

Abstract
Human testicular germ-cell tumors of young adults (TGCTs), both seminomas and nonseminomas, are characterized by 12p overrepresentation, mostly as isochromosomes, of which the biological and clinical significance is still unclear. A limited number of TGCTs has been identified with an additional high-level amplification of a restricted region of 12p including the K-RAS proto-oncogene. Here we show that the incidence of these restricted 12p amplifications is ∼8% in primary TGCTs. Within a single cell formation of i(12p) and restricted 12p amplification is mutually exclusive. The borders of the amplicons cluster in short regions, and the amplicon was never found in the adjacent carcinoma in situ cells. Seminomas with the restricted 12p amplification virtually lacked apoptosis and the tumor cells showed prolonged in vitro survival like seminoma cells with a mutated RAS gene. However, no differences in proliferation index between these different groups of seminomas were found. Although patients with a seminoma containing a homogeneous restricted 12p amplification presented at a significantly younger age than those lacking it, the presence of a restricted 12p amplification/RAS mutation did not predict the stage of the disease at clinical presentation and the treatment response of primary seminomas. In 55 primary and metastatic tumors from 44 different patients who failed cisplatinum-based chemotherapy, the restricted 12p amplification and RAS mutations had the same incidence as in the consecutive series of responding patients. These data support the model that gain of 12p in TGCTs is related to invasive growth. It allows tumor cells, in particular those showing characteristics of early germ cells (ie, the seminoma cells), to survive outside their specific microenvironment. Overexpression of certain genes on 12p probably inhibits apoptosis in these tumor cells. However, the copy numbers of the restricted amplification of 12p and K-RAS mutations do not predict response to therapy and survival of the patients.
Malignant transformation is a complex, multistep process. 1 Although involvement of several genes has been suggested in the development of testicular germ-cell tumors of young adults (TGCTs), histologically seminoma and nonseminomas, 2 evidence is lacking.
All TGCTs originate from carcinoma in situ (CIS). 3,4 CIS is frequently found in the parenchyma adjacent to an invasive TGCT, being located on the inner side of the basal membrane of the seminiferous tubules, in close association with Sertoli cells. 5 The mechanisms involved in the development from CIS, via the microinvasive stage, to overt invasive tumors are still unclear. Obviously tumor cells are selected that are capable of surviving and growing outside of the specific microenvironment of the seminiferous tubule.
The only consistent chromosomal anomaly in TGCTs is the gain of the short arm of chromosome 12, mostly because of isochromosomes of 12p. 6,7 The copy number of 12p reportedly predicts prognosis. 8-10 K-RAS has been proposed as the relevant gene on 12p, 11-13 of which the encoding protein (p21) is involved in signal transduction. Mutated RAS has been found to be correlated with poor prognosis in childhood acute lymphocytic leukemia, 14 and non-small-cell lung cancer. 15,16 Recently, activation of RAS has been shown to be involved in tumor maintenance 17,18 and in inducing anchorage-independent growth because of inhibition of apoptosis. 19,20 Indeed, a correlation between activated RAS and metastatic capacity has been reported. 21,22 Besides activation by means of mutations, RAS can also be involved in malignant transformation because of increased copy numbers of the wild-type gene, resulting in overexpression of wild-type mRNA and accumulation of the wild-type protein. 23-28
Seminomas are highly sensitive to irradiation and cisplatinum-based chemotherapy. The vast majority of patients with metastatic nonseminomas is cured by cisplatinum-based combination chemotherapy. 29 Because the clinical course of these tumors can still not be predicted for individual patients, additional prognostic markers are needed. Interestingly, activated RAS genes increase the intrinsic resistance to radiation and cisplatinum therapy. 30-34 It is unclear, however, whether amplification of wild-type RAS has the same effect.
A small percentage of TGCTs has RAS mutations, 35-37 of which the clinical relevance was not studied. We showed previously that seminomas with a mutated RAS gene have survival advantage in vitro and have reduced apoptosis in the primary tumor. 38,39 Noteworthy, in vitro survival of tumor cells correlates with poor prognosis in patients with metastatic TGCTs 40 and adult acute myeloid leukemia. 41 Recently, a number of invasive TGCTs with amplification of a restricted region of 12p have been identified. 42-45 We showed that the shortest region of overlap of amplification (SROA) is ∼1.7 Mbases, containing three known genes, ie, SOX5, JAW1, and K-RAS. 45 It is unknown so far whether amplification of wild-type K-RAS in these tumors has the same effects as RAS mutations. Finally, the clinical relevance of a restricted 12p amplification has not been conclusively investigated in TGCTs.
The goal of this study is to further investigate the biological and clinical significance of gain of 12p sequences in TGCTs. The incidence of restricted 12p amplification was studied in a consecutive series of 76 untreated primary TGCTs. The newly found six tumors, as well as the previously identified nine cases, were studied for the distribution of the restricted 12p amplification within the tumor (homogeneous or heterogeneous pattern), the borders of the SROA, the presence of i(12p), the proliferative activity, the presence of apoptosis, and capacity of the tumor cells to survive in vitro. In addition, corresponding CIS and microinvasive seminoma were tested for the presence of the restricted 12p amplification. The clinical importance of the restricted 12p amplification and RAS mutation was further studied in 44 patients who failed cisplatinum-based chemotherapy.
Materials and Methods
Samples
The freshly obtained tumor samples included in this study were collected in close collaboration with urologists and pathologists in the southwestern part of the Netherlands. All tumors were obtained before chemotherapy and/or irradiation. Directly after surgical removal, representative parts of the tumor and adjacent normal tissue (when available) were snap-frozen and other pieces were fixed overnight in 10% buffered formalin and embedded in paraffin. The sizes of the testis and the tumor were measured in three dimensions. The tumors were diagnosed according to the World Health Organization classification for testicular tumors. 46 Nonseminomas containing both a seminoma and a nonseminoma component were classified as combined tumors according to the British classification, 47 instead of as nonseminomas according to the World Health Organization classification system. Identification of CIS, seminoma, and embryonal carcinoma was aided by direct enzyme histochemical detection of alkaline phosphatase activity on representative frozen tissue sections, as reported before. 48 The consecutive series tested for the presence of a restricted 12p amplification (see below) consisted of 46 seminomas, 23 nonseminomas (14 embryonal carcinomas/yolk-sac tumors/teratomas; three teratomas; two embryonal carcinomas/yolk-sac tumors; two yolk-sac tumors, one teratoma/yolk-sac tumor; one embryonal carcinoma) and seven combined tumors (three embryonal carcinomas/seminomas; three embryonal carcinomas/yolk-sac/seminomas; one teratoma/yolk-sac tumor/seminoma). The newly identified cases with a restricted 12p amplification, and the previously found cases 45 and unpublished observations, were studied for the borders of the amplicon (see below), the distribution within the tumor (see below), presence of i(12p) by karyotyping (when available), proliferation index (see below), apoptosis, and in vitro survival, as described before (see below). 38,39 In addition, the formerly identified seminomas with a mutated RAS gene 37 were included in this analysis.
