

Abstract
SHP-1 is an important negative regulator of signaling by several receptors including receptors for interleukin-2 (IL-2R) and other cytokines. SHP-1 acts by dephosphorylating the receptors and receptor-associated kinases such as IL-2R-associated Jak3 kinase. We found that SHP-1 protein was not detectable or greatly diminished in most (six of seven) T cell lines derived from various types of T cell lymphomas and all (eight of eight) cutaneous T-cell lymphoma tissues with a transformed, large-cell morphology. All T-cell lymphoma lines tested (eight of eight) expressed diminished amounts or no detectable SHP-1 mRNA. These T cell lines did not, however, carry any mutations in the SHP-1 gene-coding, splice-junction, and promoter regions. Importantly, SHP-1 DNA promoter region in the T cell lines was resistant to digestion with three different methylation-sensitive restriction enzymes. This resistance was reversed by treatment of the cells with a demethylating agent, 5-deoxyazacytidine. The treatment resulted also in the expression of SHP-1 mRNA and, less frequently, SHP-1 protein. The expression of SHP-1 protein was associated with dephosphorylation of the Jak3 kinase. These results show that lack of SHP-1 expression is frequent in malignant T cells and results from methylation of the SHP-1 gene promoter. Furthermore, they indicate that SHP-1 loss may play a role in the pathogenesis of T cell lymphomas by permitting persistence of signals generated by IL-2R and, possibly, other receptor complexes.
SHP-1 is a member of the nontransmembrane phosphotyrosine phosphatases expressed predominantly in cells of the hematopoietic lineage. 1-5 SHP-1 is an important negative regulator involved in signaling through receptors for cytokine/growth factors such as c-kit ligand, CSF-1, erythropoietin, interleukin (IL)-3, IL-2, IL-4, and IL-13. 6-8 A variety of noncytokine receptors including B-antigen receptor, T-antigen receptor, CD22, CD72, 9-13 as well as the growing family of the inhibitory receptors expressed by natural killer and other types of cells also interact with SHP-1. 14 Association of SHP-1 with the majority of these receptors is mediated by phosphorylated tyrosine-based motifs. 15,16 SHP-1 acts by dephosphorylating the receptors and receptor-associated tyrosine kinases. 6,17 Dysfunction of SHP-1 as seen in the natural SHP-1 gene knock-out, motheaten mice, results in hyperplasia of the erythroid and lymphoid lineages. 18
Signaling through the IL-2R receptor complex is vital for proper function of normal T lymphocytes. High-affinity IL-2R receptors are composed of α, β, and γc chains. γc is shared by the receptors for IL-2, IL-4, IL-7, IL-9, and IL-15. 19 Inactivating mutations of γc result in severe combined immunodeficiency in humans and mice. 20-23 Interaction of IL-2 with IL-2R rapidly induces tyrosine phosphorylation of the IL-2R complex mediated by the receptor-associated Jak1 and Jak3 tyrosine kinases. 24-26 This leads to phosphorylation of STAT3 and STAT5 molecules which translocate to the cell nucleus and activate transcription of the IL-2 responsive proteins. 26-28 Activation of Jak3 is critical for transduction of signals mediated by IL-2R complex because mutations of Jak3 result in severe combined immunodeficiency in both humans 29,30 and mice 31,32 similar to the immunodeficiency seen in mutations of the γc chain.
Previous studies have established that a number of human T cell leukemia virus type I (HTLV-I)-positive and -negative T cell lines exhibit constitutive activation of the IL-2R Jak/STAT signaling pathway 33-35 raising the possibility that an unbalanced, permanently turned-on IL-2R/Jak signaling leads to uncontrolled growth of these cells and may play a role in the pathogenesis of various types of human T cell malignancy.
Lack of expression of SHP-1 protein has recently been identified in several HTLV-I-positive T cell lines. 7,36 This observation combined with the presence of constitutive activation of the IL-2R Jak/STAT signaling pathway, suggested that the concomitant lack of SHP-1 protein may be responsible in some instances for the unbalanced IL-2R/Jak signaling. However, the extent of the loss of SHP-1 expression in T cell lymphomas, the mechanism of such loss and the exact effect of SHP-1 on the constitutive IL-2R/Jak signaling in malignant T cells remained undefined.
Here we describe that lack of SHP-1 expression is frequent in T cell lymphomas and results from a transcriptional block of SHP-1 gene because of an extensive methylation of its promoter. Most, but not all, of the malignant T cell lines analyzed display constitutive activation of the IL-2R-associated Jak/STAT pathway. Reversal of the promoter methylation resulted in these cells in expression of SHP-1 mRNA and, less frequently, SHP-1 protein. The induced expression of SHP-1 protein correlated with dephosphorylation of the IL-2R-associated Jak3 kinase. These data demonstrate that inhibition of SHP-1 expression in malignant T cells is mediated by methylation of the SHP-1 gene promoter. Furthermore, they indicate that promoter methylation-induced transcriptional silencing of the SHP-1 gene may play a role in malignant T cell transformation by permitting persistent activation of the IL-2R/Jak signaling pathway and, possibly, other pathways regulated by SHP-1.
Materials and Methods
Cell Lines and Tissues
Most cell lines used in this study were described in detail previously. 35,36 In brief, three cell lines (PB-1, 2A, and 2B) were established from a patient with a progressive cutaneous T-cell lymphoproliferative disorder. The PB-1 cell line was obtained at a relatively early stage of the patient’s cutaneous T lymphoma whereas the 2A and 2B lines were established at a later, aggressive stage of the disease. All lines showed the same morphology, immunophenotype, T-cell receptor rearrangement, and cytogenetic abnormalities as the original, patient-derived malignant cells. Sez-4 line, kindly provided by T. Abrams (Hahnemann University), was derived from a patient with Sezary syndrome and also bears close morphological, phenotypic, and genotypic resemblance to the original tumor. ATL-2, C91PL, HUT102B, and C10MJ2 cell lines represent HTLV-I-related acute T cell lymphoma/leukemia. The SUDHL-1 line was derived from ALK+ T cell lymphoma. L540, HS455, HDLM, and KM-H2 were obtained from patients with Hodgkin’s lymphoma. 37 Whereas L540 and HDLM are of T cell origin as demonstrated by immunophenotyping and T-cell receptor gene rearrangement analysis, KM-H2 displays a B cell phenotype and genotype. We determined by flow cytometry that the HS445 cell line is also of B cell origin (kappa-restricted CD19+, CD20+, CD22+, CD23+, CD10+ B cells; MA Wasik, unpublished data). The exact nature of the HS445 line is uncertain. Although derived from a patient with Hodgkin’s lymphoma, this line may represent a nonmalignant Epstein-Barr virus-transformed lymphoblastoid B cell line (HG Drexler, personal communication). The healthy adults served as normal controls. Peripheral blood mononuclear cells (PBMC) were obtained from such individuals by centrifugation on Ficoll/Paque gradient. Phytohemagglutinin (PHA) blasts were obtained by stimulation of the PBMC with the mitogen. Cell lines and PHA-stimulated PBMC were cultured at 37°C with 5% CO2 in standard RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 1% penicillin/streptomycin/fungizone mixture, and 2 mmol/L l-glutamine. 35 Tissue sections of lymph nodes and skin were obtained from eight cases of advanced cutaneous T cell lymphoma which has undergone histologically documented transformation into a diffuse large-cell lymphoma. Before the large cell transformation the patients had a 1- to 8-year (median, 6.5 years) history of mycosis fungoides, tumor phase (1 patient), or Sezary syndrome (7 patients) diagnosed based on the combination of clinical features, biopsy and peripheral blood smear morphology, immunophenotyping, T-cell gene rearrangement, and cytogenetics. After the diagnosis of the large cell transformation most of the patients received combined chemotherapy with no or transient response; seven died within 4 to 15 months of the diagnosis.
