

Abstract
Deposition of amyloid β-peptide (Aβ) in cerebral vessel walls (cerebral amyloid angiopathy, CAA) is very frequent in Alzheimer’s disease and occurs also as a sporadic disorder. Here, we describe significant CAA in addition to amyloid plaques, in aging APP/Ld transgenic mice overexpressing the London mutant of human amyloid precursor protein (APP) exclusively in neurons. The number of amyloid-bearing vessels increased with age, from ∼10 to >50 per coronal brain section in APP/Ld transgenic mice, aged 13 to 24 months. Vascular amyloid was preferentially deposited in arterioles and ranged from small focal to large circumferential depositions. Ultrastructural analysis allowed us to identify specific features contributing to weakening of the vessel wall and aneurysm formation, ie, disruption of the external elastic lamina, thinning of the internal elastic lamina, interruption of the smooth muscle layer, and loss of smooth muscle cells. Biochemically, the much lower Aβ42:Aβ40 ratio evident in vascular relative to plaque amyloid, demonstrated that in blood vessel walls Aβ40 was the more abundant amyloid peptide. The exclusive neuronal origin of transgenic APP, the high levels of Aβ in cerebrospinal fluid compared to plasma, and the specific neuroanatomical localization of vascular amyloid strongly suggest specific drainage pathways, rather than local production or blood uptake of Aβ as the primary mechanism underlying CAA. The demonstration in APP/Ld mice of rare vascular amyloid deposits that immunostained only for Aβ42, suggests that, similar to senile plaque formation, Aβ42 may be the first amyloid to be deposited in the vessel walls and that it entraps the more soluble Aβ40. Its ability to diffuse for larger distances along perivascular drainage pathways would also explain the abundance of Aβ40 in vascular amyloid. Consistent with this hypothesis, incorporation of mutant presenilin-1 in APP/Ld mice, which resulted in selectively higher levels of Aβ42, caused an increase in CAA and senile plaques. This mouse model will be useful in further elucidating the pathogenesis of CAA and Alzheimer’s disease, and will allow testing of diagnostic and therapeutic strategies.
The pathological hallmarks of Alzheimer’s disease (AD) are amyloid β-peptide (Aβ) containing senile plaques and neurofibrillary tangles. In addition to these parenchymal lesions, 90% of AD patients show deposition of amyloid in the walls of meningeal and cerebral blood vessels. 1 However, there seems to be two extreme forms of cerebral amyloid angiopathy (CAA), one, where severe CAA is isolated from parenchymal AD lesions and, the other, where abundant parenchymal senile plaques occur in the absence of CAA. 1,2 Patients with hereditary cerebral hemorrhage with amyloidosis–Dutch type, an autosomal-dominant severe form of CAA caused by a point mutation in amyloid precursor protein (APP) do not seem to develop significantly more amyloid plaques or neurofibrillary tangles than the healthy elderly. 3 CAA also occurs as a sporadic disorder in ∼30% of people >60 years of age. 1 Cerebrovascular amyloid is deposited most commonly in meningeal and cortical arteries and arterioles, and less frequently in veins and capillaries. 4-6 Vascular amyloid deposition leads to degeneration of the vessel wall and aneurysm formation, 7,8 and may be responsible for 10 to 15% of hemorrhagic strokes in the elderly. 1,9 Many aspects of the pathogenesis of CAA, and its role in the pathogenesis of AD, are still unclear, but the fact that plaques and vascular amyloid both contain Aβ, proteolytically derived from APP, suggests that essential factors in the pathogenesis of CAA and senile plaques are similar. However, in vascular amyloid Aβ40 is the more abundant species, whereas in senile plaques Aβ42 is most abundant. 10-13
A paucity of animal models has hindered the experimental analysis of CAA. In the past, studies have primarily been based on models of naturally occurring CAA in aged nonhuman primates and dogs. 14 Several transgenic mouse models overexpressing mutant APP develop amyloid plaques, but, so far, only one appears to develop significant vascular amyloid. 15 Here, we report that, in addition to amyloid plaques, significant deposition of amyloid in blood vessels occurs with aging in the FVB/N APP/Ld and F1 (FVB/N × C57BL6) APP/Ld mice, overexpressing the London mutant of human APP under control of the neuron-specific murine thy1-gene promoter. 16 Our results suggest that neuronal production of Aβ and aging are important common pathogenetic factors in the formation of amyloid plaques and CAA. However, analysis of different areas of cerebral cortex demonstrated that other independent factors determined the local deposition of amyloid in brain parenchyma and vessels. Also, biochemical analysis of vascular amyloid in pial arterioles and plaque amyloid in neocortex showed that vascular and plaque amyloid differed considerably in the ratio of Aβ42:Aβ40, with much higher relative levels of Aβ40 in vascular amyloid. The type of vessels affected and the pattern of amyloid deposition in this model closely reproduced the pathology of CAA in patients. The effects of amyloid on vessel walls and smooth muscle, including the pathogenetic mechanisms that led to aneurysm formation, are described in detail. Exclusive neuronal expression of the transgene in the APP/Ld mice seemed sufficient to recapitulate most of the pathogenetic features of human CAA very closely. These results suggest transport of Aβ from neurons to blood vessels and drainage pathways rather than local production or uptake from blood as a primary factor in the pathogenesis of CAA. Incorporation of mutant presenilin-1 (PS1) in this model resulted in increased vascular amyloid deposition.
Materials and Methods
Animals Used for Histological, Quantitative, and Ultrastructural Analysis
Generation of the APP/Ld (thy1-APP-V717I) 16 and PS1/Mut (thy1-PS1-A246E; ID, DM, CK, IL, JVD, FC, HV, FVL, unpublished data) transgenic mice is described elsewhere. A total of 35 transgenic APP/Ld mice (21 FVB/N APP/Ld mice in a FVB/N background and 14 F1-APP/Ld mice in a hybrid FVB/N × C57BL6 genetic background) aged 15 to 24 months were used for histological and quantitative examination. Five 24-month-old wild-type (WT) FVB/N mice and five 24-month-old WT F1 littermates were used as controls. Ultrastructural and immunoelectron analyses were performed on six transgenic mice from the FVB/N APP/Ld group (20 to 24 months old) and two age-matched FVB/N WT mice. Three additional FVB/N APP/Ld mice 20 to 24 months of age were used for dissection of the arterial circle of Willis and its branches. To evaluate the effect of mutant PS1 on the amyloid deposition in plaques and vessels four double-transgenic FVB/N APP/LdxPS1/Mut mice (13.5 months old) were compared to six age-matched FVB/N APP/Ld mice. All transgenic mice were hemizygous for the APP/Ld (and/or PS1/Mut) transgene.
Histological and Quantitative Analysis
Mice were killed with chloroform and immediately decapitated. The left cerebral hemisphere was snap-frozen and stored at −70°C. The right cerebral hemisphere, brain stem, cerebellum, spinal cord, and viscera (lungs, heart, liver, spleen, and kidneys) were immersion-fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) overnight and were used for histological and quantitative analyses. Coronal vibratome sections were cut from the occipital two-thirds of the right hemisphere; the frontal part was embedded in paraffin and used for microtome sections. Brain stem, cerebellum, spinal cord, and viscera were used for vibratome sections. Thioflavine-S, Perls’ Prussian blue, and Congo red stainings were performed on vibratome or microtome sections according to standard protocols. Immunohistochemistry with the antibody FCA-18 (polyclonal, 1/100), which recognizes Aβ, was done on free-floating vibratome sections according to previously published protocols by using the avidin-biotin-peroxidase complex method with diaminobenzidine as chromogen. 17
Quantitative analysis of vascular and plaque amyloid in the brain was performed on Thioflavine-S-stained vibratome sections. Two well-defined coronal sections at bregma −1.94 mm and −3.52 mm, respectively, were selected for quantification of the number of blood vessels with amyloid and for determining the amyloid load in vessels and plaques. 18 The section at bregma −1.94 mm contained neocortex, hippocampus, amygdala, striatum, and thalamus; neocortex, hippocampus, subiculum, and brain stem were present in the section at bregma −3.52 mm. For quantification of amyloid load in vessels and plaques in the subiculum, four serial sections were used between bregma −3.40 and −3.52 mm. Aβ-immunoreactive plaque load was determined on a section at bregma −3.40 mm. Images (magnification, ×200) from these sections were collected from a 3CCD color video camera and analyzed with appropriate software (AIS/C; Imaging Research, St. Catherine, Ontario, Canada). The surface of individual amyloid deposits in vessels or plaques was measured. The total amyloid load in vessels or plaques was expressed as a percentage of the total surface of a complete section or of a neuroanatomical region.
To dissect the arterial circle of Willis, the brain was fixed overnight in 4% paraformaldehyde in PBS, and then the arterial circle of Willis and its branches were carefully separated from the brain under a dissection microscope. Thioflavine-S staining was performed and the circle of Willis was mounted in Mowiol (Calbiochem-Novabiochem, La Jolla, CA) on a glass slide.