The analysis of the possible clinical impact of the restricted 12p amplification/K-RAS mutation was studied on a series of patients who failed cisplatinum-based chemotherapy. These cases were collected in collaboration with the Departments of Hematology and Oncology, University of Tübingen, Germany, and Internal Medicine, Netherlands Cancer Institute, Amsterdam, The Netherlands. In total, samples of 44 different patients were included, of whom 22 primary TGCTs and 33 metastasis were studied (of 11 patients both the primary and metastasis was available).
Immunohistochemical Detection of Ki-67
Sections were cut from one representative paraffin block per tumor, which was mounted on 3-aminopropyl-triethoxysilane-coated slides and dried at 50°C overnight. Subsequently, the sections were heated to 120°C in sodium citrate solution (0.01 mol/L, pH 6.0) using an autoclave. 49 Endogenous peroxidase reactivity was blocked with H2O2/methanol. The Ki-67 antigen was demonstrated using the polyclonal antibody A0047 (DAKO, Glostrup, Denmark), diluted 1:100. Incubation was done for 1 hour at room temperature after blocking of nonspecific binding sites with 5% bovine serum albumin. After extensive washing, biotinylated swine anti-rabbit (1:200) (DAKO) was used as second step, which was detected using the horseradish-labeled streptavidin-biotin complex (DAKO) diluted 1:100. Peroxidase was visualized with diaminobenzidine, after which the sections were counterstained lightly with Mayer’s hemalum. Of each tumor, 3 × 50 tumor cells were independently counted by two observers in one tissue section. The results were statistically analyzed using the Student’s t-test. Positive and negative (excluding the first antibody) controls were included in each experiment.
Metaphase Preparations
After surgical removal representative parts of nonseminomas were enzymatically digested (collagenase; Sigma Chemical Co., St. Louis, MO), and cultured in T75 flasks (Corning Costar, Europe, Schiphol-Rijk, The Netherlands) for several days under standard conditions, ie, 37°C in a humidified atmosphere with 5% CO2 in air in Dulbecco’s modified Eagle’s medium/HF12 culture medium with 10% heat-inactivated fetal calf serum (Gibco-BRL, Life Technologies BV, Brueda, The Netherlands) as described before. 50 Mitotic cells were harvested after 2 to 4 hours of colcemid treatment, swollen in hypotonic solution, and fixed in methanol/acetic acid (3:1). Representative parts of seminoma were directly processed to isolate metaphase spreads as described before. 51 Briefly, the mitotic cells were, after mechanical dissociation of the tumor, directly harvested in the presence of colcemid. The cells were subsequently swollen in hypotonic KCl/EGTA/Hepes solution and fixed with methanol:acetic acid (3:1). For conventional G-band karyotyping the air-dried preparations were digested with pancreatin. The chromosome constitution was described according to the International System for Human Cytogenetic Nomenclature 1995. 52
Restricted 12p Amplification
For the random screen the in situ hybridization experiments on the methanol/acetic acid-fixed nuclei suspensions on the consecutive series were performed as described earlier 45 using YAC#5 (mapped to 12p11.2–12.1; kindly provided by Dr. B. Gemmill, Denver, CO). This probe is known to map to the SROA as determined in our earlier study. 45 To make this probe suitable for the in situ hybridization approach, it was purified by pulsed-field gel electrophoresis, amplified, and labeled with digoxigenin-11-dUTP (Roche Diagnostics Nedevland BV, Almeve, The Netherlands) using a nick-translation kit (Gibco-BRL). It was visualized with fluorescein isothiocyanate-conjugated sheep-α-digoxigenin (Roche). The presence of a 12p amplification was defined as reported before 45 as being ∼15 to 30 signals per interphase nucleus.
TGCTs identified with a restricted 12p amplification were studied in more detail regarding the breakpoints and distribution. Of these cases, frozen tissue sections of 16-μm thickness, containing tumor, microinvasive seminoma, and CIS (when available), were cut and air-dried overnight at 37°C on microscope slides treated with 3-aminopropyl-triethoxysilane. In addition, one parallel section (4-μm thickness) was stained with hematoxylin and eosin (H&E) and the other for alkaline phosphatase reactivity for histological examination. The slides for the in situ hybridization were submerged in 70% ethanol (−20°C) for 1 hour and dehydrated in an increasing ethanol series (80%, 90%, 100%, 2 minutes each) and air-dried. Subsequently, the tissue sections underwent protein digestion with 0.0005% pepsin (Sigma) in 0.01 mol/L HCl in water, 1 minute at 37°C, followed by a wash step (phosphate-buffered saline, 5 minutes) and dehydration. Hybridization was performed as described for the methanol/acetic acid fixed nuclei. 45 YAC#5 was used as control probe in combination with another probe (test probe) (Figure 1) ▶ . In particular, probes positive for sequence-taq-sites D12S1945, D12S1688, and AFM267yc9 (on the distal side) and D12S1350E, KRAS2, D12S1313, and D12S1411 (on the proximal border) were used as test probes. YAC#5 was labeled as described above with digoxigenin-11-dUTP, and visualized using fluorescein isothiocyanate-conjugated sheep anti-digoxigenin (Roche). The test probe was also labeled using nick-translation with biotin-11-dUTP and detected using avidine-CY3 (Jackson ImmunoResearch, West Grove, PA). A restricted 12p amplification was defined as the presence of nuclei with at least 15 hybridization signals. The pattern was identified as heterogeneous when the positive nuclei are scattered throughout the tissue section: both regions with and without restricted 12p amplification are present. In contrast, the pattern is defined as homogeneous when all regions showed tumor nuclei with restricted 12p amplification. Because of tissue cutting artifacts, this does not exclude the presence of single nuclei without such an amplification. The criteria used to determine the borders of the breakpoints was similar to that used in our former study. 45 Briefly, the test probe was scored as part of the amplicon when paired hybridization signals with the control probe (YAC#5) was observed, and scored as outside the amplicon when less hybridization signals were found compared to the control probe. Higher copy numbers of the test probes (see above) compared to the control probe have not been found so far.
Figure 1.

Schematic representation of the physical map of the 12p11.2-p12.1 region. Genes, STSs, and the position the YAC#5 probe (positive for STS D12S1057) are indicated. The results of the double-fluorescent in situ hybridization experiments performed on frozen tissue sections to determine the shortest region of overlap of amplification are shown. The continuous bold lines indicate the regions included in the restricted 12p amplification, and the dotted lines indicate the regions outside the amplification. The probes specific for STSs D12S1945, D12S1688, AFM267yc9, K-RAS, D12S1313, and D12S1411 are used for these analyses. The results are shown per histological subgroup (seminomas, nonseminomas, and combined tumors). The tumors studied in our previous analysis, 45 are indicated by an asterisk. Note that three breakpoints map between AFM267yc9 and D12S1688, and seven between AFM267yc9 and D12S1945 at the distal side, and six between K-RAS and D12S1313 at the proximal side. The shortest region of amplification encompasses the genomic fragment between AFM267yc9 and K-RAS.