Drug Treatment
In SHP-1 protein expression induction experiments, cell lines were treated for 72 hours with 20 ng/ml of phorbol 12-myristate 13-acetate (PMA; Sigma Chemical CO., St. Louis, MO). 38-40 In some experiments Ionomycin (Calbiochem, La Jolla, CA) was added to PMA at 1 μg/ml. In SHP-1 promoter demethylation experiments, the cell lines were cultured with 5-deoxyazacytidine (Sigma) at 1 μmol/L for 5 days.
Protein Expression and Phosphorylation
These assays were performed as described. 35,36 In brief, the cells (10 to 15 × 106) were washed and lysed in ice-cold lysis buffer. In the Jak3 phosphorylation experiments the cells were exposed for 5 minutes to 500 U of IL-2 or medium alone before lysis. The lysates were centrifuged and supernatants were precleared with protein A-Sepharose (Sigma), incubated with SHP-1 or Jak3 antiserum (Santa Cruz Biotechnology, Santa Cruz, CA) and protein A-Sepharose, washed, boiled, separated on a 10% polyacrylamide-sodium dodecyl sulfate gel, and transferred electrophoretically to hybridization transfer membranes. The membranes were blocked with 2% bovine serum albumin in tris-buffered-saline/Tween 20 (TBST) buffer. To detect protein expression the membranes were incubated with the SHP-1 or Jak3 antiserum. To detect protein phosphorylation, the membranes were incubated with anti-phosphotyrosine mAb (4G10; Upstate Biotechnology, Lake Placid, NY). Next, the membranes were incubated with an appropriate secondary, peroxidase-conjugated Ab. Blots were developed using the enhanced chemiluminescence (ECL) reagents (Amersham Life Science, Arlington Heights, IL).
Immunohistochemical Staining
The staining was performed on formalin-fixed, paraffin-embedded tissue sections of skin and lymph nodes involved by cutaneous T cell lymphoma, large-cell type using a standard streptavidin-biotin complex technique (Research Genetics, Huntsville, AL) and the following antibodies: CD3 and CD20 (L-26) (both from DAKO, Carpinteria, CA), Ki-67 (mib1; Immunotech, Westbrook, ME), and SHP-1 (Santa Cruz Technology). To detect SHP-1, heat-induced antigen retrieval was performed using Antigen Retrieval AR-10 solution (Biogenex, San Ramon, CA) and 20 minutes slide immersion into a 95°C water bath.
Nucleic Acid Analysis
Total cellular RNA was extracted by RNeasy Mini Handbook kit (Qiagen, Valencia, CA). For Northern analysis, 20 μg of total RNA samples were separated on 1% agarose-formaldehyde gels, transferred to Hybond-N+ membranes (Amersham), and hybridized with either a 563-bp or 1-kb α[32P]-dCTP-labeled, reverse transcriptase-polymerase chain reaction (RT-PCR) generated SHP-1 cDNA probe amplified from the catalytic domain of SHP-1. Hybridization with a 670-bp β-actin cDNA probe served as a control. RT-PCR was performed with 1 μg of total RNA in RT buffer mixture containing 200 U of SuperScript TM II reverse transcriptase (Life Technologies, Inc., Rockville, MD). RNase H (Promega, Piscataway, NJ) was added and cDNA was purified on Centri-Sep columns (Princeton Separations, Princeton, NJ). PCR was performed with 2 μl of cDNA in a PCR buffer mixture containing 2 U Taq polymerase (Life Technologies, Inc.) and, when developing probes for Northern blotting, 8 μCi α[32P]-dCTP (Amersham). The SHP-1-specific primers (see below) were designed based on the SHP-1 genomic DNA sequence from the GenBank database using an Oligo (National Biosciences, Plymouth, MN) computer program. The PCR reaction was performed at 94°C for 3 minutes, followed by 30 cycles: 45 seconds at 94°C, 30 seconds at 57°C, and 1.5 minutes at 72°C. The amplified products were visualized in UV light by staining with ethidium bromide after electrophoresis in the 1.5% agarose gel.
Genomic DNA extraction was performed with 2 × 10 7 cells using Qiagen Genomic DNA kit (Qiagen). For PCR, DNA was digested with methylation sensitive restriction enzymes (HpaII, EagI or NaeI; New England Biolabs, Beverly, MA) for 4 hours at 37°C. PCR was performed with primers which cover SHP-1 promoter 2 to exon 3 region (see below). For Southern blotting, DNA was digested with MobI and the methylation-sensitive enzymes. The DNA digests were purified by phenol/chloroform extraction, ethanol precipitated, separated on 0.8% agarose gel, and blotted on Hybond N+ nylon membranes. The membrane-bound DNA was hybridized to 50 ng of the digoxigenin-labeled, PCR-generated SHP-1 DNA probe. Membranes were washed in buffer containing 0.2× standard saline citrate, 0.1% sodium dodecyl sulfate twice at 24°C and once at 68°C and exposed to autoradiographic film. To determine the nucleic acid sequence of the SHP-1 genomic DNA, we used the seven primer pairs to cover the entire gene region used by hematopoietic cells spanning from promoter 2 (P2) to exon 16 (E16). The primers were designed to include ∼60 to 80 bp of exon adjacent intronic DNA containing splice junction sequences. The primer pairs and DNA sequences analyzed were as follows: 1) 5′-cactgcagctgactcactgat and 5′-acggggaaccaggaatgagtg (P50 -E50), 2) 5′-tgctgtgctctaaaacgagaa and 5′-gtggaaagggtggtaggttag (E1-E3), 3) 5′-cacagtaggtgcttgatttcc and 5′-gggtggagacctgtgagatga (E4-E7), 4) 5′-cactccctccatacagatgat and 5′-agccctcagtttcccaacaag (E8-E9), 5) 5′-caggcactcagaacatagagc and 5′-aagaggaggaatggggagcac (E10-E11), 6) 5′-cggtgaccctgggcacattcc and 5′-gctacatctcatacacgagtg (E12-E14), and 7) 5′-gtccccctgtgctgtctcctg and 5′-actgtgcccgtcttatcgtca (E15-E16). The PCR amplification products were separated on agarose gel, visualized, and purified using the QIAEX gel purification kit (Qiagen). The nucleotide sequence of the PCR products was determined using ABI PRISM Ready Reaction Dye Deoxy Terminator Cycle Sequencing Kit (Perkin-Elmer, Emeryville, CA). The nucleotide sequence was determined on an automated ABI DNA sequencer (Applied Biosystems, Foster City, CA) and compared to the SHP-1 DNA sequence of a normal donor and the one available from GenBank.