Ultrastructural and Immunoelectron Analysis
For transmission electron microscopy, areas of cerebral neocortex with covering leptomeninges and hippocampus were excised from 40-μm-thick vibratome sections and fixed in 4% paraformaldehyde and 0.1% glutaraldehyde in PBS. After fixation with OsO4, tissue samples for routine electron microscopy were embedded in epon or LR-white (Agar Scientific, Stansted, UK). For immunogold labeling, ultrathin sections from LR-white-embedded tissues on formvar-coated nickel grids were treated with 6% sodium metaperiodate for 10 minutes and with 5% normal goat serum in PBS for 30 minutes. This was followed by incubating the sections with specified monoclonal or polyclonal antibody in 1% normal goat serum in PBS at a dilution of 1/50 for 2 hours. After washing, 10-nm colloidal gold-tagged secondary antibody in Tris-buffered saline (goat anti-mouse and goat ant-rabbit; British Biocell, Cardiff, UK) was applied for 1 hour. Then, after washing, sections were stained with lead citrate and uranyl acetate. Control sections were stained following the same procedure, but with omission of the primary antibody. The following antibodies were used: FCA-18, 17 NCL-βA (monoclonal; Novocastra, Newcastle, UK), WO2, 19 and JRF/Aβtot/14 (monoclonal-specific for human Aβ; Janssen Research Foundation (JRF), Beerse, Belgium) against Aβ; FCA-40 (polyclonal) 17 and JRF/cAβ40/10 (monoclonal; JRF) against Aβ40; FCA-42 (polyclonal) 17 and JRF/cAβ42/8 (monoclonal; JRF) against Aβ42; and anti-glial fibrillary acidic protein (polyclonal; DAKO, Glostrup, Denmark).
Expression of Human APP
Expression of human APP was examined by immunohistochemistry and in situ hybridization in four hemizygous FVB/N APP/Ld mice 2 to 3 months of age and in three age-matched WT mice. Mice were anesthetized and perfused with 4% paraformaldehyde in PBS. Brains were postfixed overnight and manually embedded in paraffin. Immunohistochemistry with the antibody 1G5 (monoclonal, dilution 1/50; Athena Neurosciences, San Francisco, CA) specific for human APP was performed by using the avidin-biotin-peroxidase complex method. Sense and anti-sense human APP RNA probes were synthesized from a pGEM-T vector (Promega, Madison, WI) in which a 263-bp polymerase chain reaction fragment from the human APP gene was cloned. The sequences of the forward and reverse primer were respectively 5′-GACTCATGGTGGGCGGTGTTGT-3′ and 5′-CCGATGGGTAG-TGAAGC-AATGGTT-3′. The plasmid was linearized with either NotI or SphI and transcribed with T7 and SP6 RNA polymerase, respectively, in the presence of [33P]UTP. After rehydration, sections for in situ hybridization were digested with proteinase K (20 μg/ml), postfixed in 4% paraformaldehyde, and treated with 0.25% acetic anhydride in 0.1 mol/L triethanolamine-HCL. Sections were hybridized overnight in 50% deionized formamide, 0.3 mol/L NaCl, 20 mmol/L Tris-HCl (pH 7.6), 5 mmol/L ethylenediaminetetraacetic acid (pH 8.0) with 10% dextran sulfate, 1× Denhardt’s solution, 0.5 mg/ml yeast RNA, and 100 mmol/L dithiothreitol and supplemented with the appropriate probe. After stringency washes and ribonuclease-A treatment, sections were dehydrated, dipped in photographic emulsion (LM-1; Amersham, Buckinghamshire, UK), and exposed for 3 weeks.
Determination of Cerebral Blood Flow (CBF)
Mice were anesthetized with urethane (1.4 mg/g body weight i.p.), tracheotomized, intubated, and artificially ventilated (small animal respirator, model 683; Harvard Apparatus, Holliston, MA) with a tidal volume of 1.5 ml at a respiratory rate of 100/minutes. Core body temperature was measured and maintained at 36.5 to 37.5°C. CBF was determined by laser-Doppler flowmetry (ML192 dual blood flow meter; AD Instruments, Castle Hill, Australia). Because the mouse skull is very thin, the laser-Doppler probe was placed directly over the intact parietal bone after reflection of the skin and subcutaneous tissue. The probe was positioned stereotaxically 1.5 mm lateral to the midline and 1.5 mm caudal from bregma. Twenty minutes after the start of ventilation basal CBF was measured. Hypercapnia was induced for 2 minutes by inhalation of 7% CO2 balanced with O2 and N2. The data were stored on a computer and analyzed using MacLab/8 data acquisition and analysis system (AD Instruments). In this experiment four FVB/N APP/Ld mice (20 to 24 months old) were compared to five age-matched FVB/N WT mice. After the measurements, the animals were perfused with 4% paraformaldehyde in PBS and their brains were analyzed by Thioflavine-S staining of vibratome sections to verify the presence and extent of CAA in the transgenic mice.
Detection of Aβ in Cerebrospinal Fluid (CSF) and Plasma
CSF and plasma were collected from four hemizygous FVB/N APP/Ld mice (4 months of age) anesthetized with urethane. CSF was collected with a fine glass pipette from the surgically exposed cisterna and immediately frozen. Analysis was performed only on CSF without blood. Blood samples from the same mice were taken by transcardial puncture. Plasma was obtained from the blood samples after centrifugation (3,000 rpm) at 4°C for 10 minutes. Protein electrophoresis was performed on 4 to 12% Nu polyacrylamide gel electrophoresis gels (MES-sodium dodecyl sulfate buffer; Novex, San Diego, CA). Samples corresponding to 5 μl of CSF and plasma were denatured and reduced, loaded, electrophoresed, and transferred to nitrocellulose membrane. Incubation with mouse monoclonal antibody specific for the N-terminal region of Aβ (WO2) was followed by horseradish peroxidase-labeled goat anti-mouse secondary antibody (BioRad, Hercules, CA) and chemiluminescence (ECL; Amersham). For quantification 300, 100, 30, and 10 pg of synthetic peptide (Aβ40; Peninsula, San Carlos, CA) were used in the same Western blot analysis.
Determination of the Ratio of Insoluble Aβ42 to Aβ40 in Neocortex and Meningeal Blood Vessels
For this experiment, nine snap-frozen left hemispheres from 24-month-old hemizygous F1-APP/Ld mice (F1-APP/Ld group) were transferred to cold saline and the leptomeninges including the leptomeningeal blood vessels covering the superolateral cerebral surface were carefully separated from the cortex. Then, a small sample (7 to 9 mg) of inferolateral temporooccipital neocortex was dissected from the cerebral hemisphere. Great care was taken to obtain leptomeninges free of cortex and cortex free of leptomengeal blood vessels. Aβ was extracted from leptomeningeal and cortical samples in 5.0 mol/L guanidinium-chloride (pH 8) for 3 hours at room temperature. Dilutions were used to measure levels of insoluble (amyloid-associated) Aβ40 and Aβ42 by sandwich enzyme-linked immunosorbent assays using, respectively, JRF/cAβ40/10 and 21F12 (monoclonal; Innogenetics, Gent, Belgium) as capture antibodies, and JRF/cAβtot/14 and 3D6 (monoclonal; Innogenetics) as detecting antibodies. Both enzyme-linked immunosorbent assays are human-specific. Standard curves of Aβ40 (Bachem, Torrence, CA) and Aβ42 (Innogenetics) were based on dilutions of the peptides in guanidinium-chloride-containing buffer. The ratio of Aβ42 to Aβ40 was calculated for leptomeningeal blood vessels and neocortex.
Statistical Analysis
Statistical analysis was conducted with the SAS 6.12 computer package (SAS, Cary, NC). The Wilcoxon/Mann-Whitney test was used to compare plaque load or vascular amyloid between different age groups. To explore the relation between plaque load and vascular amyloid the Spearman correlation was calculated. Because age might affect this correlation also a partial correlation was performed with age held constant. To compare vascular and amyloid load in the neocortex, hippocampus, and subiculum analysis of variance for repeated measurements was performed. The Wilcoxon signed-rank test was used to compare the difference in ratio of Aβ42:Aβ40 between vascular amyloid in pial arterioles and amyloid in plaques. The reported P values are two-sided and were considered statistically significant when P < 0.05. Data in the text and figures are expressed as mean ± SE.
Results
Plaque Load in Aged APP/Ld Mice
All APP/Ld mice used in this study (n = 35; 15 to 24 months old) contained neuritic as well as diffuse plaques in their brain (Figures 1 and 2) ▶ ▶ . Plaque formation was first and most obvious in the subiculum and entorhinal cortex, but was also present in the neocortex, hippocampus, and thalamus. Rare small plaques were seen in the striatum and brain stem. In the oldest APP/Ld mice, small amyloid depositions were present in some sections through the spinal cord, but the cerebellum was always free of plaques. Estimations of the plaque load varied strongly with the staining technique used. Thioflavine-S stained only fibrillary Aβ containing amyloid cores of neuritic plaques (Figure 1, A and E) ▶ , which occupied 0.17 ± 0.03% (Figure 3A ▶ ; FVB/N APP/Ld mice) or 0.32 ± 0.04% (Figure 3D ▶ ; F1-APP/Ld mice) of the cortical surface in the oldest APP/Ld groups. By immunohistochemical staining for Aβ, neuritic as well as diffuse plaques were detected (Figure 2B) ▶ , resulting in an estimated total plaque load of 3.6 ± 0.7% (Figure 3C ▶ ; 20 to 24-month-old FVB/N APP/Ld mice). This demonstrates that APP/Ld mice, as Alzheimer’s patients, 20 developed diffuse plaques as a quantitatively important component.