The paraffin-embedded tissue sections of the treatment-resistant TGCTs (4-μm thickness) were preincubated overnight at 50°C, and subsequently baked for 10 minutes at 80°C. The sections were deparaffinized using xylene, washed in 100% methanol four times at room temperature, and air-dried. Sodium thiocyanate (1 mol/L) pretreatment was done for 10 minutes at 80°C, after which the slides were thoroughly rinsed in water. Digestion was done using 8000 U pepsin (Sigma) in 0.2 mol/L HCl in phosphate-buffered saline at 37°C for 30 to 40 minutes, depending on the tissue under investigation. After rinsing in water at 4°C, dehydration was done using a series of increasing ethanol concentrations (70%, 80%, 90%, and 100%). The hybridization with a K-RAS-specific digoxigenin-labeled probe, washing, and detection procedures were performed as described for the methanol/acetic acid-fixed nuclei (see above). A restricted 12p amplification was defined as the presence of at least 10 hybridization signals of the K-RAS probe. Because of the thickness of the tissue sections used, the tumors were not scored for the presence of a homogeneous or heterogeneous pattern.
Visualization was done with a Zeiss Axioskop epifluorescence microscope (Carl Zeiss, BV, Weesp, The Netherlands) with a Pinkel filter in combination with a triple band-pass filter, which enabled the observation of fluorescein isothiocyanate, CY3, and DAPI in one view.
Comparative Genomic Hybridization (CGH)
For CGH, metaphase spreads were prepared using standard procedures from in vitro phytohemagglutinin-stimulated peripheral blood lymphocyte cultures of a healthy male as reported previously. 53 High molecular weight DNA was isolated from the snap-frozen tissue samples (test DNA) and from peripheral blood of a normal male (reference DNA) using standard procedures. 54 The CGH procedure and analysis were performed as described before. 55 Both the control male DNA and the tumor DNA were directly labeled by nick-translation with lissamine and fluorescein isothiocyanate, respectively. The data were analyzed using Quips XL software from Vysis (Downers Grove, IL). Normalization was performed using the average of the green-to-red fluorescent intensities for the entire metaphase. At least 10 metaphases were studied for each case. Losses of DNA sequences were defined as chromosomal regions where the average green-to-red ratio and its 95% confidence intervals are below 0.9 whereas gains are >1.1. 56 The heterochromatic blocks of chromosomes 1, 9, 16, and Y, and the immediate telomeric regions were excluded from the analysis because these regions present variable results in experiments with normal control DNAs.
Spectral Karyotyping
Spectral karyotyping was performed on a single seminoma with a restricted 12p amplification, demonstrated both by CGH and in situ hybridization, using a spectral karyotyping kit and analysis system (Vysis) with a slightly modified procedure as described by the supplier. In particular, proteinase K digestion was performed in 1 mol/L Tris-HCl, pH 7.5, and after denaturation of the probe, the mix was kept on ice. Washing during the detection procedure was performed in 55% formamide at 39°C instead of 45°C. In addition, 0.05% instead of 0.1% Tween was used in 4× standard saline citrate, and the last washing step was performed at room temperature.
RAS Gene Mutation Detection
Mutations in codon 12 or 13 of the N- and K-RAS gene were analyzed by direct sequencing (Amplicycle; Amersham, Arlington Heights, IL), using primer NA (5′-GACTGAGTACAAACTGGTGG-3′)/NB (5′-CTCTATGGTGGGATCATATT-3′) and KA (5′-GACTGAATATAAACTTGTGG-3′)/KB (5′-CTATTGTTGGATCATATTCC-3′), respectively, on DNA isolated from snap-frozen seminomas tested for in vitro survival and presence of apoptosis. DNA was isolated as described above. Only histological areas containing >70% tumor cells were used. In addition, the presence of K-RAS codon 12 mutations was investigated in the series of paraffin-embedded tumors of the nonresponding patients as follows: two 15-μm thickness sections from each sample were first deparaffinized with xylene and then dehydrated with absolute ethanol. A parallel section (4-μm thickness) was stained with H&E to confirm the presence of tumor and to check for histology. Only sections with a major tumor component were used for DNA isolation. DNA was eluted in 30 μl of water by heating the air-dried tissues at 95°C for 5 minutes. Typically 50 to 100 ng of DNA was used for each polymerase chain reaction (PCR). Each sample was first tested with HLA-dQ primers 57 to assess the quality of the eluted DNA. Only samples showing proper amplification were subsequently used. The PCR reactions to detect mutations in codon 12 of K-RAS were performed essentially as described before 58 with the following modifications. The K-RAS 5′ primer used in the two rounds of amplifications carried a biotin label at the 5′ end. After the first MvaI digestion, the PCR products were affinity purified on streptavidin-coated paramagnetic beads (Dynal AS, Oslo, Norway) and the bound PCR products were redigested on the beads in 50 μl of MvaI buffer containing 10 U of enzyme for 3 hours at 37°C. Subsequently the beads were affinity purified and washed on the magnet to remove the buffer and the unbound fraction. Finally, single-stranded DNA was eluted from the bound fraction by boiling the beads with 10 μl of water. One half of each fraction was used in the second amplification step. Samples positive for mutations in codon 12 were further characterized by cycle sequencing the single-stranded DNA obtained by affinity purification of the PCR products retained on the beads after the final MvaI digestion. The procedure was controlled at every stage with DNA samples obtained from archival paraffin-embedded primary TGCTs with and without codon 12 mutations. 37
Detection of DNA Laddering
High molecular weight DNA was isolated from snap-frozen histologically checked samples using standard procedures (see above). DNA laddering was visualized using ethidium bromide staining after electrophoresis of 1 μg as described previously. 39
Results
Restricted 12p Amplification and i(12p) in Primary Invasive TGCTs
The incidence of a restricted 12p amplification in primary untreated TGCTs was determined by analysis of methanol/acetic acid-fixed nuclei suspensions of 76 surgically removed, consecutively collected TGCTs, including 46 seminomas, 23 nonseminomas, and seven combined tumors (see Table 1 ▶ and Materials and Methods for histology). Double-fluorescent in situ hybridization showed that all tumor nuclei had more than four copies of 12p, and at least three chromosome 12 centromeres (not shown). Six tumors (7.9%), ie, four seminomas (8.7%), one nonseminoma (4.3%), and one combined tumor (14.2%) showed the restricted 12p amplification. So far, in total 15 primary TGCTs with such a restricted 12p amplification are available (the six identified in this consecutive study and nine previously found by us, of which six have been included in our previous study 45 ): nine seminomas, four nonseminomas, and two combined tumors. Karyotyping (Table 1) ▶ revealed the presence of i(12p) in two of the six successfully analyzed seminomas with a restricted 12p amplification. However, the presence of an i(12p) could not be confirmed by in situ hybridization on matched metaphase spreads, although the amplification was found (not shown). Only one of the four karyotyped 12p amplification-positive nonseminomas contained an i(12p) in some of the metaphases studied, whereas the only karyotyped combined tumor lacked it. Because of inappropriate quality of this material no in situ hybridization could be performed on these cases. In the informative cases, it was found that the additional 12p sequences visualized by in situ hybridization were predominantly (five of the nine cases) present as tandemly organized units as add (12)(p11), add(p12), or add(p13), although other sites were also found to contain 12p-specific sequences, including parts of chromosomes 6, 8, and 11 (not shown).