Results
T Cell Lymphomas Frequently Fail to Express SHP-1 Protein
Previous studies 7,36 have shown that most (five of seven) of the HTLV-I-positive T cell lines tested displayed either a lack or marked decrease in expression of the SHP-1 protein. To determine better the frequency of the SHP-1 protein loss in T cell malignancies, we analyzed seven additional T cell lines derived from various types of T cell lymphoma unrelated to the HTLV-I infection. These lymphomas represented aggressive types of non-Hodgkin’s T cell lymphoma and Hodgkin’s lymphoma with a T cell phenotype. As shown in Figure 1 ▶ , six out of the seven T cell lines showed greatly diminished to undetectable expression of SHP-1 protein. This finding suggests that lack of the SHP-1 protein expression is frequent in T-cell lymphoma cells regardless of their HTLV-I status. Because three of the SHP-1-negative cell lines (Sez-4, SUDHL-1, and HDLM) did not display constitutive IL-2R/Jak3 signaling as determined by the lack of Jak3 phosphorylation (Q Zhang and MA Wasik, unpublished data), 36 this indicates that lack of SHP-1 expression may play a role in the malignant transformation of T cells by affecting also other receptor complexes controlled by SHP-1. Noteworthy, the PB-1 cell line showed only a mild decrease in expression of SHP-1 protein compared to the control T-cell-rich PBMC and PHA blasts. This was in striking contrast to the SHP-1-negative 2A and 2B lines derived from the same patient as PB-1 at a more advanced stage of the T cell lymphoma. 35 This finding suggests that loss of SHP-1 protein expression may be progressive and lymphoma stage-dependent.
Figure 1.

Absent or markedly diminished SHP-1 protein expression in several cell lines representing various types of malignant T cell lymphoma. T cell lines derived from a progressive cutaneous T cell lymphoma (PB-1, 2A, and 2B), Sezary syndrome (Sez-4), ALK+ T cell lymphoma (SUDHL-1), and Hodgkin’s lymphoma (HDLM and L540) were evaluated for expression of the SHP-1 protein by immunoprecipitation and immunoblotting with an anti-SHP-1 antibody. T-cell-rich PBMC, mitogen (PHA)-stimulated T cell blasts (PHA bl), and HS445 cell line (see Materials and Methods) served as positive controls.
To determine whether a lack of SHP-1 expression is present not only in cultured cells but also in lymphoma tissues, we tested biopsy-derived lymphoma tissue samples from several patients. Because our cell lines studied in the greatest detail 35 (see below) were obtained from an advanced, transformed cutaneous T cell lymphoma, we tested tissue samples from patient with cutaneous T cell lymphoma (mycosis fungoides and its leukemic form Sezary syndrome) which has undergone transformation to a diffuse large-cell lymphoma. These transformed lymphomas represented either skin lesions (three samples) or involved lymph nodes (five samples) obtained from eight different patients. When analyzed by immunohistochemistry, large lymphoma cells but not small, presumably mainly residual normal cells showed lack of SHP-1 expression in all cases. Representative results from one such patient are shown in Figure 2 ▶ . The lymph node showed almost total effacement of normal architecture by the greatly expanded interfollicular T cell zones (Figure 2A) ▶ comprised of a rather homogeneous population of large cells (Figure 2D) ▶ with a high proliferative rate (Figure 2E) ▶ and a T cell phenotype (Figure 2, B and C) ▶ with deletions of CD2 and CD5 antigens as determined by flow cytometry (data not presented). The lymphoma cells were completely devoid of any SHP-1 staining (Figure 2F) ▶ . This contrasted with strong staining of residual normal cells, mostly mantle B cells from involuted follicles and scattered small T cells. These findings indicate that the loss of SHP-1 expression is common, if not universal, in at least one type of T cell lymphoma, ie, transformed large-cell cutaneous T cell lymphoma.
Figure 2.
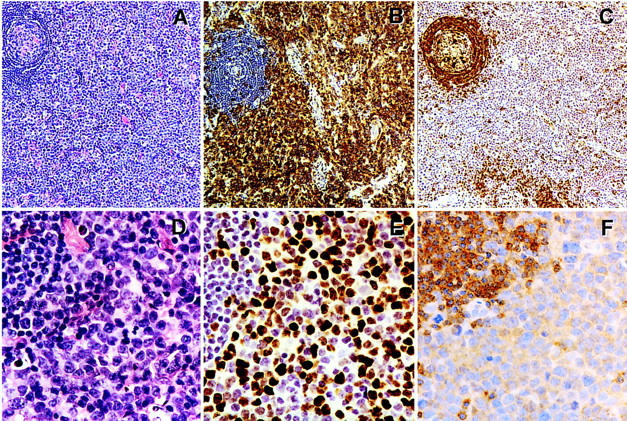
Lack of SHP-1 expression by a transformed large-cell cutaneous T cell lymphoma involving lymph node. The images represent ×100 low-power (A–C) and ×400 high-power (D–F) magnifications. Methylene blue was used as a counterstain. A and D: H&E staining. B: Anti-CD3 antigen staining (T-cell receptor-associated antigen). C: Anti-CD79a staining (immunoglobulin-associated B cell antigen). E: Anti-mib1 (Ki67) staining (antigen expressed in cycling but not resting cells). F: anti-SHP-1 staining. Note a residual nonmalignant, partially involuted follicle with preserved to mildly expanded mantle zone in the upper left corners of all photographs.