Figure 1.
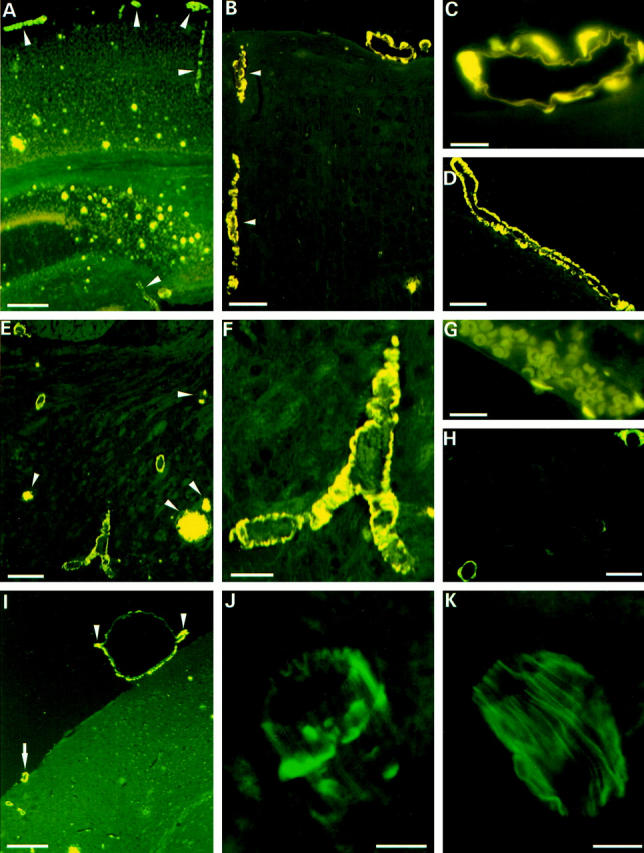
Thioflavine-S stainings of vascular amyloid in APP/Ld mice. A: Vibratome section from the brain of a 24-month-old APP/Ld mouse showing fluorescence in vessel walls (arrowheads) and plaque cores. B: Microtome section from a 24-month-old APP/Ld mouse demonstrating prominent fluorescence in vessel walls. A cortical arteriole is indicated by arrowheads. C: High magnification of leptomeningeal arteriole with amyloid depositions. D: Longitudinal section through artery with CAA. E: Thalamus of 20-month-old transgenic animal demonstrating several blood vessels with amyloid. Plaques are indicated by arrowheads. F: High magnification of thalamic blood vessel loaded with amyloid. G: Leptomeningeal vein with small focal amyloid depositions. H: Three capillaries showing deposition of amyloid. The capillary in the upper right-hand corner shows amyloid extending into the neuropil. I: Aneurysm of leptomeningeal arteriole. The aneurysm has a diameter of 460 μm, which is approximately half the thickness of the cortex. Compare with the leptomeningeal arteriole (arrow) in the same figure and with the leptomeningeal arterioles in A. The nondilated part of the aneurysmally dilated vessel is indicated by arrowheads. J: Confocal image of leptomeningeal blood vessel with focal amyloid deposits. K: Confocal image of leptomeningeal blood vessel from WT mouse. Scale bars: 300 μm (A and I), 70 μm (B and F), 20 μm (C and H), 100 μm (D), 200 μm (E), 15 μm (G), and 10 μm (J and K).
Figure 2.
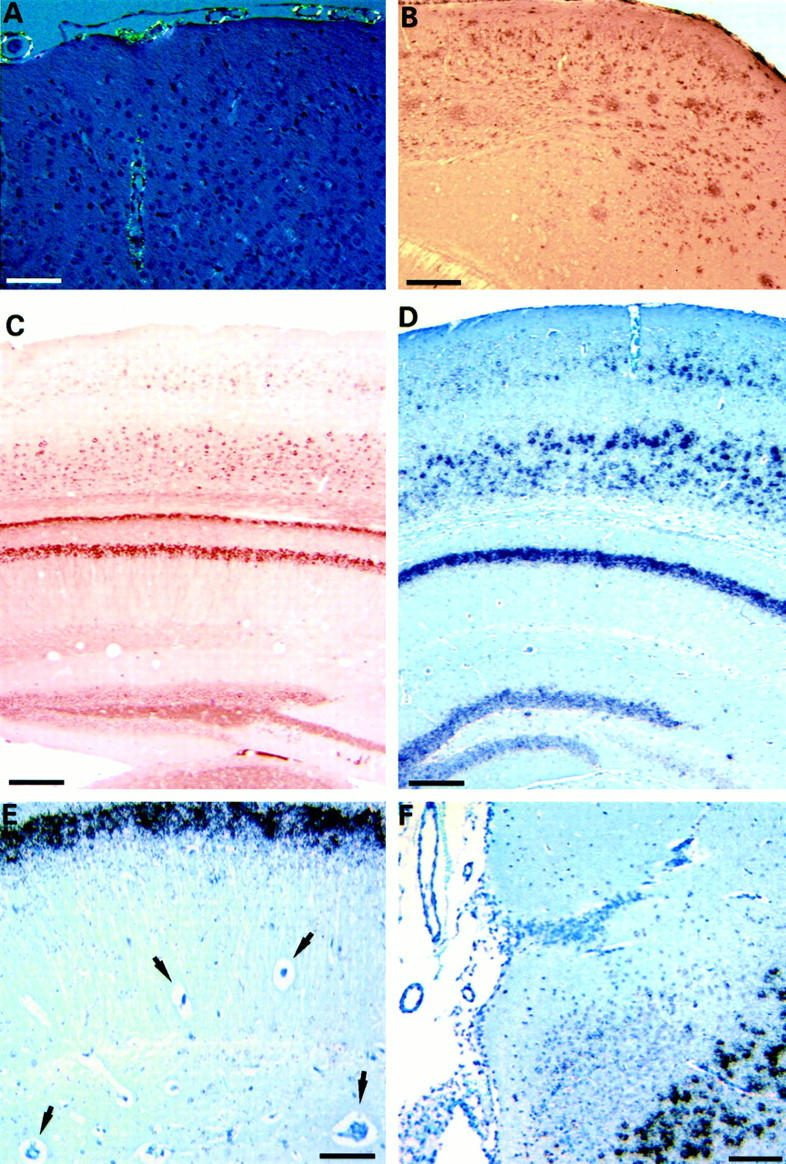
Congo red staining of vascular amyloid, Aβ-immunostaining of plaques, and APP-expression in APP/Ld mice. A: Congo red staining of blood vessels with amyloid depositions reveals a yellow-green birefringence, indicative of the presence of amyloid fibers. B: Aβ-immunostaining of the neocortex of a 20-month-old APP/Ld mouse demonstrates numerous diffuse plaques in addition to neuritic plaques. C: Immunohistochemistry for human APP shows expression in neurons of the neocortex, hippocampus, and dentate gyrus, but not in vessel walls. D: Similarly, in situ hybridization for human APP reveals neuronal labeling in the same regions. E: Higher magnification of the hippocampus showing strong labeling in pyramidal cells, but not in vessels (arrows). F: High magnification of subiculum with overlying leptomeninges and blood vessels demonstrating expression only in neurons. Scale bars: 75 μm (A), 250 μm (B), 200 μm (C and D), 60 μm (E), and 125 μm (F).
Figure 3.

Plaque core load (A and D), total plaque load (C), number of vessels with CAA (B and E), and vascular amyloid load (F) increase with age in the APP/Ld mice. All differences between age groups are statistically significant (P < 0.05). For plaque core and vascular amyloid load the surface of the Thioflavine-S fluorescent signal in two sections is expressed as a percentage of cortical surface or total surface, respectively (see Materials and Methods). The number of vessels in B and E is the total number of vessels with CAA in two sections stained with Thioflavine-S. Total plaque load, measured by immunohistochemistry for Aβ in one section, is expressed as a percentage of cortical surface. PL, plaque; NUM, number; AA, amyloid angiopathy; and VA, vascular.
Vascular Amyloid in APP/Ld Mice
The APP/Ld mice (15 to 24 months old) developed significant amyloid depositions primarily in pial (leptomeningeal), cortical, thalamic, and hippocampal vessels (Figures 1 and 2) ▶ ▶ . Vascular amyloid was present in 33 of the 35 mice studied and both vascular and plaque amyloid increased with age (Figure 3) ▶ . Amyloid accumulation in vessels might occur somewhat later than the first plaques, because in two FVB/N APP/Ld mice of 15 months, some plaques, but no vascular amyloid depositions, were observed. This finding was confirmed in 13.5-month-old APP/Ld mice. As in patients with CAA, individual vessels had a varying extent of amyloid deposition. Severely affected vessels stained with Thioflavine-S exhibited a pattern of fluorescence in concentric rings (Figure 4C) ▶ , whereas less affected vessels showed focal accumulations (Figure 1, J and K ▶ , and Figure 4B ▶ ), similar to human CAA. 7,20 Congo red staining of affected vessels revealed yellow-green birefringence, indicative of the presence of amyloid fibers (Figure 2A) ▶ . Based on anatomical inspections, arteries seemed to be more frequently affected than veins, which showed only small focal accumulations (Figure 1G) ▶ . Capillaries were rarely affected, but some showed amyloid penetrating into the neuropil (dyshoric amyloid; Figure 1H ▶ ). 1
Figure 4.