Table 1.
Results of Detection of the Restricted 12p Amplification Using Methanol/Acetic Acid-Fixed Nuclei Suspensions of a Consecutive Series of Primary Seminomas, Nonseminomas, and Combined Tumors as Detected by Double-Fluorescent In Situ Hybridization
| Consecutive series | i(12p) + | Heterogeneity | |||
|---|---|---|---|---|---|
| Tested | +12p amplification | Karyotyping | In situ hybridization | ||
| Seminoma | 46 | 4 (9) | 2/6 | 0/6 | 3/9 |
| Nonseminoma | 23 | 1 (4) | 1/4 | NA | 4/4 |
| Combined tumor | 7 | 1 (2) | 0/1 | NA | 2/2 |
| Total | 76 | 6 (15) | |||
The total numbers of cases with a restricted 12p amplification identified so far are indicated between brackets. The results of karyotyping and in situ hybridization on matched frozen tissue sections are also shown. Abbreviations: +12p amplification, with restricted 12p amplification; NA, not available.
Breakpoints Involved in the Restricted 12p Amplification
In addition to our earlier report consisting of six cases, 45 three seminomas, two nonseminomas, and one combined tumor (indicated by an asterisk in Figure 1 ▶ ), in situ hybridization was applied on the newly identified TGCTs (n = 9) with a restricted 12p amplification. Because this analysis was done on frozen tissue sections instead of methanol/acetic acid-fixed nuclei suspension, as in our earlier study, all cases could be analyzed, including the two previously identified noninformative cases. 45 Again YAC#5 was used in combination with probes specific for the more proximal and distal regions of the contig (see Figure 1 ▶ and Materials and Methods section). In accordance with our previous findings, the amplified region always includes the genomic fragment between STS AFM267yc9 at the distal end and K-RAS at the proximal end. Therefore, K-RAS, JAW1, and SOX5 are consistently amplified in all TGCTs with a restricted 12p amplification, irrespective of histology. In addition to this finding, the borders of the amplicon appeared to cluster in narrow regions: 40% between K-RAS and STS D12S1313 at the proximal side (44.4% for seminoma and 25% for nonseminoma), and 20% between AFM267yc9 and D12S1688 at the distal side (11% for seminoma and 50% for nonseminoma). In addition, 46.7% of the breakpoints map between AFM267yc9 and D12S1945 (44.4% for seminoma and 75% for nonseminoma) (Figure 1) ▶ .
Intratumor 12p Amplification and Tumor Heterogeneity
In situ hybridization results on the suspensions of nuclei already indicated that the restricted 12p amplification can be heterogeneously distributed in one tumor (not shown). This was verified by in situ hybridization on frozen tissue sections. Six out of the nine seminomas showed the amplification homogeneously throughout the tumor (Table 1) ▶ . The other three cases showed regions with and without amplification, of which two contained i(12p) by karyotyping. All nonseminomas and combined tumors showed a heterogeneous pattern. In two cases the amplification was present in a subpopulation of cells of the yolk-sac tumor component, and in one in a subpopulation of both the embryonal carcinoma and yolk-sac tumor component. One of the combined tumors with the restricted 12p amplification was a mixture of seminoma and embryonal carcinoma. The amplification was found in a subpopulation of both components. No i(12p) was identified in this case. The other combined tumor showed amplification in a minority of the seminoma cells only and karyotyping failed.
One of the nonseminomas, initially diagnosed as mixed nonseminoma showed no i(12p) by karyotyping. This tumor was xenografted (orthotopically into a nude rat), and after ∼10 months, a tumor completely composed of yolk sac tumor was formed. Karyotyping after direct harvesting revealed i(12p) in seven out of 14 metaphase spreads, confirmed by in situ hybridization (not shown). These metaphase spreads did not contain the 12p amplification. However, a limited number of interphase nuclei isolated from the original tumor and the xenograft showed the presence of 12p amplification by in situ hybridization (not shown). In situ hybridization on frozen tissue sections of these tumors demonstrated that <5% of the tumor cells from the primary tumor showed the 12p amplification, preferentially in the embryonal carcinoma and yolk-sac tumor component. This percentage was constant during subsequent xenograft passages.
CGH was performed on three seminomas with homogeneous restricted 12p amplification, three of the heterogeneous nonseminomas, and on both combined tumors. No other high-level amplifications, besides the one on 12p, were found in these cases, and a similar pattern of gains and losses was identified as reported before (not shown). 43,56 Every tumor with a restricted 12p amplification also showed gain of the complete short arm of chromosome 12, of which representative examples are illustrated in Figure 2A ▶ . Spectral karyotyping was done on one seminoma, of which a sufficient number of metaphase spreads containing the restricted 12p amplification was available. The results confirm the presence of chromosome 12 material in the aberrant chromosome 12 derivatives and in the most telomeric region a part of chromosome 20 (Figure 2B) ▶ .
Figure 2.
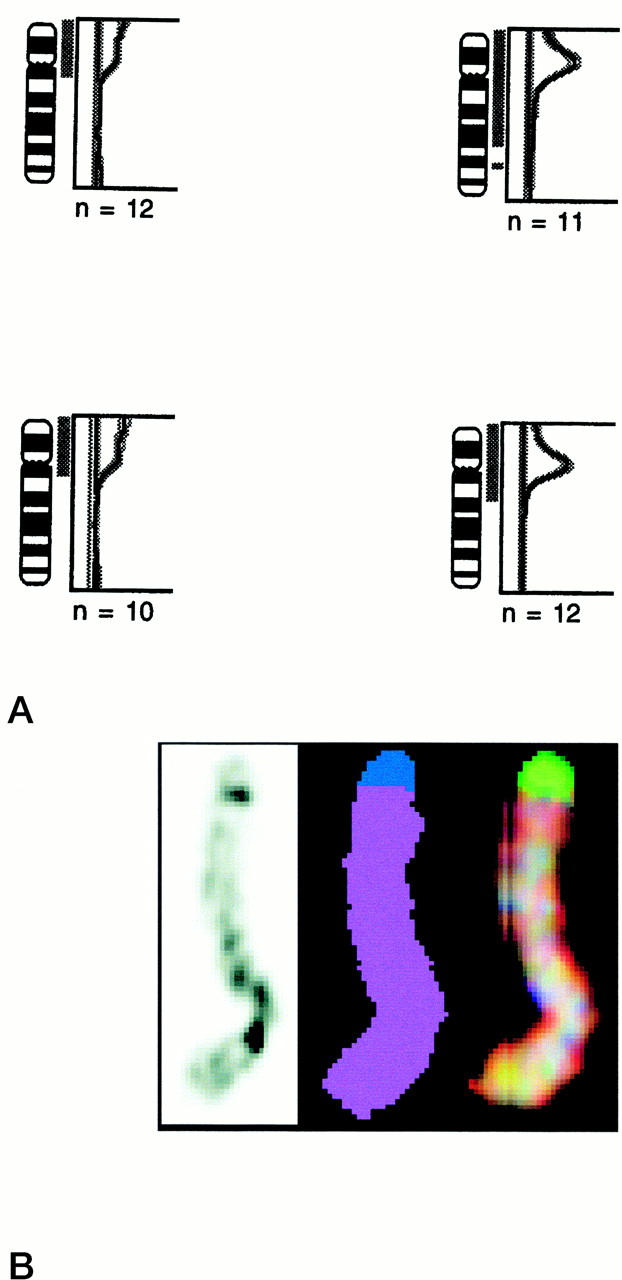
A: Representative examples of CGH for chromosome 12 on two seminomas without 12p amplification (left) and two with a homogeneous restricted 12p amplification (right). Note that the seminomas with an amplification also show gain of the complete short arm of this chromosome. The number of metaphase spreads analyzed is indicated. B: Representative example of banding (left) and spectral karyotyping (real color at the right, and false color in the middle) of a chromosome 12 derivative known to contain restricted 12p amplification (as determined by in situ hybridization and CGH). Note that the chromosome is composed of chromosome 12 material (indicated in purple), except the most distal region of the p arm, showing chromosome 20-specific material (indicated in blue).