Malignant T Cells Frequently Fail to Express SHP-1 Transcript
To determine whether lack of the SHP-1 protein expression represents a pre- or posttranscriptional event, several cell lines were tested for expression of SHP-1 mRNA. Beside PB-1, 2A, 2B, and Sez-4 cell lines, we also analyzed four HTLV-I-positive, malignant T cell lines derived from an adult T cell leukemia/lymphoma. Three of these lines (C91PL, ATL-2, and C10MJ2) did not and one (HUT102B) did express SHP-1 protein. 36 Northern blot analysis (Figure 3A) ▶ showed that almost all SHP-1 protein-negative lines failed to express SHP-1 mRNA with the C91PL cell line being the only exception. Interestingly, three cell lines which expressed SHP-1 mRNA (PB-1, C91PL, and HUT102B) contained less SHP-1 mRNA than the control PBMC. This indicates that SHP-1 concentration in such cells may be relatively decreased. Similar results were obtained using RT-PCR (data not presented). The above findings indicate that transcription of the SHP-1 gene is frequently, if not universally, inhibited in malignant T cells.
Figure 3.

Absent or markedly decreased constitutive SHP-1 mRNA expression and PMA-induced SHP-1 protein expression in malignant T cells. T cell lines from cutaneous T cell lymphoma (PB-1, 2A, 2B, and Sez-4) and HTLV-I-positive adult-type T cell leukemia/lymphoma (C91PL, ATL-2, HUT102B, and C10MJ2), and control PBMC from healthy donors and HS445 cells were examined for expression of the SHP-1 mRNA (A). Detection of actin mRNA (B) served as a control of the quality and quantity of mRNA in the samples. The T cell lines were stimulated with phorbol ester (PMA) and examined for expression of SHP-1 protein (C). (−)*, denotes the control, PMA-unstimulated cells
Stimulation with Phorbol Ester Does Not Induce SHP-1 Protein Expression in Malignant T Cells
Several groups have shown that expression of SHP-1 protein may be induced by cell stimulation with PMA and other agents. 38-40 To determine whether SHP-1 protein expression can be induced in the SHP-1-negative malignant T cells, we exposed the cells to PMA in the presence (Figure 3C) ▶ or absence (not shown) of a calcium ionophore, Ionomycin A. Only one cell line, C91PL, which constitutively expressed SHP-1 mRNA (Figure 3A) ▶ , was able to express SHP-1 protein on such stimulation. Five other SHP-1-negative lines including 2A, 2B, and Sez-4, failed to express SHP-1 protein. Furthermore, none of the lines was able to express SHP-1 mRNA on PMA stimulation (data not presented). These findings indicate that lack of SHP-1 in most T-cell lymphoma cells is not reversible by cell stimulation.
Sequence of the SHP-1 Gene Is Intact in T-Cell Lymphoma Cells
To determine whether loss of the SHP-1 expression results from structural abnormalities of genomic DNA, we analyzed the sequence of the coding region and splice junctions of the SHP-1 gene. Although the entire SHP-1 gene consists of 17 exons, we examined the structure of exons 2 to 16, because exon 1 is not expressed in hematopoietic cells and exon 17 contains untranslated sequences. 41 We evaluated nine T-cell lymphoma lines (PB-1, 2A, 2B, Sez-4, C91PL, ATL-2, C10MJ2, L540, and SUDHL-1) from which only two (PB-1 and C91PL) showed, respectively, constitutive and inducible expression of SHP-1 protein (Figure 3C) ▶ . Using the sequence deposited in GenBank and the one from normal PBMC as reference, we did not identify any DNA deletions or single-base mutations, either nonsense or missense, in any of the cell lines analyzed in either coding or splice junction regions (data not presented). Because PB-1 and C91PL expressed SHP-1 mRNA, albeit at a rather low concentration (Figure 3A) ▶ , we sequenced also cDNA from these two cell lines. In agreement with results of the genomic DNA analysis, we found no mutations in the cDNA. Next, we analyzed the sequence of SHP-1 promoter region to determine whether alterations therein may be responsible for lack of the SHP-1 transcript in malignant T cells. Transcription of the SHP-1 gene is processed by two alternative promoters. 41 Promoter 2 is active in hematopoietic cells, whereas promoter 1 is functional in cells of nonhematopoietic origin, such as epithelial cells. With this in mind, we sequenced 520 bp upstream of the transcription initiation site of promoter 2, a region that contains two GATA motifs as well as single CCAAT, AP-2, and Sp-1 sites. As was the case with the gene coding and splice junction regions, the sequence of promoter 2 in all 10 malignant T cell lines tested was identical to that of normal PBMC and the publicly available sequence.
Methylation of the SHP-1 Promoter Region in Malignant T Cells
Transcriptional gene silencing may result from methylation of cytosine residue in CpG islands at the 5′ end of the gene. 42 The region encompassing promoter 2 and exon 2 of SHP-1 is particularly rich in the CpG islands. 43 To determine whether methylation of the CpG island within this region may be responsible for lack of the SHP-1 expression in malignant T cells, we analyzed the region using cytosine methylation-sensitive restriction enzymes. To accomplish this, we digested the genomic DNA with either HpaII, EagI, or NaeI which have from five to one recognition sites within the promoter region of the SHP-1 gene and performed PCR amplification of the DNA fragment spanning the promoter 2/exon 3 region. As shown in Figure 4B ▶ , the 1,004-bp PCR product was still present in the HpaII-digested DNA from all 10 malignant T cell lines tested but not the control, normal PBMC and PHA blasts. Similar results were obtained when either EagI or NaeI methylation-sensitive enzymes were used instead of HpaII for DNA digestion (data not presented). Because these three restriction enzymes recognize different cytosine residues, this finding indicates an extensive methylation of the SHP-1 promoter in the malignant T cells. In addition, we also performed Southern blot analysis of the promoter region by using combination of the flanking, methylation-resistant MobI, and methylation-sensitive HpaII restriction enzymes and promoter 2-exon 3 region-specific probe. As expected from the PCR-based studies, HpaII digestion was effective in PBMC but not the T-cell lymphoma lines (2A, 2B, C10MJ2, ATL-2, and SUDHL-1; data not shown). The above results document extensive methylation of the CpG island within the SHP-1 promoter region and indicate that lack of SHP-1 expression in malignant T cells is because of the promoter methylation-mediated transcriptional silencing of the SHP-1 gene.
Figure 4.