Thioflavine-S staining of the arterial circle of Willis from a 24-month-old APP/Ld mouse. A: The main branches of the arteries at the base of the brain are almost completely free of amyloid, whereas the smaller branches show prominent amyloid deposition. B: High magnification of a branch of the middle cerebral artery showing focal amyloid deposits. C: Branch of middle cerebral artery with a pattern of fluorescence in concentric rings. D: No amyloid deposition in the internal carotid artery. ICA, internal carotid artery; MCA, middle cerebral artery; ACA, anterior cerebral artery; and PCA, posterior communicating artery. Scale bars, 200 μm (A), 80 μm (B), 100 μm (C), and 90 μm (D).
Thioflavine-S-stained sections of 24-month-old F1-APP/Ld mice (n = 9) showed plaque cores and vascular amyloid accounting for, respectively, 71 ± 2% and 29 ± 2% of the surface of the fluorescent signal, indicating that vascular amyloid was an important component of the amyloid load in these animals. Vascular amyloid appeared first and was most pronounced in pial arterioles: in F1-APP/Ld mice (24 months old) 70 ± 4% of the vascular amyloid was present in pial blood vessels, whereas amyloid in intracerebral vessels accounted only for 30 ± 4% of the vascular amyloid. In the same animals 89 ± 3% of pial arterioles in the leptomeninges surrounding the cerebrum contained amyloid. In none of the animals examined was amyloid present in the leptomeninges surrounding the cerebellum or the spinal cord. To see whether also the large cerebral arteries were affected, we dissected the arterial circle of Willis and its branches of three APP/Ld mice (20 to 24 months old) (Figure 4) ▶ . Thioflavine-S staining showed that the main branches of the middle and anterior cerebral artery and of the posterior communicating artery were almost free of amyloid, whereas the smaller branches were loaded with amyloid. In the APP/Ld mice with the highest amyloid load, obviously dilated and even true saccular aneurysms of pial arterioles were seen (Figure 1I) ▶ . The largest aneurysm measured 460 μm in diameter, which is two to three times the diameter of the internal carotid artery. Despite the presence of aneurysms, hemorrhages were not found and Perls’ Prussian blue staining did not show deposition of iron. Heart, lungs, kidneys, spleen, and liver were examined to exclude extracerebral amyloid, but all these organs were free of vascular amyloid, which was also never present in WT mice.
Correlations between Plaques and Vascular Amyloid
In both the FVB/N APP/Ld (n = 21) (Figure 5A) ▶ and F1-APP/Ld (n = 14) transgenic mice plaque core load correlated with the number of vessels containing vascular amyloid (respectively, r = 0.89, P = 0.0001 and r = 0.77, P = 0.001). With total vascular amyloid load used, instead of the number of vessels containing amyloid, a comparable correlation was found (r = 0.83, P = 0.0002; Figure 5B ▶ ). Also total plaque load correlated well with the number of vessels with amyloid (FVB/N APP/Ld group; r = 0.84, P = 0.0001). Because both plaque load and vascular amyloid were strongly dependent of age, we used partial correlation to examine the importance of aging in the correlation found between total vascular amyloid and plaque core load in the F1-APP/Ld mice with age held constant. Although partial correlation statistics were lower (r = 0.68), a significant relationship remained between plaques and CAA (P = 0.01). Thus, although age was a clear risk factor for amyloid deposition, other pathogenetic factors linked plaques and vascular amyloid. To see whether the same local factors determine the deposition of amyloid in plaques and in intracerebral blood vessels, three areas of cortex were examined: neocortex (six-layered), hippocampus (three-layered), and subiculum (transitional cortex). The subiculum had a much higher plaque load than the neocortex and the hippocampus, but its vascular amyloid load did not differ (Figure 5C) ▶ . This suggests that pathogenetic factors, other than age and expression of the transgene, determine the local deposition of amyloid in brain parenchyma and vessels.
Figure 5.

The number of blood vessels with CAA (A) and the vascular amyloid load (B) correlate with the plaque core load (P = 0.0001 and P = 0.0002, respectively). Plaque load in the subiculum is much higher than in the neocortex and hippocampus (P = 0.0001 for both comparisons), whereas the vascular amyloid load is not significantly different (C). NUM, number; AA, amyloid angiopathy; PL, plaque; VA, vascular; neocx, neocortex; hippoc, hippocampus; and subic, subiculum.
Effect of Mutant PS1 on Plaques and Vascular Amyloid
CAA has been linked to familial forms of AD involving mutations in presenilin genes. 21-23 To see whether mutant PS1 has an effect on CAA in the APP/Ld mice, double-transgenic APP/LdxPS1/Mut mice were compared to single APP/Ld mice. Expression of PS1/Mut resulted in a significant increase in both plaque and vascular amyloid (Figure 6, A–C) ▶ .
Figure 6.

Expression of PS1/Mut in APP/Ld mice 13.5 months old results in an increase in plaque core load (A) (P = 0.02) as well as in the number of blood vessels with CAA (B) (P = 0.02) and vascular amyloid load (C) (P = 0.02). PL, plaque; NUM, number; AA, amyloid angiopathy; and VA, vascular.
Analysis of Expression of Transgenic Human APP/Ld
Analysis of the expression of human APP in the transgenic mice was performed with antibody 1G5 specific for human APP and with a specific RNA probe that did not react with mouse APP mRNA. Expression of human APP protein and of its mRNA was strong in the hippocampus, subiculum, amygdala, and in the third, fifth, and sixth layer of the neocortex (Figure 2, C–F) ▶ . Expression was also evident in the dentate gyrus and in some thalamic and brain stem nuclei, whereas it was practically absent in the cerebellum. The transgene was expressed exclusively in neurons; expression was absent in smooth muscle and endothelium of vessel walls and in glial cells.
Ultrastructural Analysis and Immunogold Labeling
In WT mice, leptomeningeal, cortical, and hippocampal arterioles showed a layer of endothelial cells, an internal elastic lamina, an uninterrupted thick layer of smooth muscle cells (media), and an external elastic lamina (Figure 7A) ▶ . In addition, larger arterioles were surrounded by collagen fibers (adventitia). In the APP/Ld transgenic mice, the degree of amyloid deposition in the leptomeningeal, cortical, and hippocampal arterioles was dependent on their diameter. Almost all leptomeningeal arterioles were affected, whereas amyloid deposition in smaller cortical and hippocampal arterioles was less prominent, correlating well with the light microscopic findings. In larger arterioles, deposition of amyloid fibers was circumferential (Figure 8) ▶ or focal. In contrast, in the smallest arterioles amyloid fiber deposition was usually focal (Figure 7, B and C) ▶ . The focal amyloid depositions were situated in the outermost (abluminal) part of the media around intact smooth muscle cells. Often the external elastic lamina was interrupted by the amyloid foci. In larger amyloid depositions, ie, presumably more advanced stages, amyloid fibers spread toward the internal elastic lamina and interrupted the smooth muscle layer. Most smooth muscle cells surrounded by amyloid fibers seemed well preserved. However, vessels with important amyloid deposition showed loss of smooth muscle cells (Figure 8, A and C) ▶ . Some affected large arterioles were dilated and their walls showed large segments where the smooth muscle of the media was replaced by amyloid fibers (Figure 8, C and D) ▶ . These dilated vessels had an attenuated endothelium and their internal elastic lamina was stretched. In rare vessels, the amyloid depositions encroached on the internal elastic lamina, which seemed thinned and irregular. Even large amyloid deposits did not seem to cause any narrowing of the vessel lumen. In some cortical and hippocampal arterioles the amyloid extended into the perivascular space.
Figure 7.