Restricted 12p Amplification and Tumor Development
Matched CIS cells and microinvasive seminoma of the TGCTs with 12p amplification were studied by in situ hybridization on tissue sections. The cells of interest were visualized by enzyme histochemistry for alkaline phosphatase reactivity. Out of the eight seminomas studied, three showed the presence of both CIS and microinvasive seminoma, and one only of CIS. The amplification was found in all microinvasive components, but never in CIS (summarized in Table 2 ▶ ), of which examples are shown in Figure 3 ▶ . Also no restricted 12p amplification was found in the matched CIS of one nonseminoma and one combined tumor. The microinvasive seminoma cells adjacent to the combined tumor showed also a heterogeneous pattern of restricted 12p amplification as detected in the matched invasive tumor (not shown).
Table 2.
Summary of the Presence of the Restricted 12p Amplification in the Different Development Stages of Testicular Germ Cell Tumors of Young Adults, as Determined by In Situ Hybridization on Frozen Tissue Sections
| Carcinoma in situ | Micro-invasive seminoma | Invasive tumor | |
|---|---|---|---|
| n = 3 | − | + | + (SE) |
| n = 1 | − | NA | + (SE) |
| n = 1 | − | NA | ± (NS) |
| n = 1 | − | ± | ± (CT) |
Abbreviations: SE, seminoma; NS, nonseminoma; CT, combined tumor; NA, not available; −, absence of restricted 12p amplification; +, presence of restricted 12p- amplification; ±, heterogeneous presence of 12p amplification.
Figure 3.
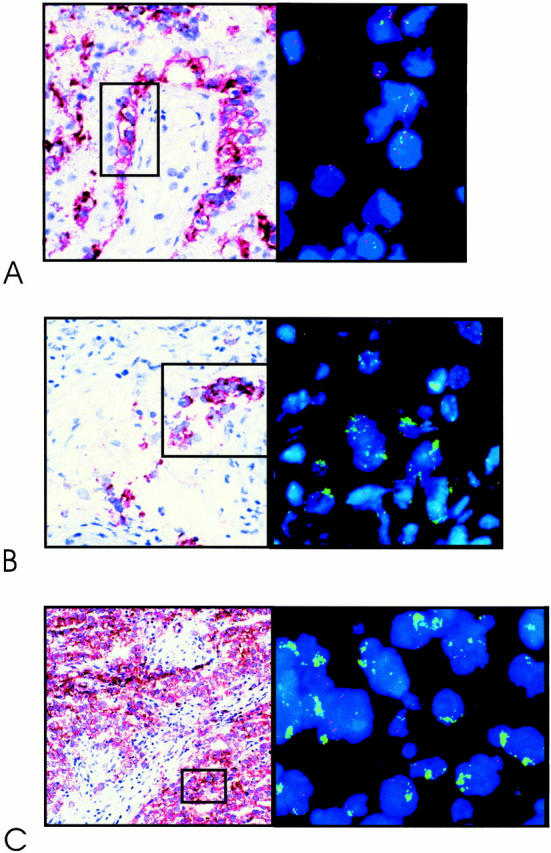
Representative examples of double-fluorescent in situ hybridization on frozen tissue sections, 4-μm thick, using a probe specific for the centromeric region of chromosome 12 (red), and YAC#5 (green), mapped within the shortest region of overlap of amplification (see Figure 1 ▶ ). Shown are carcinoma in situ (A); microinvasive seminoma (B); and invasive seminoma (C) . The tumor cells are identified by the direct enzymatic alkaline phosphatase detection method (stained in red) on a parallel tissue section.
12p Amplification/RAS Mutation, Proliferation, Apoptosis, and in Vitro Survival
Immunohistochemistry for ki-67 on histological sections was performed to investigate the proliferation index in seminomas containing 12p amplification (n = 8) or RAS mutation (n = 4; of two no appropriate material was available anymore), or neither of these (n = 13). As indicated in Table 3 ▶ , no differences between these groups were observed. However, all nine seminomas with 12p amplification, as well as five of the six with a mutated RAS gene, demonstrated low levels of apoptosis in the primary tumor, indicated by the lack of profound DNA laddering after electrophoresis. In contrast, nine of 13 tested seminomas without any of these features showed apoptosis (Table 3 ▶ and Figure 4 ▶ ).
Table 3.
Summary of the Results on Proliferation Index, Apoptosis, In Vitro Survival, and Volume of the Tumor (cm3) in Seminomas With and Without a Restricted 12p Amplification/RAS Mutation
| Wild-type RAS | Mutant RAS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Proliferation index | Apoptosis | In vitro survival | Tumor volume | Proliferation index | Apoptosis | In vitro survival | Tumor volume | |||||
| + | − | + | − | + | − | + | − | |||||
| 12p-ampl. | ||||||||||||
| − | 34.5 (10.5) | 9 | 4 | 1 | 11 | 256 (173) | 30.8 (3.1) | 1 | 5 | 6 | 0 | 121 (88) |
| + | 29.7 (15.3) | 0 | 9 | 4 | 0 | 246 (294) | NA | NA | NA | NA | NA | NA |
Abbreviations: 12p-ampl., restricted 12p amplification; +, present; −, absent; NA, not available. Standard deviations are indicated between brackets.
Figure 4.

Representative examples of the presence of DNA laddering after electrophoresis (indicative for apoptosis) of 1-μg high molecular weight DNA isolated from primary seminomas with and without a restricted 12p amplification/RAS mutation. Note the correlation between less apoptosis and the presence of a restricted 12p amplification or RAS mutation. In addition, the results of in vitro culture of the tumor cells are shown (see also Table 3 ▶ ).