Methylation of the SHP-1 promoter region in malignant T cells. T-cell lymphoma lines from cutaneous anaplastic large T cell lymphoma (PB-1, 2A, and 2B), HTLV-I+ adult-type T cell leukemia/lymphoma (C10MJ2 and ATL-2), ALK+ T cell lymphoma line (SUDHL-1), and two Hodgkin’s lymphoma cell lines (L540 and KMH2) were tested for methylation status of the 5′ CpG island-rich DNA within the SHP-1 promoter. PBMC and PHA blasts served as positive controls. A: PCR-amplified DNA fragment of the SHP-1 gene promoter region. B: SHP-1 promoter DNA treated with methylation-sensitive enzyme HpaII before the PCR amplification. C: SHP-1 promoter DNA from cell lines precultured with 5-deoxyazacytidine. D: HpaII-digested SHP-1 promoter DNA from 5-deoxyazacytidine-cultured cells. E: SHP-1 mRNA expression in untreated cells. F: Expression of control actin mRNA in untreated cells. G: SHP-1 mRNA expression in cells treated with 5-deoxyazacytidine detected by RT-PCR. H: Expression of SHP-1 mRNA in 5-deoxyazacytidine-treated malignant T cells detected by Northern blot (amount of SHP-1 mRNA in normal PBMC is used as a reference).
Demethylation of the SHP-1 Promoter Region Removes the Transcriptional Block
To determine whether removal of the methylation would confer on malignant cells sensitivity to the methylation-sensitive enzymes and result in expression of SHP-1 mRNA, we treated the cells with a demethylating agent, 5-deoxyazacytidine. As shown in Figure 4D ▶ , treatment with the drug rendered genomic DNA from the SHP-1 promoter region sensitive to HpaII digestion in all eight cell lines tested. Furthermore, exposure to 5-deoxyazacytidine induced transcription of the SHP-1 gene yielding the RT-PCR detectable SHP-1 mRNA in all of the cell lines (Figure 4G) ▶ . To determine better the amount of the expressed mRNA, we tested four of the 5-deoxyazacytidine-treated lines by Northern blot (Figure 4H) ▶ . The amount of SHP-1 mRNA in these drug-treated malignant cells was similar to the control, normal PBMC indicating complete reversal of the transcriptional inhibition.
Expression of the SHP-1 Protein Results in Decreased Phosphorylation of Jak3
We explored next if expression of the SHP-1 mRNA leads to expression of the SHP-1 protein. As shown in Figure 5A ▶ , the 2A T cell line displayed expression of the SHP-1 protein after treatment with 5-deoxyazacytidine. Three other lines, including the 2B line which is clonally related to the 2A cell line and shares with it a number of other features, 35 failed to express SHP-1 protein despite expressing SHP-1 mRNA. This indicates the presence of additional, posttranscriptional blocks in the SHP-1 synthesis pathway in some of the malignant T cell lines.
Figure 5.

Expression of SHP-1 protein leads to dephosphorylation of Jak3. T-cell lymphoma lines treated with 5-deoxycitidyne were examined for expression of SHP-1 protein (A) and tyrosine phosphorylation of IL-2R-associated Jak3 kinase before (−) and after (+) stimulation with IL-2 (B). Detection of Jak3 kinase expression (C) served as control for the latter experiment.
Because both 2A and 2B cell lines display a spontaneous, constitutive tyrosine phosphorylation of the IL-2R-associated Jak3 kinase 35 and SHP-1 acts by dephosphorylating receptor/kinase complexes including the IL-2R/Jak3 complex, 7 we examined an effect of 5-deoxyazacytidine treatment on the phosphorylation status of Jak3 in these two lines. There was a profound difference between the 2A and 2B lines which, respectively, expressed and failed to express SHP-1 protein in response to the drug. Whereas the 2B line continued to display maximal constitutive phosphorylation of Jak3 which could not by augmented by IL-2, the 2A line showed complete inhibition of the constitutive phosphorylation of Jak3 which was reversed to only a minor degree by cell stimulation with IL-2 (Figure 5B) ▶ . As expected, expression of the Jak3 protein was not affected by 5-deoxyazacytidine in either 2A or 2B (Figure 5C) ▶ . These findings indicate that expression of SHP-1 protein can result in dephosphorylation and, presumably, inactivation, of Jak3 kinase in malignant T cells.
Discussion
Here we report that lack of SHP-1 protein expression was frequently found in the T-cell lymphoma cells both cultured and derived directly from patients. The SHP-1 protein-negative T-cell lymphoma cells failed to express SHP-1 mRNA (see Table 1 ▶ for the data summary) even when stimulated, but did not harbor any mutations in the coding, splice junction, and promoter regions of the SHP-1 gene. However, these malignant T cells displayed extensive methylation of the SHP-1 gene promoter as demonstrated by resistance to digestion with methylation-sensitive restriction enzymes. This resistance was reversible by treatment of the cells with a demethylating agent 5-deoxyazacytidine. Furthermore, the drug treatment induced expression of SHP-1 mRNA, and, less frequently, SHP-1 protein. The expression of SHP-1 protein correlated with dephosphorylation of the IL-2R-associated Jak3. These results indicate that lack of SHP-1 expression may be frequent in various types of malignant T cell lymphomas and is primarily because of the extensive methylation of the SHP-1 gene promoter. In addition, these data provide additional evidence that lack of SHP-1 expression may play a role in malignant transformation of T cells by permitting constitutive activation of the IL-2R/Jak3 complex and, possibly, other cell-surface receptor complexes.
Table 1.
SHP-1: Methylation of the Gene Promoter and Expression of mRNA and Protein in Malignant T Cell Lymphoma Cell Lines (without and with Exposure to 5-Deoxyazacytidine)*
| Lymphoma type/ cell line 5-deoxyazacytidine | Promoter methylation | mRNA expression | Protein expression | |||
|---|---|---|---|---|---|---|
| − | + | − | + | − | + | |
| CTCL (MF and SS): | ||||||
| PB-1 | + | − | + | + | + | + |
| 2A | + | − | − | + | − | + |
| 2B | + | − | − | + | − | − |
| Sez-4 | − | − | ||||
| HTLV-I+(ATLL): | ||||||
| ATL-2 | + | − | − | + | − | |
| C91PL | + | − | ||||
| HUT102B | + | + | + | |||
| C10MJ2 | + | − | − | + | − | − |
| ALCL | ||||||
| SUDHL-1 | + | − | − | |||
| Hodgkin’s lymphoma (T cell) | ||||||
| L540 | + | − | − | + | − | − |
| HDLM | + | |||||
The failure to express SHP-1 in response to PMA +/− Ionomycin A stimulation in all but one malignant T cell line contrasted with the previous findings in normal and malignant germinal center B cells 40 and other types of cells 38,39 in which PMA and several other stimuli induced expression of the protein. This finding suggested the presence of a structural abnormality of the SHP-1 gene in the malignant T cells. SHP-1 and other inhibitory phosphatases such as PTEN 44-46 and PIP-B1 47 may be viewed as a novel class of oncosuppressor genes. Mutations are the major mechanism of inactivation in this broad category of genes. Accordingly, the best examples of the SHP-1 dysfunction which were identified in the motheaten (me/me) and viable motheaten (mev/mev) mice, are because of, respectively, null and phosphatase-domain deletional mutation of the SHP-1 gene. 17,18 Furthermore, structural analysis of PTEN phosphatase which is missing in a high proportion of gliomas, breast, prostate, and kidney cancers, and melanomas, revealed that its absence is also because of mutations. 44 However, our finding of intact SHP-1 gene indicated that a different mechanism has to be responsible for lack of SHP-1 expression in the malignant T cells.