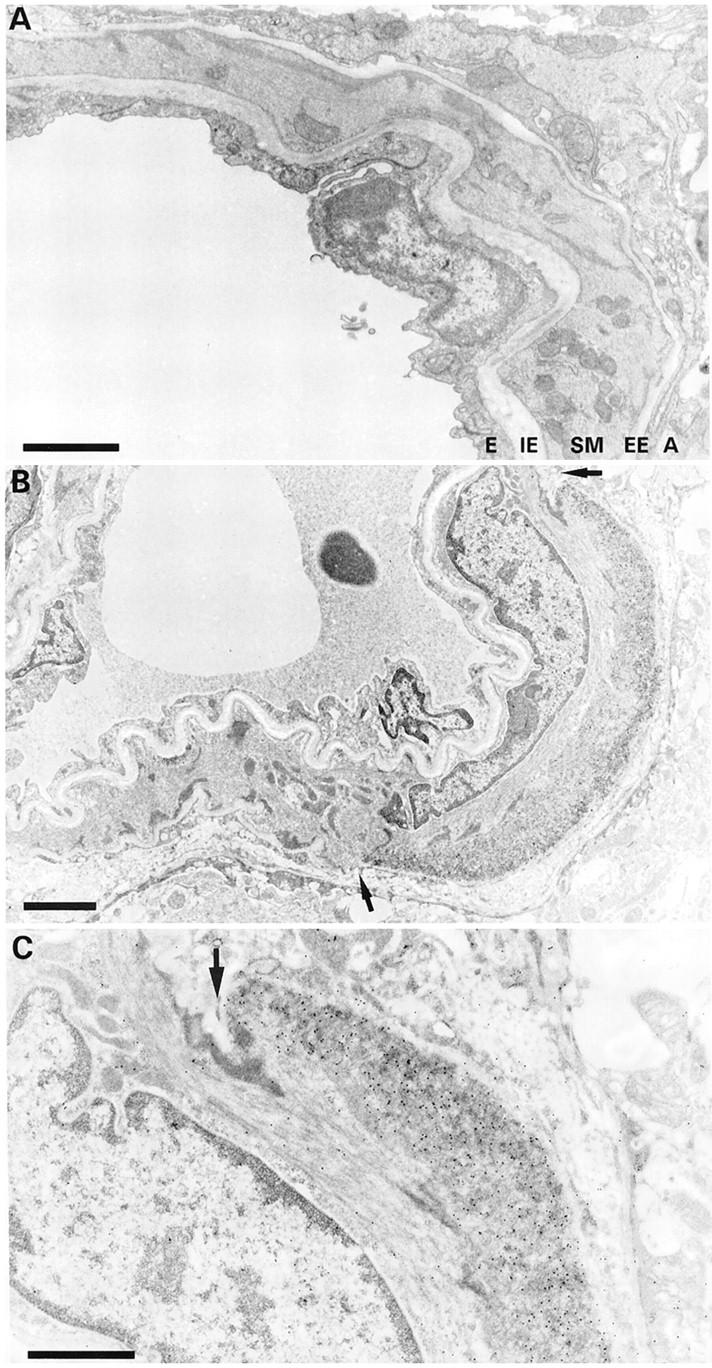
Ultrastructural aspect of focal amyloid deposition. A: Leptomeningeal arteriole of WT mouse showing the endothelium (E), internal elastic lamina (IE), smooth muscle layer (SM), external elastic lamina (EE), and adventitia (A). B: Focal amyloid deposition in the abluminal part of the smooth muscle layer (immunogold staining for Aβ40). The external elastic lamina is disrupted (arrows). C: High magnification showing the amyloid deposition decorated by 10 nm gold particles. An arrow indicates the interrupted external elastic lamina. Scale bars, 2 μm (A and B) and 1 μm (C).
Figure 8.
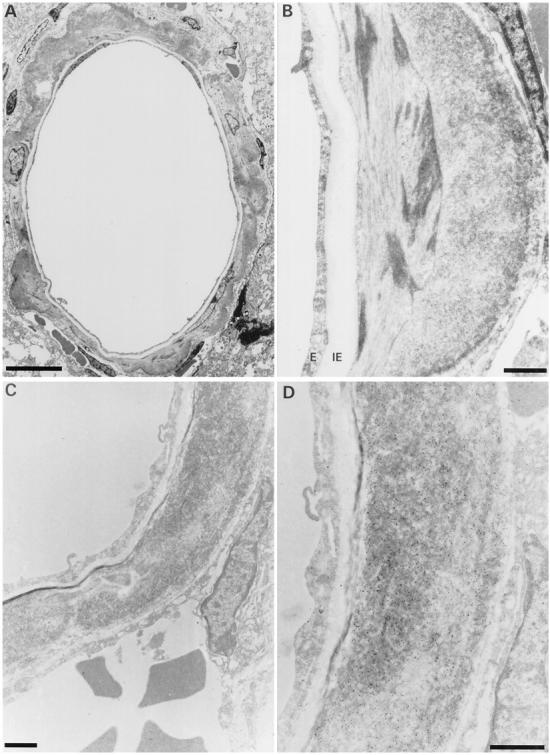
Ultrastructural aspect of dilated cerebral blood vessel from an unperfused FVB/N APP/Ld mouse. A: Dilated vessel with circumferential amyloid deposition. The endothelial layer is thinned and the internal elastic lamina is stretched. B: Higher magnification of amyloid deposit. The amyloid deposit is situated in the outermost part of the smooth muscle layer. E, endothelium; IE, internal elastic lamina. C: Serial section of same blood vessel stained for Aβ40. The smooth muscle layer is replaced by a large mass of amyloid fibers. D: High magnification showing the gold particles on the amyloid fibers. The endothelium and the internal elastic lamina are intact, whereas the smooth muscle layer and the external elastic lamina are disrupted. Scale bars, 10 μm (A) and 1 μm (B–D).
The vascular amyloid depositions consisted of randomly oriented slightly curved fibers with a thickness of 8 to 10 nm (Figure 9E) ▶ . In comparison, amyloid fibers in senile plaques were less curved and formed rigid bundles (Figure 9F) ▶ . The diameter of both fiber types was comparable. The amyloid deposits in the vessel walls were acellular and devoid of macrophages and did not cause an obvious increase in deposition of collagen fibers. In contrast, senile plaques in these transgenic mice, elicited an obvious microglial and astroglial reaction (not shown).
Figure 9.
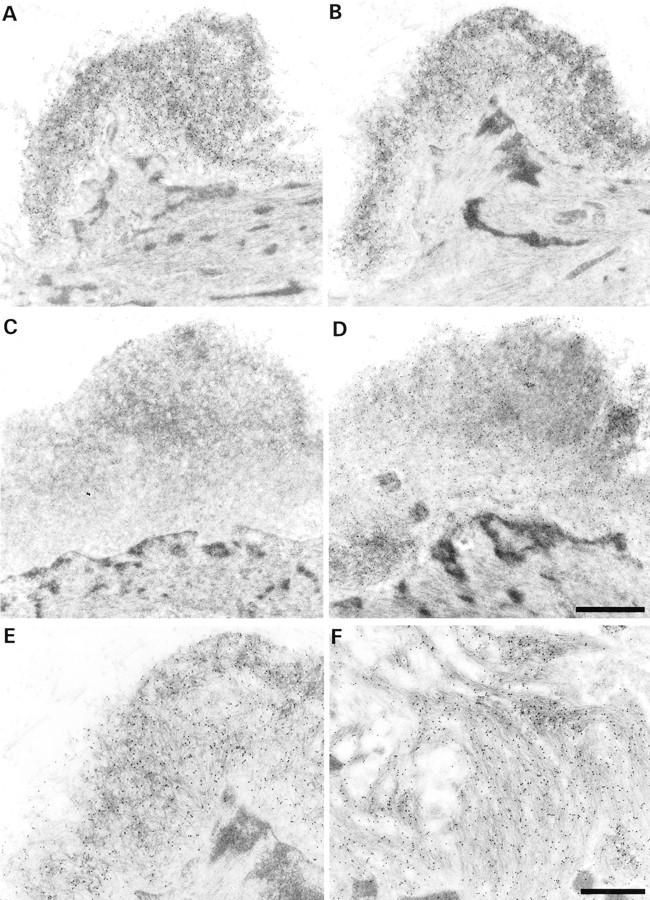
Immunogold stainings for Aβ40 and Aβ42. A and B: Serial sections showing a vascular amyloid deposit staining for Aβ40 (A) and Aβ42 (B). C and D: Amyloid focus staining for Aβ42 (D), but not for Aβ40 (C). The existence of foci that contain Aβ42, but not Aβ40 suggests that Aβ42 is the initially deposited form. A and C and B and D are from the same ultrathin section. E: Higher magnification of B. The amyloid deposit consists of randomly oriented slightly curved fibers with a thickness of 8 to 10 nm. F: In comparison, amyloid fibers in plaques are straight and form rigid bundles (immunogold for Aβ42). Scale bars, 1 μm (A–D) and 0.5 μm (E and F).
Immunogold labeling gave signals well above background labeling with all antibodies tested. Background labeling was low, especially with the monoclonal antibodies, which reacted almost exclusively with amyloid fibers (Figure 9) ▶ . The antibody to glial fibrillary acidic protein labeled exclusively the glial fibrillary acidic protein-intermediate filaments in astrocytes. Most amyloid deposits in the arteriolar walls and the senile plaques reacted with antibodies against Aβ, Aβ40, and Aβ42. However, rare small deposits in the arteriolar walls contained only Aβ42, but not Aβ40.
Quantitative Analysis of Aβ Levels in CSF and Blood, and in Plaques and Vascular Amyloid
High levels of Aβ were detected in the CSF of APP/Ld mice by Western blotting (≈20 pg/μl) (Figure 10A) ▶ . Using the same techniques, Aβ was never detected in the plasma of APP/Ld mice. This indicates that blood is unlikely as the source of the Aβ deposited in vessel walls. Also, in AD patients and healthy patients CSF levels are much higher than plasma levels. 9,24
Figure 10.