The capacity of the tumor cells to survive in vitro after disruption of their microenvironment to generate single cell suspensions was studied as described before, 38 of which the results are also summarized in Table 3 ▶ . We excluded that one of the newly identified seminomas in this study contained a mutation of either the N- or K-RAS gene in codons 12 and 13 (not shown). All six seminomas with a RAS mutation and all four tested seminomas with a restricted 12p amplification showed prolonged in vitro survival (at least up to 10 days). Eleven seminomas without a restricted 12p amplification or RAS mutation of the 12 tested could not be maintained in vitro. Nine of these showed profound apoptosis. The seminoma that showed in vitro survival for several days did not show obvious apoptosis.
The nonseminomas, with or without restricted 12p amplification all lacked significant apoptosis, as was found for both combined tumors. All nonseminomas and one combined tumor were successfully karyotyped after short-term in vitro culturing, indicating that these tumors allowed short-term culturing in vitro (see Materials and Methods).
12p Amplification, RAS Mutation, and Clinical Behavior
The age at clinical diagnosis of patients with a seminoma with and without a restricted 12p amplification/RAS mutation is indicated in Figure 5 ▶ . No age differences exist between seminoma and nonseminoma patients with and without a RAS mutation (Figure 5 ▶ and not shown). Despite the small number of cases, seminoma patients with a restricted 12p amplification showed a trend toward a younger age at clinical presentation than those without (P = 0.055, Student’s t-test). When only the seminomas with a homogeneous pattern of 12p amplification were included (n = 6) (see above), the age difference was significant: 36.7 years without a restricted 12p amplification and 26.8 years with a restricted 12p amplification (Figure 5 ▶ ; P = 0.023, Student’s t-test). No differences were found between these different groups regarding stage of the disease (they all presented as stage I or II), size of the tumor (see Table 3 ▶ ), and clinical response (all had a complete response). In addition, no correlation was found between outcome and age, stage, proliferation index, or apoptosis. Also no correlation was detected between the presence of 12p amplification/RAS mutation and proliferation index.
Figure 5.

Schematical representation of the ages of patients at clinical presentation with a seminoma with and without 12p amplification and RAS mutation (mean, average, and standard deviations are indicated). In addition, the ages of the subgroup of patients with a homogeneous 12p amplification are shown. No differences were found between the ages of patients with a seminoma without either of these aberrations and those with a RAS mutation. However, those with a 12p amplification-positive seminoma showed a borderline significant difference compared to the control group, whereas a significant difference was found in case only patients with a homogeneous 12p amplification were included (P = 0.023). Abbreviations: RAS+, RAS mutation; RAS−/12p−, no RAS mutation or 12p amplification; 12p+, 12p amplification both heterogeneously and homogeneously present; 12p+ homo, homogeneous 12p amplification.
To extend the analysis of the clinical significance of K-RAS mutations/12p amplifications, we retrospectively investigated a series of 22 primary tumors and 33 surgical specimens from metastasis from a total of 44 different patients who failed cisplatinum-based chemotherapy. No K-RAS mutations were identified. The sensitive detection method (see Materials and Methods), makes it highly unlikely that mutated genes were obscured by a predominance of wild-type alleles. A restricted 12p amplification was detected once in a metastatic tumor (primary not available).
Discussion
Although gain of the short arm of chromosome 12 has already been recognized for more than 25 years as a characteristic chromosomal anomaly in TGCTs, 6,7 the biological significance still remains unknown, and the clinical importance is a matter of debate. Because overrepresentation of 12p is found in all TGCTs, 7,59 it is most likely crucial in the development of this cancer. Here we show that the actual incidence of a restricted 12p amplification is around 8% for primary TGCTs. Our earlier observation that the three known genes JAW1, SOX5, and K-RAS map in this region, 45 is confirmed in the study presented here. We hypothesized that if important genes for the development of this cancer reside in this region of 12p, a comparative study of TGCTs with and without this type of amplification could be meaningful. In addition, because K-RAS maps within the SROA, these tumors were compared with previously published seminomas with a RAS mutation. 37
Although we were unable to reduce the SROA, as compared to our previous findings, 45 it was found that the breakpoints of the amplicon cluster in rather narrow regions (40% between D12S1313 and K-RAS at the proximal end and 53% between AFM267yc9 and D12S1945 at the distal end). Assuming that the STSs are spread over the genome with intervals of ∼250 kb, 160 fragments are to be expected on the short arm of chromosome 12 (around 40 Mbases). The change that one breakpoint maps within the region between STS AFM267yc and D12S1945 is estimated to be three times 6.3E-3. The change that six additional breakpoints map to that particular region is around 5.7E-9. In addition, the change that six breakpoints map between K-RAS and D12S1313 is around 1.3E-11 (binomial distribution). This indicates a strong positive selection toward these particular regions. Because no fragile sites have been identified at these regions, the closest fragile site has been reported on 12p11, 60 which might be related to the development of the isochromosome 12p, the borders may be determined by the presence of genes which confer selective advantage to the tumors. If this is true, the amplicon contains at least two genes of interest, a hypothesis that is currently under investigation.
All TGCTs with 12p amplification identified so far also show gain of the complete short arm of chromosome 12. Interestingly, seminomas with a homogeneous 12p amplification, ie, present in all cells, lack i(12p), whereas those showing a heterogeneous pattern could contain an isochromosome. The restricted 12p amplification and i(12p) were never found in the same cell. A similar pattern was found for the nonseminomas and combined tumors. Interestingly, the seminomatous components of the combined tumors showed a similar pattern of restricted 12p amplification as the pure seminomas. Clearly, 12p amplification is found predominantly in TGCTs without i(12p). These results suggest that there are at least two mechanisms leading to extra copies of the short arm of chromosome 12 in TGCTs: i(12p) formation, and an alternative way, also leading to extra copies of the complete short arm of chromosome 12, which can be followed by amplification of a restricted region of 12p.
The homogeneous presence of 12p amplification in most of the identified seminomas (and not in nonseminomas) implies that increased copy numbers of genes present in the amplified region result in a selective advantage for these tumor cells containing this specific anomaly. We demonstrated that this is related to a reduced apoptosis and not because of enhanced proliferation. A similar lack of apoptosis was observed previously by us in seminomas containing a mutated RAS gene, 38,61 which also showed an enhanced in vitro survival and reduced apoptosis. These results are in accordance with the idea that activation of RAS by mutation or amplification can cause inhibition of apoptosis.
Despite data on the role of activated RAS in resistance to irradiation and chemotherapy and induction of metastatic potential 21,22,30-34 and our results on in vitro survival and apoptosis, 38,39 no differences were found regarding stage of the primary seminoma at clinical presentation and treatment response based on the presence of a RAS mutation or restricted 12p amplification. This is supported by an observation in two patients: 62 the restricted 12p amplification (detected by CGH) was present in both a responding and a nonresponding tumor. In addition, we observed no predictive value of K-RAS mutations and restricted 12p amplification in chemoresistance of patients with extensively pretreated TGCTs. Patients having a seminoma with a homogeneous restricted 12p amplification were, however, younger at clinical presentation than those lacking it. This is most likely explained by an early development of the restricted 12p amplification in these tumors, leading to reduced apoptosis. This combined with an unchanged proliferation index results in faster growth of the tumor and an earlier clinical presentation. Such an age difference was not observed in the case of a RAS mutation, which is likely because mutation of RAS is a relatively late event in the pathogenesis of TGCTs. This is supported by our observation that TGCTs with a mutated RAS gene also show gain of the complete short arm of chromosome 12, although no information about the presence of i(12p) is available (not shown).