Methylation of the 5′ CpG islands in the gene promoter region represents another mechanism of gene inactivation, alternative to mutation. Indeed, promoter methylation which results in transcriptional repression of the affected genes, has been identified in a number of oncosuppressors involved mainly in the control of cell cycle in a variety of epithelial and nonepithelial tumors. 48,49 To our knowledge, the reported methylation in this study of the SHP-1 promoter is the first example to demonstrate that this mechanism of transcriptional gene silencing affects phosphatase involved in a negative regulation of signal transduction. Noteworthy, the ability to reverse the transcriptional block and restore expression of functional SHP-1 protein (Figure 5) ▶ by treatment with demethylating agents may have implications for therapy of T cell lymphomas and other malignant tumors. Indeed, treatment with 5-deoxyazacytidine and its analogue, 5-azacytidine, has already been applied clinically with some success in acute myelogeneous leukemia 50-53 and myelodysplastic syndrome. 54,55 However, our finding that reversal of the promoter methylation leads invariably to expression of SHP-1 mRNA but much less frequently to expression of the SHP-1 protein, suggests the existence of additional, posttranscriptional blocks in the SHP-1 synthesis pathway in malignant T cells. These blocks may need to be identified before applying demethylating agents to therapy of T cell lymphomas and other malignancies on a larger scale.
The mechanism of the methylation-mediated gene inactivation is poorly understood but involves proteins such as MeCP1 and MeCP2 that bind to methyl-CpG and, therefore, contribute to inhibition of transcription. MeCP1 binds to DNA containing at least 12 symmetrically methylated CpGs, 56 whereas MeCP2 is relatively sequence-independent and binds to a single methylated CpG pair. 57 In addition to the MeCP-mediated repression, transcription can also be silenced by direct interference of site-specific CpG methylation with binding of transcription factors. Recent identification of the family of DNA methyltransferases 58 capable of de novo methylation and of CpG demethylase 59 indicates that imbalance between these two types of enzymes with the opposite function may play a role in carcinogenesis. 60,61
There are several possible explanations for the persistence of constitutive phosphorylation of the IL-2R/Jak3 complex in a few malignant T cell lines despite co-expression by these cells of the SHP-1 protein. 7,36 First, malignant T cells may produce cytokine which interacts with IL-2R. This interaction may lead to a steady, repetitive rephosphorylation of the IL-2R complex even in the presence of SHP-1. In fact, one such double-positive line, HUT102B, secretes large amount of IL-15. However, there is so far no convincing evidence to support the autocrine effect of IL-15 in these cells because a blocking anti-IL-15 antibody had no effect on their growth rate. 62 Alternatively, and perhaps more likely, the expressed SHP-1 protein is rendered nonfunctional in such malignant T cells. SHP-1 usually interacts via its two SH2 domains with the target protein by recognizing phosphotyrosine residues surrounded by specific sequence motifs; 15,16 the exact mechanism of SHP-1 interaction with the IL-2R complex is currently less understood. 7 Although we found no mutations in the SHP-1 gene sequence, it is possible that mutation in IL-2R or Jak3 may preclude proper interaction of SHP-1 with the IL-2R complex. Accordingly, mutations that affect the cytoplasmic domain of receptor for erythropoietin lead to the receptor hypersensitivity which is resistant to the SHP-1-mediated inhibition. 6,63 Finally, SHP-1 may not be available to IL-2R by being tightly bound to other types of receptors such as members of the inhibitory-receptor superfamily. 14 Further studies are needed to resolve the apparent paradox of the co-expression of SHP-1 protein and constitutively activated IL-2R complex in the malignant T cells.
Footnotes
Address reprint requests to Mariusz A. Wasik, M.D., 7.106 Founders Bldg., Department of Pathology and Laboratory Medicine, University of Pennsylvania Medical Center, 3400 Spruce St., Philadelphia, PA 19104. E-mail: wasik@mail.med.upenn.edu.
Supported in part by grants from National Cancer Institute (CA76627) and the University of Pennsylvania Research Foundation.
References
- 1.Plutzky J, Neel BG, Rosenberg RD: Isolation of a src homology 2-containing tyrosine phosphatase. Proc Natl Acad Sci USA 1992, 89:1123-1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen S-H, Bastien L, Posner BI, Chre′tien P: A protein-tyrosine phosphatase with sequence similarity to the SH2 domain of the protein-tyrosine kinases. Nature 1991, 352:736-739 [DOI] [PubMed] [Google Scholar]
- 3.Yi T, Cleveland JL, Ihle JN: Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12–p13. Mol Cell Biol 1992, 12:836-846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthews RJ, Bowne DB, Flores E, Thomas ML: Characterization of hematopoietic intracellular protein tyrosine phosphatases: description of a phosphatase containing an SH2 domain and another enriched in proline-, glutamic acid-, serine-, and threonine-rich sequences. Mol Cell Biol 1992, 12:2396-2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ulyanova T, Blasioli J, Thomas ML: Regulation of cell signaling by the protein tyrosine phosphatases, CD-45 and SHP-1. Immunol Res 1997, 16:101-113 [DOI] [PubMed] [Google Scholar]
- 6.Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF: Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of Jak2 and termination of proliferative signals. Cell 1995, 80:729-738 [DOI] [PubMed] [Google Scholar]
- 7.Migone TS, Calacano NA, Migone TS, Cacalano NA, Taylor N, Yi T, Waldmann TA, Johnston JA: Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc Natl Acad Sci USA 1998, 95:3845-3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haque SJ, Harbor P, Tabrizi M, Yi T, Williams BR: Protein-tyrosine phosphatase Shp-1 is a negative regulator of IL-4- and IL-13-dependent signal transduction. J Biol Chem 1998, 273:33893-33896 [DOI] [PubMed] [Google Scholar]