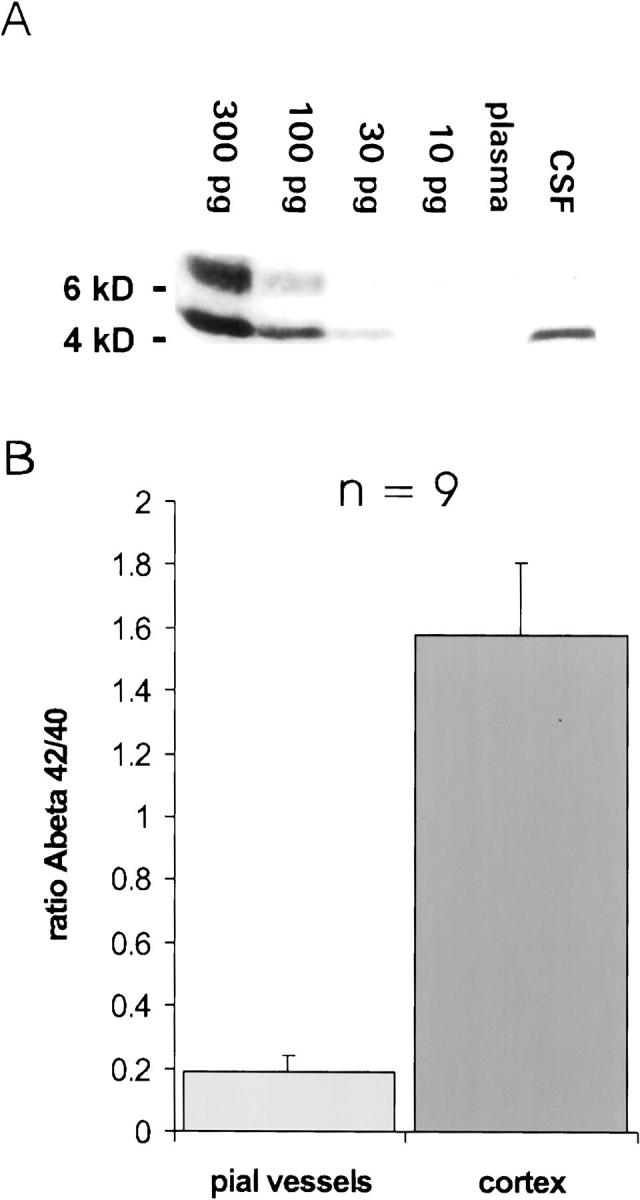
Levels of Aβ in CSF and plasma, and the ratio of insoluble Aβ42:Aβ40 in pial vessels and neocortex. A: Western blot for Aβ in CSF (5 μl) and plasma (5 μl) from a 4-month-old APP/Ld mouse. Aβ could be detected in CSF (level ≈ 20 pg/μl), but not in plasma. Using a longer exposure time (not shown), it was possible with this method to detect 10 pg of Aβ, but in plasma Aβ remained undetectable (level < 2pg/μl). The band above the 6-kd marker probably represents polymerized Aβ. B: The ratio of Aβ42:Aβ40 is eight times lower in vascular amyloid (0.19 ± 0.04) than in plaque amyloid (1.58 ± 0.23, P = 0.003), showing that Aβ40 is much more abundant relative to Aβ42 in vascular amyloid.
In all APP/Ld mice (n = 9) the ratio of insoluble Aβ42 to Aβ40 (Figure 10B) ▶ was much higher in the neocortex (1.58 ± 0.23) than in the leptomeningeal blood vessels (0.19 ± 0.04, P = 0.003). Thus, relative to Aβ42 there was approximately eight times more Aβ40 in the amyloid deposits in leptomeningeal blood vessels than in the neocortical plaques.
CBF and Response to Hypercapnia
To determine whether CAA in the APP/Ld mice affected cerebral perfusion, laser-Doppler flowmetry was used. The resting CBF in the APP/Ld transgenic mice and the WT mice was similar (WT: 303.8 ± 24.7; APP/Ld: 301.7 ± 11.7, arbitrary perfusion units, P > 0.05). Because in the APP/Ld mice the vascular amyloid deposition was mainly in the smooth muscle layer, we evaluated smooth-muscle cell function by inducing hypercapnia, which is a strong endothelium-independent stimulus for smooth muscle relaxation. 25 In the APP/Ld mice, hypercapnia resulted in a 40.7 ± 3.4% increase of CBF. Surprisingly, the increase was very similar to that in the WT mice (41 ± 3.7%, P > 0.05). Thus, the vasodilatory capacity of the cerebral vasculature seemed well preserved, despite the obvious amyloid load in the blood vessels of the APP/Ld mice.
Discussion
We report a transgenic mouse model that, in addition to amyloid plaques, develops prominent cerebral amyloid angiopathy. The CAA in the APP/Ld mice exhibited a striking similarity to that observed in aged individuals and in AD patients. The morphological pattern, ultrastructural aspects, and biochemical composition of the vascular amyloid deposition described in human CAA are closely reproduced in this model. Also, the localization and the types of vessels affected recapitulate very well the specific neuroanatomical pattern of human CAA. Vascular amyloid in these mice led to progressive vessel wall damage and aneurysm formation, a factor predisposing to hemorrhage. 7,8
Pathogenesis and Pathways of Vascular Aβ Deposition
In the brain, as well as in other tissues, APP is ubiquitously expressed by many cell types, including by those of the vasculature, and it has been hypothesized that vascular Aβ in the vessel walls is derived from vascular smooth muscle cells and/or pericytes. 26,27 In vitro studies suggest that Aβ can induce its own production in cultured human degenerating cerebrovascular smooth muscle cells. 28 The observation that pial vessels, which are apart from the neuropil, are more often affected would also suggest that local production may be an important source. However, these hypotheses fail to explain the specific neuroanatomical pattern of CAA and its exclusive localization in intracranial vessels. The APP/Ld mice were generated by using the murine thy1-gene promoter element, 16,29,30 limiting transgenic expression to brain and, more specifically, to neurons. In this model, we have shown that a neuronal source of mutant APP is sufficient to very closely mimic human CAA, suggesting that production of Aβ within the vasculature is not a necessary event for the formation of CAA. The formation of amyloid fibers is concentration-dependent in vitro. 31 Thus, for CAA to form in vessels that do no express the transgene, APP or Aβ must be either transported to that localization or must circulate through an other mechanism: for instance, CSF, brain interstitial fluid, or blood. One mechanism that can be excluded is a direct synaptic source for APP or Aβ, because in our model, amyloid depositions were first apparent in the outer portion of the smooth muscle layer, an area poor in synaptic contacts. In addition, the predilection of CAA for pial arterioles argues against this hypothesis, although transport from vasomotor nuclei cannot completely be excluded as a synaptic source of Aβ. The blood transport hypothesis as the cause of CAA, suggests Aβ uptake and blood brain barrier transport. The lack of significant amounts of Aβ in the blood of APP/Ld mice and the fact that transgenic mice with high levels of Aβ do not develop CAA, but peripheral amyloid pathology, 32,33 argue against this mechanism.
It has been shown that Aβ is present in the CSF of normal and AD individuals. 19,24 However, the mere presence of Aβ in the CSF cannot explain the presence of CAA in the pial vessels in the leptomeninges surrounding the cerebrum and its absence in those surrounding the cerebellum, brain stem, or spinal cord, as it was seen in the APP/Ld mice. In addition, in the APP/Ld mice small leptomeningeal branches of the cerebral arteries were much more affected than the larger main branches, yet they are all surrounded by CSF. The same difference in vascular amyloid load between small and large arteries in the subarachnoidal space is seen in patients with CAA. 6 Small intracortical arterioles in the APP/Ld mice were also affected, but they are at some distance from the CSF, separated from it by the pia mater. 34 Thus, gradients of Aβ or specific drainage pathways must be involved. Brain interstitial fluid can drain along perivascular spaces around intracortical and leptomeningeal arteries; these channels eventually connect with nasal lymphatics, which drain to the cervical lymph nodes. 20 Furthermore, it has been suggested that significant amounts of Aβ drain along this pathway in humans. 6
In AD patients, the ratio of Aβ42 to Aβ40 is higher in plaques than in vascular amyloid. 10-13 In the past, this finding has been interpreted as being consistent with the hypothesis that neurons are the source of Aβ42 and that the vasculature produces mainly Aβ40. 10 However, this hypothesis is challenged by the demonstration in the APP/Ld mice of a much higher ratio of Aβ42:Aβ40 in plaque than in vascular amyloid, and in this model, vessels, which do not express the transgene, are not likely an important source of Aβ. In addition, others demonstrated that elimination of endogenous APP/Aβ did not affect CAA in transgenic mice overexpressing the APP/Swedish mutant in neurons. 15 In patients with CAA, vascular amyloid depositions can consist solely of Aβ42, whereas depositions consisting of Aβ40 alone have not been reported. 35 Despite the much higher levels of Aβ40 in vascular amyloid, we observed rare vascular deposits that consisted only of Aβ 42 in the APP/Ld mice. Similar to senile plaque formation, 36 Aβ42 could be the first amyloid deposited in the vessel walls, subsequently entrapping the more soluble Aβ40, as also suggested by in vitro experiments. 31 The increase in CAA and in senile plaques noted in the APP/LdxPS1/Mut mice, in which PS1/Mut results selectively in higher levels of Aβ42 (50% increase of Aβ42 in APP/LdxPS1/Mut mice, 6 to 8 weeks old, compared to APP/Ld mice; ID, DM, CK, IL, JVD, FC, HV, FVL, unpublished data), supports this hypothesis. The interstitial fluid pathway is in close proximity to the vascular smooth muscle layer, which may facilitate amyloid deposition. 37,38 In addition, substances thought to bind amyloid and increase fibrillization such as proteoglycans are abundantly present in the basal lamina of smooth muscle cells. 39 Selective deposition of Aβ in vascular malformations and in vessels after radiation therapy suggests that local alterations in vessel walls or changes in the vascular basement membrane could play a role in Aβ-deposition. 40,41 The solubility properties of Aβ40 may permit this molecule to drain from the brain along perivascular spaces more easily than the less soluble Aβ42, resulting in a lower ratio of Aβ42:Aβ40 in vascular amyloid. Consistent with this hypothesis is the demonstration in AD patients of higher ratios of Aβ42:Aβ40 in intracerebral vessels than in leptomeningeal vessels, 42 which are more distal along the interstitial fluid drainage pathway. Although APP/Ld mice express APP and Aβ at significantly higher levels than humans, 16 the striking similarities to human CAA, suggest a similar mechanism of vascular amyloid deposition.
