00180-4. [DOI] [PubMed] [Google Scholar]
- Einat H, Yuan P, Gould TD, Li J, Du JH, Zhang L, Manji H, Chen G. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci. 2003;23:7311–7316. doi: 10.1523/JNEUROSCI.23-19-07311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier LL, Brunjes PC. Unilateral odor deprivation: early postnatal changes in olfactory bulb cell density and number. J Comp Neurol. 1988;269:355–370. doi: 10.1002/cne.902690304. [DOI] [PubMed] [Google Scholar]
- Garden GA, Redeker-DeWulf V, Rubel EW. Afferent influences on brainstem auditory nuclei of the chicken: regulation of transcriptional activity following cochlea removal. Comp Neurol. 1995;359:412–423. doi: 10.1002/cne.903590305. [DOI] [PubMed] [Google Scholar]
- Gould TD, Chen G, Manji HK. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology. 2003;29:32–38. doi: 10.1038/sj.npp.1300283. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Hashisaki GT, Rubel EW. Effects of unilateral cochlea removal on anteroventral cochlear nucleus neurons in developing gerbils. J Comp Neurol. 1989;283:65–73. doi: 10.1002/cne.902830402. [DOI] [PubMed] [Google Scholar]
- Hyde GE, Durham D. Increased deafferentation-induced cell death in chick brainstem auditory neurons following blockade of mitochondrial protein synthesis with chloramphenicol. J Neurosci. 1994;14:291–300. doi: 10.1523/JNEUROSCI.14-01-00291.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyson RL, Rubel EW. Transneuronal regulation of protein synthesis in the brainstem auditory system of the chick requires synaptic activation. J Neurosci. 1989;9:2835–2845. doi: 10.1523/JNEUROSCI.09-08-02835.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyson RL. Transneuronal regulation of ribosomes after blockade of ionotropic excitatory amino acid receptors. Brain Res. 1997;749:61–70. doi: 10.1016/s0006-8993(96)01160-2. [DOI] [PubMed] [Google Scholar]
- Hyson RL. Activation of metabotropic glutamate receptors is necessary for transneuronal regulation of ribosomes in chick auditory neurons. Brain Res. 1998;809:214–220. doi: 10.1016/s0006-8993(98)00873-7. [DOI] [PubMed] [Google Scholar]
- Kopinsky KL, Chalecka-Franaszek E, Gonzalez-Zulueta M, Chuang DM. Chronic lithium treatment antagonizes glutamate-induced decrease of phosphorylated CREB in neurons via reducing protein phosphatase 1 and increasing MEK activities. Neuroscience. 2003;116:425–435. doi: 10.1016/s0306-4522(02)00573-0. [DOI] [PubMed] [Google Scholar]
- Manji HK, Chen G. PKC and MAP kinases and the bcl-2 family of proteins as long term targets for mood stabilizers. Mol Psychiatry. 2002;7:546–556. doi: 10.1038/sj.mp.4001018. [DOI] [PubMed] [Google Scholar]
- Meisami E, Safari L. A quantitative study of the effects of early unilateral olfactory deprivation on the number and distribution of mitral and tufted cells and of glomeruli in the rat olfactory bulb. Brain Res. 1981;221:81–107. doi: 10.1016/0006-8993(81)91065-9. [DOI] [PubMed] [Google Scholar]
- Mostafapour SP, Mae del Puerto N, Rubel EW. Bcl-2 overexpression eliminates deprivation-induced cell death of brainstem auditory neurons. J Neurosci. 2002;22:4670–4674. doi: 10.1523/JNEUROSCI.22-11-04670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas AH, Hyson RL. Group I and group II metabotropic glutamate receptors are necessary for the activity-dependent regulation of ribosomes in chick auditory neurons. Brain Res. 2004;1014:110–119. doi: 10.1016/j.brainres.2004.03.066. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport. 1998;9:2081–2084. doi: 10.1097/00001756-199806220-00031. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Hough C, Chuang DM. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Neurobiology. 1998;95:2642–2647. doi: 10.1073/pnas.95.5.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of REL/NFkappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Pardo R, Andreolotti AG, Ramos B, Picatoste F, Claro E. Opposed effects of lithium on the MEK-ERK pathway in neural cells: Inhibition in astrocytes and stimulation in neurons by GSK3 independent mechanisms. J Neurochem. 2003;87:417–426. doi: 10.1046/j.1471-4159.2003.02015.x. [DOI] [PubMed] [Google Scholar]