- 9.Yeung Y-G, Berg KL, Pixley FJ, Angeletti RH, Stanley ER: Protein tyrosine phosphatase-1C is rapidly phosphorylated in tyrosine in macrophages in response to colony stimulating factor-1. J Biol Chem 1992, 267:23447-23450 [PubMed] [Google Scholar]
- 10.Yi T, Ihle JN: Association of hematopoietic cell phosphatase with c-Kit after stimulation with c-Kit ligand. Mol Cell Biol 1993, 13:3350-3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, Thomas ML, Beier DR: Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell 1993, 73:1445-1454 [DOI] [PubMed] [Google Scholar]
- 12.Tsui HW, Siminovitch KA, de Souza L, Tsui FW: Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet 1993, 4:124-129 [DOI] [PubMed] [Google Scholar]
- 13.Wu Y, Nadler MJ, Brennan LA, Gish GD, Timms JF, Fusaki N, Jongstra-Bilen J, Tada N, Pawson T, Wither J, Neel BG, Hozumi N: The B-cell transmembrane protein CD72 binds to and is an in vivo substrate of the protein tyrosine phosphatase SHP-1. Curr Biol 1998, 10:1009-1017 [DOI] [PubMed] [Google Scholar]
- 14.Long EO: Regulation of immune responses through inhibitory receptors. Annu Rev Immunol 1999, 17:875-904 [DOI] [PubMed] [Google Scholar]
- 15.Burshtyn DN, Yang W, Yi T, Long EO: A novel phosphotyrosine motif with a critical amino acid at position-2 for the SH2 domain-mediated activation of the tyrosine phosphatase SHP-1. J Biol Chem 1997, 272:13066-13072 [DOI] [PubMed] [Google Scholar]
- 16.Burshtyn DN, Lam AS, Weston M, Gupta N, Warmerdam PA, Long EO: Conserved residues amino-terminal of cytoplasmic tyrosines contribute to the SHP-1-mediated inhibitory function of killer cell Ig-like receptors. J Immunol 1999, 162:897-902 [PubMed] [Google Scholar]
- 17.Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, Thomas ML, Beier DR: Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell 1993, 73:1445-1454 [DOI] [PubMed] [Google Scholar]
- 18.Shultz LD, Rajan TV, Greiner DL: Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotech 1997, 15:302-307 [DOI] [PubMed] [Google Scholar]
- 19.Leonard WJ, O’Shea JJ: JAKS AND STATS: biological implications. Annu Rev Immunol 1998, 16:293-322 [DOI] [PubMed] [Google Scholar]
- 20.Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, McBride OW, Leonard WJ: Interleukin-2 receptor γ chain mutations result in X-linked severe combined immunodeficiency in humans. Cell 1993, 73:147-157 [DOI] [PubMed] [Google Scholar]
- 21.Ohbo K, Suda T, Hashiyama M, Mantani A, Ikebe M, Moriyama M, Nakamura M, Katsuki M, Takahashi K, Yamamura K, Sugamura K: Modulation of hematopoiesis in mice with a truncated mutant of the interleukin-2 receptor γ chain. Blood 1996, 87:956-967 [PubMed] [Google Scholar]
- 22.DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K: Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor γ chain. Proc Natl Acad Sci USA 1995, 92:377-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, Drago J, Noguchi M, Grinberg A, Bloom ET, Paul WE, Katz SI, Love PI, Leonard WJ: Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity 1995, 2:223-238 [DOI] [PubMed] [Google Scholar]
- 24.Johnston JA, Kawamura M, Kirken RA, Chen YQ, Blake TB, Shibuya K, Ortaldo JR, McVicar DW, O’Shea JJ: Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2. Nature 1994, 370:151-153 [DOI] [PubMed] [Google Scholar]
- 25.Russell SM, Johnston JA, Noguchi M, Kawamura M, Bacon CM, Friedmann M, Berg M, McVicar DW, Witthun BA, Silvennoinen OAS, Goldman AS, Schmaisteg FC, Ihle JN, O’Shea JJ, Leonard WJ: Interaction of IL-1R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science 1994, 266:1042-1045 [DOI] [PubMed] [Google Scholar]
- 26.Beadling C, Guschin D, Witthuhn BA, Ziemiecki A, Ihle JN, Kerr IM, Cantrell DA: Activation of JAK kinases and STAT proteins by interleukin-2 and interferon alpha, but not the T cell antigen receptor, in human T lymphocytes. EMBO J 1994, 13:5605-5615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujii H, Nakagawa Y, Schindler U, Kawahara A, Mori H, Gouilleux F, Groner B, Ihle JN, Minami Y, Miyazaki T, Taniguchi T: Activation of Stat5 by interleukin 2 requires a carboxyl-terminal region of the interleukin 2 receptor beta chain but is not essential for the proliferative signal transmission. Proc Natl Acad Sci USA 1995, 92:5482-5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen M, Svejgaard A, Skov S, Odum N: Interleukin-2 induces tyrosine phosphorylation and nuclear translocation of STAT3 in human T lymphocytes. Eur J Immunol 1994, 24:3082-3086 [DOI] [PubMed] [Google Scholar]
- 29.Macchi P, Villa A, Gillani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O’Shea JJ, Vezzoni P, Notarangelo LD: Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature 1995, 377:65-68 [DOI] [PubMed] [Google Scholar]
- 30.Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, Migone TS, Noguchi M, Markert ML, Buckley RH, O’Shea JJ, Leonard WJ: Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science 1995, 270:797-800 [DOI] [PubMed] [Google Scholar]
- 31.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ: Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science 1995, 270:794-797 [DOI] [PubMed] [Google Scholar]
- 32.Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Dohrty PC, Grosveld GC, Ihle JN: Defective lymphoid development in mice lacking Jak3. Science 1995, 270:800-802 [DOI] [PubMed] [Google Scholar]
- 33.Migone TS, Lin JX, Cereseto A, Mulloy JC, O’Shea JJ, Franchini G, Leonard WJ: Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 1995, 269:79-81 [DOI] [PubMed] [Google Scholar]
- 34.Xu X, Kang S-H, Heidenreich O, Okerholm M, O’Shea JJ, Nerenberg MI: Constitutive activation of different Jak tyrosine kinases in human T cell leukemia virus type 1 (HTLV-1) Tax protein or virus-transformed cells. J Clin Invest 1995, 96:1548-1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q, Nowak I, Vonderheid EC, Rook AH, Kadin ME, Nowell PC, Shaw LM, Wasik MA: Activation of Jak/STAT proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic large T-cell lymphoma and Sezary syndrome. Proc Natl Acad Sci USA 1996, 93:9148-9153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Q, Lee B, Korecka M, Li G, Weyland C, Eck S, Gessain A, Arima N, Shaw L, Luger S, Kamoun M, Wasik MA: Differences in phosphorylation of the IL-2R associated Jak/STAT proteins between HTLV-I(+), IL-2-independent and IL-2-dependent cell lines and uncultured leukemic cells from patients with adult T-cell lymphoma/leukemia (ATLL). Leuk Res 1999, 23:373-384 [DOI] [PubMed] [Google Scholar]