Vessel Wall Damage and Aneurysm Formation
CAA is associated with intracerebral hemorrhage, and brains of patients with CAA and intracerebral hemorrhage contain microaneurysms that are implicated in vessel wall rupture and hemorrhage. 7,8 In the APP/Ld mice we have examined in detail which factors contribute to vessel damage and aneurysm formation. The first amyloid depositions were situated in the outermost part of the media around intact smooth muscle cells and were often associated with a disruption of the external elastic lamina. In more severely affected vessels the amyloid depositions spread toward the inner part of the media and interrupted the smooth muscle layer. Smooth muscle cells were morphologically intact in most vessels, but vessels with important amyloid deposition showed loss of smooth muscle. This in vivo toxic effect of fibrillary Aβ is consistent with the toxic effects on smooth muscle cells that have been described in vitro. 43 Some vessels in which the complete thickness of the media was replaced by amyloid also showed a thinned and irregular internal elastic lamina. In the APP/Ld model, disruption of the external elastic lamina, thinning of the internal elastic lamina, interruption of the smooth muscle layer, and loss of smooth muscle cells led to weakening of the vessel wall, dilatation, and, finally, aneurysm formation. In patients with CAA, the effect of vascular amyloid on vessel walls is very similar. 8,44 Small amyloid depositions were seen in the outer part of the smooth muscle layer at the media-adventitia junction, whereas larger depositions caused loss of elastic lamina and smooth muscle cells. Spindle-shaped dilations and microaneurysms have been shown by computer-assisted three-dimensional image. 8 Despite the obvious vascular pathology, cerebral hemorrhages were never observed in the APP/Ld mice. This suggests that in humans other factors may contribute to rupture of the vessel wall. For instance, age-related pathological changes, such as thickening of the intima, atherosclerosis, and hypertension could aggravate the vessel wall damage.
In patients, CAA is associated with infarcts and leukoencephalopathy, 45,46 a generalized abnormality of the white matter, thought to be because of hypoperfusion. In addition, CAA may also play a role in the pathogenesis of Alzheimer’s dementia. Recently, it has been found that in transgenic mice overexpressing human APP soluble Aβ has a profound and selective impairment on endothelium-dependent regulation of the neocortical circulation, but it has no effect on vascular smooth muscle cell function. 47 However, these animals did not have plaque or vascular amyloid; therefore, we investigated the possibility that fibrillary Aβ has a functional effect on vascular smooth muscle, a predilection site for amyloid deposition. Hypercapnia was used to induce smooth muscle relaxation, because it acts in an endothelium-independent manner. The increase in CBF produced by inhalation of 7% CO2 was well preserved in the APP/Ld mice, showing that the vasodilatory capacity of the cerebral arterioles was not reduced. In view of the effects of vascular amyloid on vessel walls described ultrastructurally, this seemed surprising. However, a possible explanation could be that, in the APP/Ld mice, the vessels or segments of vessels that are most affected are also dilated, thereby decreasing vascular resistance, whereas the less affected vessels remain functionally intact. These data will have to be completed by additional experiments that test the hemodynamic effects of CAA in the APP/Ld mice. The pathogenetic similarities to human CAA suggest that this model will be of value in unraveling mechanisms that play a role in the cerebrovascular dysfunction seen in CAA and AD.
In Vivo Models of CAA
A variety of nonhuman species naturally manifest CAA as they age, most notably nonhuman primates and dogs, 14 but a mouse model of CAA offers distinct advantages. Two other mouse models have been reported that show deposition of amyloid in vessels. In one of these models, co-expression of TGF-β1 in transgenic mice overexpressing APP accelerated the deposition of amyloid, and induced amyloid deposition in cerebral vessels and meninges, suggesting that TFG-β1 may promote or initiate amyloidogenesis in plaques and blood vessels. 48 The second mouse model, overexpressing the Swedish mutant of APP under control of the murine thy1 promoter (APP23), showed vascular amyloid deposition very similar to the CAA in our APP/Ld model. 15 The analysis of the APP/Ld mice allowed progress toward understanding the pathogenesis of CAA and allowed identification of specific characteristics and factors contributing to vessel wall damage and aneurysm formation. These factors very likely play a role also in human CAA.
The APP/Ld model can be used to further characterize underlying factors and mechanisms in the pathogenesis of CAA and hemorrhage. In addition, the same transgenic approach may be used to study the effect of overexpression of the hereditary cerebral hemorrhage with amyloidosis–Dutch type mutant of APP, allowing differentiation of factors responsible for Aβ deposition in plaques versus vessels. A better understanding of factors that influence amyloid deposition in vessels also will have implications for treating CAA and AD. The progression of CAA from mild (asymptomatic) to severe (associated with hemorrhage) represents an accumulation of amyloid fibers in already affected vessels rather than an increase in the number of vessels affected. 49 Thus, therapeutic interventions that can inhibit the deposition of Aβ onto existing vascular amyloid depositions would be expected to prevent the development of hemorrhagic stroke. These avenues of new research will be in the position to address long-standing questions in the pathogenesis of CAA and AD, and, hopefully, will lead to new diagnostic and therapeutic strategies.
Acknowledgments
We thank K. Beyreuther, C. Armée, R. Renwart, W. Annaert, B. Van der Schueren, and R. Vlietinck for their intellectual, material, and technical contributions and C. Vochten for the help with administration.
Footnotes
Address reprint requests to Fred Van Leuven, Ph.D., Dr. Sc, Experimental Genetics Group, Flemish Institute for Biotechnology, Center for Human Genetics, K. U. Leuven, Gasthuisberg O&N 06, B-3000, Leuven, Belgium. E-mail: fredvl@med.kuleuven.ac.be.
Supported by the Fonds voor Wetenschappelijk Onderzoek (FWO-Vlaanderen), the Interuniversity-Network for Fundamental Research (IUAP), by the special Action Program for Biotechnology of the Flemish government (VLAB/IWT, COT-008), by the Rooms-fund, by Janssen Research Foundation, and by K.U. Leuven Research and Development.