Our data suggest that RAS mutation and restricted 12p amplification are related to invasive growth of TGCTs. It allows tumors cells to survive outside the specific microenvironment of the CIS cells by inhibition of induction of apoptosis, known as “anoikis.” 20 The reason that homogeneous 12p amplification is predominantly found in seminomas can be related to the diminished requirements of nonseminomatous tumor cells for survival. This idea is supported by the relative ease of short-term culture for karyotyping, 59 and the success rate of establishing nonseminoma cell lines. 63 It remains to be established whether involvement of RAS causes down-regulation of FAS, as suggested recently, 64,65 and whether activation of the serine/threonine kinase AKT is involved. 66,67 In addition, it is unknown thus far which genes in the SROA are in fact involved. The absence or at least lower incidence of gain of 12p sequences in CIS as reported before 68-70 and unpublished observations support this model.
Acknowledgments
We thank the pathologists and urologists in the southwestern part of The Netherlands for their help in collecting the tumor samples; Dr. D. de Jong and Prof. S. Rodenhuis (Pathology and Experimental Therapy, Netherlands Cancer Institute, Amsterdam, The Netherlands) for their help in providing part of the tumors of patients that failed cisplatinum-based chemotherapy; and Dr. R. Slater and E. van Drunen for their help in doing the spectral karyotyping.
Footnotes
Address reprint requests to L. H. J. Looijenga, Ph.D., Pathology/Laboratory for Experimental Patho-Oncology, University Hospital Rotterdam/ Daniel, Josephine Nefkens Institute, FGG/EUR Building Be, Room 430b, P. O. Box 1738, 3000 DR Rotterdam, The Netherlands. E-mail: looijenga@leph.azr.nl.
Supported by Dutch Cancer Society grant KWF, DDHK 94 836 and a fortune grant of Tübingen University.
References
- 1.Vogelstein B, Kinzler KW: The multistep nature of cancer. Trends Genet 1993, 9:138-141 [DOI] [PubMed] [Google Scholar]
- 2.Mostofi FK, Sesterhenn IA, Davis CJJ: Immunopathology of germ cell tumors of the testis. Semin Diagn Pathol 1987, 4:320-341 [PubMed] [Google Scholar]
- 3.Skakkebæk NE: Possible carcinoma-in-situ of the testis. Lancet 1972, 2:516-517 [DOI] [PubMed] [Google Scholar]
- 4.Jørgensen N, Rajpert-De Meyts E, Graem N, Müller J, Giwercman A, Skakkebæk NE: Expression of immunohistochemical markers for testicular carcinoma in situ by normal fetal germ cells. Lab Invest 1995, 72:223-231 [PubMed] [Google Scholar]
- 5.Gondos B: Ultrastructure of developing and malignant germ cells. Eur Urol 1993, 23:68-75 [DOI] [PubMed] [Google Scholar]
- 6.Atkin NB, Baker MC: i(12p): specific chromosomal marker in seminoma and malignant teratoma of the testis? Cancer Genet Cytogenet 1983, 10:199-204 [DOI] [PubMed] [Google Scholar]
- 7.Sandberg AA, Meloni AM, Suijkerbuijk RF: Reviews of chromosome studies in urological tumors. 3. Cytogenetics and genes in testicular tumors. J Urol 1996, 155:1531-1556 [PubMed] [Google Scholar]
- 8.Bosl GJ, Dmitrovsky E, Reuter VE, Samaniego F, Rodriguez E, Geller NL, Chaganti RSK: Isochromosome of chromosome 12: clinically useful marker for male germ cell tumors. J Natl Cancer Inst 1989, 81:1874-1878 [DOI] [PubMed] [Google Scholar]
- 9.Bosl GJ, Chaganti RSK: The use of tumor markers in germ cell malignancies. Hematol Oncol Clin North Am 1994, 8:573-587 [PubMed] [Google Scholar]
- 10.Malek NP, Casper J, Looijenga LHJ, Strohmeyer T, Schmoll H-J, Nordheim A, Janknecht R: Quantification of additional short arms of chromosome 12 in germ cell tumors using polymerase chain reaction. Eur J Cancer 1997, 33:1488-1494 [DOI] [PubMed] [Google Scholar]
- 11.Dmitrovsky E, Murty VVVS, Moy D, Miller WH, Jr, Nanus D, Albino AP, Samaniego F, Bosl G, Chaganti RSK: Isochromosome 12p in non-seminoma cell lines: karyologic amplification of c-ki-ras2 without point-mutational activation. Oncogene 1990, 5:543-548 [PubMed] [Google Scholar]
- 12.Houldsworth J, Reuter V, Bosl GJ, Chaganti RSK: Aberrant expression of cyclin D2 is an early event in human male germ cell tumorigenesis. Cell Growth Dev 1997, 8:293-299 [PubMed] [Google Scholar]
- 13.Murty VVVS, Chaganti RSK: A genetic perspective of male germ cell tumors. Semin Oncol 1998, 25:133-144 [PubMed] [Google Scholar]
- 14.Lübbert M, Mirro J, Miller CW, Kahan J, Isaac G, Kitchingman G, Mertelsmann R, Herrmann F, McCormick F, Koeffler HP: N-Ras gene point mutations in childhood acute lymphocytic leukemia correlate with a poor prognosis. Blood 1990, 75:1163-1169 [PubMed] [Google Scholar]
- 15.Slebos RJC, Kibbelaar RE, Dalesio O, Kooistra A, Stam J, Meijer CJLM, Wagenaar SS, Vanderschueren RGJRA, van Zandwijk N, Mooi WJ, Bos JL, Rodenhuis S: K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N Engl J Med 1990, 323:561-565 [DOI] [PubMed] [Google Scholar]
- 16.Mitsudomi T, Steinberg SM, Oie HK, Mulshine JL, Phelps R, Viallet J, Pass H, Minna JD, Gazdar AF: Ras gene mutations in non-small cell lung cancers are associated with shortened survival irrespective of treatment intent. Cancer Res 1991, 51:4999-5002 [PubMed] [Google Scholar]
- 17.Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O’Hagan R, Pantginis J, Zhou H, Horner JW, Jr, Cordon-Cardo C, Yancopoulos GD, DePinho RA: Essential role for oncogenic Ras in tumour maintenance. Nature 1999, 400:468-472 [DOI] [PubMed] [Google Scholar]
- 18.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA: Creation of human tumour cells with defined genetic elements. Nature 1999, 400:464-468 [DOI] [PubMed] [Google Scholar]