- 37.Drexler HG: Recent results on the biology of Hodgkin and Reed-Sternberg cells. II. Continuous cell lines. Leuk Lymphoma 1993, 9:1-25 [DOI] [PubMed] [Google Scholar]
- 38.Uchida T, Matozaki T, Matsuda K, Suzuki T, Matozaki S, Nakano O, Wada K, Konda Y, Sakamoto C, Kasuga M: Phorbol ester stimulates the activity of a protein tyrosine phosphatase containing SH2 domains (PTP1C) in HL-60 leukemia cells by increasing gene expression. J Biol Chem 1993, 268:11845-11850 [PubMed] [Google Scholar]
- 39.Zhao Z, Shen SH, Fischer EH: Phorbol ester-induced expression, phosphorylation, and translocation of protein-tyrosine-phosphatase 1C in HL-60 cells. Proc Natl Acad Sci USA 1994, 91:5007-5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delibrias CC, Floettmann JE, Rowe M, Fearon DT: Down-regulated expression of SHP-1 in Burkitt lymphomas and germinal center B lymphocytes. J Exp Med 1997, 186:1575-1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banville D, Stocco R, Shen SH: Human protein tyrosine phosphatase 1C (PTPN6) gene structure: alternate promoter usage and exon skipping generate multiple transcripts. Genomics 1995, 27:165-173 [DOI] [PubMed] [Google Scholar]
- 42.Kundu TK, Rao MR: CpG islands in chromatin organization and gene expression. J Biochem 1999, 125:217-222 [DOI] [PubMed] [Google Scholar]
- 43.Asimakopoulos FA, Hinshelwood S, Gilbert JG, Delibrias CC, Gottgens B, Fearon DT, Green AR: The gene encoding hematopoietic cell phosphatase (SHP-1) is structurally and transcriptionally intact in polycythemia vera. Oncogene 1997, 14:1215-1222 [DOI] [PubMed] [Google Scholar]
- 44.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 1947, 275:1943. [DOI] [PubMed] [Google Scholar]
- 45.Cantley B, Neel BG: New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA 1999, 13:4240-4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP: Impaired Fas response and autoimmunity in Pten+/mice. Science 1999, 285:2122-2125 [DOI] [PubMed] [Google Scholar]
- 47.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan C-C, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP: Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283:1544-1548 [DOI] [PubMed] [Google Scholar]
- 48.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP: Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 1998, :141-196 [PubMed] [Google Scholar]
- 49.Jones PA, Laird PW: Cancer epigenetics comes of age. [Review] [74 refs]. Nat Genet 1999, 21:163-167 [DOI] [PubMed] [Google Scholar]
- 50.Gattei V, Aldinucci D, Petti MC, Da Ponte A, Zagonel V, Pinto A: In vitro and in vivo effects of 5-aza-2′-deoxycytidine (Decitabine) on clonogenic cells from acute myeloid leukemia patients. Leukemia 1993, 7(Suppl 1):S42-S48 [PubMed] [Google Scholar]
- 51.Petti MC, Mandelli F, Zagonel V, De Gregoris C, Merola MC, Latagliata R, Gattei V, Fazi P, Monfardini S, Pinto A: Pilot study of 5-aza-2′-deoxycytidine (Decitabine) in the treatment of poor prognosis acute myelogenous leukemia patients: preliminary results. Leukemia 1993, 7(Suppl 1):S36-S41 [PubMed] [Google Scholar]
- 52.Willemze R, Suciu S, Archimbaud E, Muus P, Stryckmans P, Louwagie EA, Berneman Z, Tjean M, Wijermans P, Dohner H, Jehn U, Labar B, Jaksic B, Dardenne M, Zittoun R: A randomized phase II study on the effects of 5-aza-2′-deoxycytidine combined with either amsacrine or idarubicin in patients with relapsed acute leukemia: an EORTC Leukemia Cooperative Group phase II study (06893). Leukemia 1997, 11(Suppl 1):S24-S27 [PubMed] [Google Scholar]
- 53.Willemze R, Archimbaud E, Muus P: Preliminary results with 5-aza-2′-deoxycytidine(DAC)-containing chemotherapy in patients with relapsed or refractory acute leukemia. The EORTC Leukemia Cooperative Group. Leukemia 1993, 7(Suppl 1):S49-S50 [PubMed] [Google Scholar]
- 54.Zagonel V, Lo Re G, Marotta G, Babare R, Sardeo G, Gattei V, De Angelis V, Monfardini S, Pinto A: 5-Aza-2′-deoxycytidine (Decitabine) induces trilineage response in unfavourable myelodysplastic syndromes. Leukemia 1993, 7(Suppl 1):S30-S35 [PubMed] [Google Scholar]
- 55.Silverman LR, Holland JF, Weinberg RS, Alter BP, Davis RB, Ellison RR, Demakos EP, Cornell CJ, Jr, Carey RW, Schiffer C, Frei E, McIntyre OR: Effects of treatment with 5-azacytidine on the in vivo and in vitro hematopoiesis in patients with myelodysplastic syndromes. Leukemia 1993, 7(Suppl 1):S21-S29 [PubMed] [Google Scholar]
- 56.Meehan RR, Lewis JD, Mckay S, Kleiner EL, Bird AP: Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 1989, 58:499-507 [DOI] [PubMed] [Google Scholar]
- 57.Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A: Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69:905-914 [DOI] [PubMed] [Google Scholar]
- 58.Okano M, Xie S, Li E: Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 1998, 19:219-220 [DOI] [PubMed] [Google Scholar]
- 59.Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M: A mammalian protein with specific demethylase activity for mCpG DNA [see comments]. Nature 1999, 397:579-583 [DOI] [PubMed] [Google Scholar]
- 60.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA: The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 1999, 27:2291-2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Marzo AM, Marchi VL, Yang ES, Veeraswamy R, Lin X, Nelson WG: Abnormal regulation of DNA methyltransferase expression during colorectal carcinogenesis. Cancer Res 1999, 59:3855-3860 [PubMed] [Google Scholar]
- 62.Takemoto S, Mulloy JC, Cereseto A, Migone TS, Patel BK, Matsuoka M, Yamaguchi K, Takatsuki K, Kamihira S, White JD, Leonard WJ, Waldmann T, Franchini G: Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc Nat Acad Sci USA 1997, 94:13897-13902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de la Chapelle A, Traskelin AL, Juvonen E: Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Nat Acad Sci USA 1993, 90:4495-4499 [DOI] [PMC free article] [PubMed] [Google Scholar]