References
- 1.Vinters HV: Cerebral amyloid angiopathy a critical review. Stroke 1987, 18:311-324 [DOI] [PubMed] [Google Scholar]
- 2.Yoshimura M, Yamanouchi H, Kuzuhara S, Mori H, Sugiura S, Mizutani T, Shimada H, Tomonaga M, Toyokura Y: Dementia in cerebral amyloid angiopathy: a clinicopathological study. J Neurol 1992, 239:441-450 [DOI] [PubMed] [Google Scholar]
- 3.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B: Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248:1124-1126 [DOI] [PubMed] [Google Scholar]
- 4.Mandybur T: The incidence of cerebral amyloid angiopathy in Alzheimer’s disease. Neurology 1975, 25:120-126 [DOI] [PubMed] [Google Scholar]
- 5.Gilbert JJ, Vinters HV: Cerebral amyloid angiopathy: incidence and complications in the aging brain. I. Cerebral hemorrhage. Stroke 1983, 14:915-923 [DOI] [PubMed] [Google Scholar]
- 6.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE: Cerebral amyloid angiopathy: amyloid β accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 1998, 153:725-733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okazaki H, Reagan TJ, Campbell RJ: Clinicopathologic studies of primary cerebral amyloid angiopathy. Mayo Clin Proc 1979, 54:22-31 [PubMed] [Google Scholar]
- 8.Maeda A, Yamada M, Itoh Y, Otomo E, Hayakawa M, Miyatake T: Computer-assisted three-dimensional image analysis of cerebral amyloid angiopathy. Stroke 1993, 24:1857-1864 [DOI] [PubMed] [Google Scholar]
- 9.Itoh Y, Yamada M, Hayakawa M, Otomo E, Miyatake T: Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J Neurol Sci 1992, 116:135-141 [DOI] [PubMed] [Google Scholar]
- 10.Prelli F, Castano E, Glenner GG, Frangione B: Differences between vascular and plaque core amyloid in Alzheimer’s disease. J Neurochem 1988, 51:648-651 [DOI] [PubMed] [Google Scholar]
- 11.Joachim CL, Duffy LK, Morris JH, Selkoe DJ: Protein chemical and immunocytochemical studies of meningovascular β-amyloid protein in Alzheimer’s disease and normal aging. Brain Res 1988, 474:100-111 [DOI] [PubMed] [Google Scholar]
- 12.Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, Reardon IM, Zürcher-Neely HA, Heinrikson RL, Ball MJ, Greenberg BD: Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J Biol Chem 1993, 268:3072-3083 [PubMed] [Google Scholar]
- 13.Iwatsubo T, Mann DMA, Odaka A, Suzuki N, Ihara Y: Amyloid β protein (Aβ) deposition: Aβ42(43) precedes Aβ40 in Down syndrome. Ann Neurol 1995, 37:294-299 [DOI] [PubMed] [Google Scholar]
- 14.Walker LC: Animal models of cerebral β-amyloid angiopathy. Brain Res Brain Res Rev 1997, 25:70-84 [DOI] [PubMed] [Google Scholar]
- 15.Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M: Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 1999, 96:14088-14093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Van den Haute C, Checler F, Godaux E, Cordell B, Van Leuven F: Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 1999, 274:6483-6492 [DOI] [PubMed] [Google Scholar]
- 17.Barelli H, Lebeau A, Vizzavona J, Delaere P, Chevallier N, Drouot C, Marambaud P, Ancolio K, Buxbaum JD, Khorkova O, Heroux J, Sahasrabudhe S, Martinez J, Warter JM, Mohr M, Checler F: Characterization of new polyclonal antibodies specific for 40 and 42 amino-acid long amyloid beta peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer’s disease and cerebral amyloid angiopathy cases. Mol Med 1997, 3:695-707 [PMC free article] [PubMed] [Google Scholar]
- 18.Franklin KBJ, Paxinos G: The Mouse Brain in Stereotaxic Coordinates. 1997. G Paxinos. San Diego, Academic Press, Edited by KBJ Franklin
- 19.Ida N, Hartmann T, Pantel J, Schröder J, Zerfass R, Förstl H, Sandbrink R, Masters CL, Beyreuther K: Analysis of heterogeneous βA4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem 1996, 271:22908-22914 [DOI] [PubMed] [Google Scholar]
- 20.Weller RO: Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer’s disease, prion disorders and multiple sclerosis. J Neuropathol Exp Neurol 1998, 57:885-894 [DOI] [PubMed] [Google Scholar]
- 21.Yasuda M, Maeda K, Ikejiri Y, Kawamata T, Kuroda S, Tanaka C: A novel missense mutation in the presenilin gene in a familial Alzheimer’s disease pedigree with abundant amyloid angiopathy. Neurosci Lett 1997, 232:29-32 [DOI] [PubMed] [Google Scholar]
- 22.Nochlin D, Bird TD, Nemens EJ, Ball MJ, Sumi SM: Amyloid angiopathy in a Volga family with Alzheimer’s disease and a presenilin-2 mutation (N141I). Ann Neurol 1998, 43:131-135 [DOI] [PubMed] [Google Scholar]
- 23.Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H, Farrer M, Hutton M, Lincoln S, Hardy J, Gwinn K, Somer M, Paetau A, Kalimo H, Ylikoski R, Poyhonen M, Kucera S, Haltia M: A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med 1998, 4:452-455 [DOI] [PubMed] [Google Scholar]
- 24.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D: Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature 1992, 359:325-327 [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Pelligrino DA, Koenig HM, Albrecht RF: The role of endothelium and nitric oxide in rat arteriolar dilatory responses to CO2 in vivo. J Cereb Blood Flow Metab 1994, 14:944-951 [DOI] [PubMed] [Google Scholar]
- 26.Kalaria RN, Premkumar DR, Pax AB, Cohen DL, Lieberburg I: Production and increased detection of amyloid beta protein and amyloidogenic fragments in brain microvessels, meningeal vessels and choroid plexus in Alzheimer’s disease. Brain Res Mol Brain Res 1996, 35:58-68 [DOI] [PubMed] [Google Scholar]
- 27.Natte R, de Boer WI, Maat-Schieman ML, Baelde HJ, Vinters HV, Roos RA, Van Duinen SG: Amyloid beta precursor protein-mRNA is expressed throughout cerebral vessel walls. Brain Res 1999, 828:179-183 [DOI] [PubMed] [Google Scholar]
- 28.Davis-Salinas J, Saporito-Irwin SM, Cotman CW, Van Nostrand WE: Amyloid β-protein induces its own production in cultured degenerating cerebrovascular smooth muscle cells. J Neurochem 1995, 65:931-934 [DOI] [PubMed] [Google Scholar]
- 29.Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F: Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol 1999, 155:2153-2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tesseur I, Van Dorpe J, Spittaels K, Van den Haute C, Moechars D, Van Leuven F: Expression of human apolipoprotein in neurons causes hyperphosphorylation of protein Tau in the brain of transgenic mice. Am J Pathol 2000, 156:951-964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lansbury PT: Structural neurology: are seeds at the root of neuronal degeneration? Neuron 1997, 19:1151-1154 [DOI] [PubMed] [Google Scholar]
- 32.Kawarabayashi T, Shoji M, Sato M, Sasaki A, Ho L, Eckman CB, Prada CM, Younkin SG, Kobayashi T, Tada N, Matsubara E, Iizuka T, Harigaya Y, Kasai K, Hirai S: Accumulation of beta-amyloid fibrils in pancreas of transgenic mice. Neurobiol Aging 1996, 17:215-222 [DOI] [PubMed] [Google Scholar]
- 33.Fukuchi K, Ho L, Younkin SG, Kunkel DD, Ogburn CE, LeBoef RC, Furlong CE, Deeb SS, Nochlin D, Wegiel J, Wisniewski HM, Martin GM: High levels of circulating β-amyloid peptide do not cause cerebral β-amyloidosis in transgenic mice. Am J Pathol 1996, 149:219-227 [PMC free article] [PubMed] [Google Scholar]
- 34.Hutchings M, Weller RO: Anatomical relationship of the pia mater to cerebral blood vessels in man. J Neurosurg 1986, 63:316-325 [DOI] [PubMed] [Google Scholar]
- 35.Shinkai Y, Yoshimura M, Ito Y, Odaka A, Suzuki N, Yanagisawa K, Ihara Y: Amyloid β-proteins 1–40 and 1–42 (43) in the soluble fraction of extra- and intracranial blood vessels. Ann Neurol 1995, 38:421-428 [DOI] [PubMed] [Google Scholar]
- 36.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y: Visualization of Aβ 42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ 42(43). Neuron 1994, 13:45-53 [DOI] [PubMed] [Google Scholar]
- 37.Prior R, D’Urso D, Frank R, Prikulis I, Pavlakovic G: Experimental deposition of Alzheimer amyloid beta-protein in canine leptomeningeal vessels. Neuroreport 1995, 6:1747-1751 [DOI] [PubMed] [Google Scholar]
- 38.Van Nostrand WE, Melchor JP, Ruffini L: Pathologic amyloid beta-protein cell surface fibril assembly on cultured human cerebrovascular smooth muscle cells. J Neurochem 1998, 70:216-223 [DOI] [PubMed] [Google Scholar]
- 39.Snow AD, Mar H, Nochlin D, Kimata K, Kato M, Suzuki S, Hassell J, Wight TN: The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. Am J Pathol 1988, 133:456-463 [PMC free article] [PubMed] [Google Scholar]
- 40.Hart MN, Merz P, Bennett-Gray J, Menezes AH, Goeken JA, Schelper RL, Wisniewski HM: β-amyloid protein of Alzheimer’s disease is found in cerebral and spinal cord vascular malformations. Am J Pathol 1988, 132:167-172 [PMC free article] [PubMed] [Google Scholar]
- 41.Sugihara S, Ogawa A, Nakazato Y, Yamaguchi H: Cerebral β amyloid deposition in patients with malignant neoplasms: its prevalence with aging and effects of radiation therapy on vascular amyloid. Acta Neuropathol 1995, 90:135-141 [DOI] [PubMed] [Google Scholar]
- 42.Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ: β-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer’s disease. Proc Natl Acad Sci USA 1993, 90:10836-10840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis J, Cribbs DH, Cotman CW, Van Nostrand WE: Pathogenic amyloid beta-protein induces apoptosis in cultured human cerebrovascular smooth muscle cells. Amyloid 1999, 6:157-164 [DOI] [PubMed] [Google Scholar]
- 44.Yamaguchi H, Yamazaki T, Lemere CA, Frosch MP, Selkoe DJ: Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer’s disease: an immunoelectron microscopic study. Am J Pathol 1992, 141:249-259 [PMC free article] [PubMed] [Google Scholar]
- 45.Haan J, Algra PR, Roos RAC: Hereditary cerebral hemorrhage with amyloidosis—Dutch type: clinical and computed tomographic analysis of 24 cases. Arch Neurol 1990, 47:649-653 [DOI] [PubMed] [Google Scholar]
- 46.Loes DJ, Biller J, Yuh WTC, Hart MN, Godersky JC, Adams HP, Keefauver SP, Tranel D: Leukoencephalopathy in cerebral amyloid angiopathy: MR imaging in four cases. Am J Neuroradiol 1990, 11:485-488 [PMC free article] [PubMed] [Google Scholar]
- 47.Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA: SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci 1999, 2:157-161 [DOI] [PubMed] [Google Scholar]
- 48.Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L: Amyloidogenic role of cytokine TGF-β1 in transgenic mice and in Alzheimer’s disease. Nature 1997, 389:603-606 [DOI] [PubMed] [Google Scholar]
- 49.Alonzo NC, Hyman BT, Rebeck GW, Greenberg: Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol 1998, 57:353-359 [DOI] [PubMed] [Google Scholar]