- Parks TN, Rubel EW. Organization and development of the brainstem auditory nuclei of the chicken: primary afferent projections. J Comp Neurol. 1978;180:439–448. doi: 10.1002/cne.901800303. [DOI] [PubMed] [Google Scholar]
- Rubel EW. Ontogeny of structure and function in the vertebrate auditory system. In: Jacobson M, editor. Handbook of sensory physiology. IX. New York: Springer-Verlag; 1978. pp. 135–237. Development of sensory systems. [Google Scholar]
- Silverstone T, Romans S. Long term treatment of bipolar disorder. Drugs. 1996;5:367–382. doi: 10.2165/00003495-199651030-00003. [DOI] [PubMed] [Google Scholar]
- Solum D, Hughes D, Major MS, Parks TN. Prevention of normally occurring and deafferentation-induced neuronal death in chick brainstem auditory neurons by periodic blockade of AMPA/kainate receptors. J Neurosci. 1997;17:4744–4751. doi: 10.1523/JNEUROSCI.17-12-04744.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Rubel EW. Afferent influences on brain stem auditory nuclei of the chicken: presynaptic action potentials regulate protein synthesis in nucleus magnocellularis neurons. J Neurosci. 1985;231:385–395. doi: 10.1523/JNEUROSCI.08-03-00901.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamatani M, Che Y, Matsukai H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces bcl-2 and bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- Tierney TS, Russell FA, Moore DR. Susceptibility of developing cochlear nucleus neurons to deafferentation-induced death abruptly ends just before the onset of hearing. J Comp Neurol. 1997;378:295–306. doi: 10.1002/(sici)1096-9861(19970210)378:2<295::aid-cne11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Wei H, Qin ZH, Senatorov VV, Wei W, Wang Y, Qian Y, Chuang DM. Lithium suppresses excitotoxicity-induced striatal lesions in a rat model of Huntington’s disease. Neuroscience. 2001;106:603–612. doi: 10.1016/s0306-4522(01)00311-6. [DOI] [PubMed] [Google Scholar]
- Wilkinson BL, Sadler KA, Hyson RL. Rapid deafferentation-induced upregulation of Bcl-2 mRNA in the chick cochlear nucleus. Mol Brain Res. 2002;99:67–74. doi: 10.1016/s0169-328x(02)00113-4. [DOI] [PubMed] [Google Scholar]
- Yamagata T, Miller J, Ulfendahl M, Olivius P, Altschuler R, Pyykko I, Bredberg G. Delayed neurotrophic treatment preserves nerve survival and electrophysiological responsiveness in neomycin-deafened guinea pigs. J Neurosci Res. 2004;78:75–86. doi: 10.1002/jnr.20239. [DOI] [PubMed] [Google Scholar]
- Zirpel L, Lachicha EA, Lippe WR. Deafferentation increases the intracellular calcium of cochlear nucleus neurons in the embryonic chick. J Neurophysiol. 1995;74:1355–1357. doi: 10.1152/jn.1995.74.3.1355. [DOI] [PubMed] [Google Scholar]
- Zirpel L, Rubel EW. Eighth nerve activity regulates intracellular calcium concentration of avian cochlear nucleus neurons via a metabotropic glutamate receptor. J Neurophysiol. 1996;76:4127–4137. doi: 10.1152/jn.1996.76.6.4127. [DOI] [PubMed] [Google Scholar]
- Zirpel L, Janowiak MA, Veltri CA, Parks TN. Developmental changes in metabotropic glutamate receptor-mediated calcium homeostasis. J Comp Neurol. 2000;421:95–106. [PubMed] [Google Scholar]