- 19.Arends MJ, McGregor AH, Toft NJ, Brown EJH, Wyllie AH: Susceptibility to apoptosis is differentially regulated by c-myc and mutated Ha-ras oncogenes and is associated with endonuclease availability. Br J Cancer 1993, 68:1127-1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frisch SM, Francis H: Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 1994, 124:619-626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ananthaswamy HN, Price JE, Tainsky MA, Goldberg LH, Bales ES: Correlation between Ha-ras gene amplification and spontaneous metastasis in NIH 3T3 cells transfected with genomic DNA from human skin cancers. Clin Exp Metastasis 1989, 7:301-313 [DOI] [PubMed] [Google Scholar]
- 22.Takiguchi Y, Takahashi Y, Kuriyama T, Miyamoto T: NIH3T3 transfectant containing human K-ras oncogene shows enhanced metastatic activity after in vivo tumor growth or co-culture with fibroblasts. Clin Exp Metastasis 1992, 10:351-360 [DOI] [PubMed] [Google Scholar]
- 23.Pulciani S, Santos E, Long LK, Sorrentino V, Barbacid M: Ras gene amplification and malignant transformation. Mol Cell Biol 1985, 5:2836-2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKay IA, Marshall CJ, Cales C, Hall A: Transformation and stimulation of DNA synthesis in NIH-3T3 cells are a titratable function of normal p21N-ras expression. EMBO J 1986, 5:2617-2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujita K, Ohuchi N, Yao T, Okumura M, Fukushima Y, Kanakura Y, Kitamura Y, Fujita J: Frequent overexpression, but not activation by point mutation, of ras genes in primary human gastric cancers. Gastroenterology 1987, 93:1339-1345 [DOI] [PubMed] [Google Scholar]
- 26.Radinsky R, Kraemer PM, Raines MA, Kung HJ, Culp LA: Amplification and rearrangement of the Kirsten ras oncogene in virus-transformed BALB/c 3T3 cells during malignant tumor progression. Proc Natl Acad Sci USA 1987, 84:5143-5147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coleman WB, Throneburg DB, Grisham JW, Smith GJ: Overexpression of c-K-ras, c-N-ras and transforming growth factor beta co-segregate with tumorigenicity in morphologically transformed C3H 10T1/2 cell lines. Carcinogenesis 1994, 15:1005-1012 [DOI] [PubMed] [Google Scholar]
- 28.Zhang B, Shih JW, Wear DJ, Tsai S, Lo SC: High-level expression of H-ras and c-myc oncogenes in mycoplasma-mediated malignant cell transformation. Proc Soc Exp Biol Med 1997, 214:359-366 [DOI] [PubMed] [Google Scholar]
- 29.Bokemeyer C, Schmoll HJ: Treatment strategies in metastatic testicular cancer. Eur Urol 1993, 23:50-52 [DOI] [PubMed] [Google Scholar]
- 30.Sklar MD: Increased resistance to cis-diamminedichloroplatinum(II) in NIH3T3 cells transformed by ras oncogenes. Cancer Res 1988, 48:793-797 [PubMed] [Google Scholar]
- 31.Sklar MD: The ras oncogenes increase the intrinsic resistance of NIH3T3 cells to ionizing radiation. Science 1988, 239:645-647 [DOI] [PubMed] [Google Scholar]
- 32.Isonishi S, Hom DK, Thiebaut FB, Mann SC, Andrews PA, Basu A, Lazo JS, Eastman A, Howell SB: Expression of the c-Ha-ras oncogene in mouse NIH 3T3 cells induces resistance to cisplatin. Cancer Res 1991, 51:5903-5909 [PubMed] [Google Scholar]
- 33.Holford J, Rogers P, Kelland LR: Ras mutation and platinum resistance in human ovarian carcinomas in vitro. Int J Cancer 1998, 77:94-100 [DOI] [PubMed] [Google Scholar]
- 34.Kinashi Y, Akaboshi M, Masunaga S, Ono K, Watanabe M: Resistance to 195mPt-radiolabeled cis-diaminedichloroplatinum (II) of SHOK cells transfected with various oncogenes. Radiat Med 1998, 16:233-237 [PubMed] [Google Scholar]
- 35.Ganguly S, Murty VV, Samaniego F, Reuter VE, Bosl GJ, Chaganti RS: Detection of preferential NRAS mutations in human male germ cell tumors by the polymerase chain reaction. Genes Chromosom Cancer 1990, 1:228-232 [DOI] [PubMed] [Google Scholar]
- 36.Moul JW, Theune SM, Chang EH: Detection of ras mutations in archival testicular germ cell tumors by polymerase chain reaction and oligonucleotide hybridization. Genes Chromosom Cancer 1992, 5:109-118 [DOI] [PubMed] [Google Scholar]
- 37.Olie RA, Looijenga LHJ, Boerrigter L, Top B, Rodenhuis S, Mulder MP, Oosterhuis JW: N- and KRAS mutations in human testicular germ cell tumors: incidence and possible biological implications. Genes Chromosom Cancer 1995, 12:110-116 [DOI] [PubMed] [Google Scholar]
- 38.Olie RA, Looijenga LHJ, Dekker MC, De Jong FH, De Rooy DG, Oosterhuis JW: Heterogeneity in the in vitro survival and proliferation of human seminoma cells. Br J Cancer 1995, 71:13-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olie RA, Boersman AWM, Dekker MC, Nooter K, Looijenga LHJ, Oosterhuis JW: Apoptosis of human seminoma cells upon disruption of their micro-environment. Br J Cancer 1996, 73:1031-1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otto T, Virchow S, Fuhrmann C, Steinberg F, Streffer C, Goepel M, Rubben H: Detection of vital germ cell tumor cells in short-term cell cultures of primary tumors and of retroperitoneal metastasis—clinical implications. Urol Res 1997, 25:121-124 [DOI] [PubMed] [Google Scholar]
- 41.Löwenberg B, Van Putten WLJ, Touw IP, Delwel R, Santini V: Autonomous proliferation of leukemic cells in vitro as a determinant of prognosis in adult acute myeloid leukemia. N Engl J Med 1993, 328:614-619 [DOI] [PubMed] [Google Scholar]
- 42.Suijkerbuijk RF, Sinke RJ, Olde Weghuis DEM, Roque L, Forus A, Stellink F, Siepman A, Van de Kaa C, Soares J, Geurts van Kessel A: Amplification of chromosome subregion 12p11.2-p12.1 in a metastasis of an i(12p)-negative seminoma: relationship to tumor progression? Cancer Genet Cytogenet 1994, 78:145-152 [DOI] [PubMed] [Google Scholar]
- 43.Mostert MMC, Van de Pol M, Olde Weghuis D, Suijkerbuijk RF, Geurts van Kessel A, Van Echten-Arends J, Oosterhuis JW, Looijenga LHJ: Comparative genomic hybridization of germ cell tumors of the adult testis: confirmation of karyotypic findings and identification of a 12p-amplicon. Cancer Genet Cytogenet 1996, 89:146-152 [DOI] [PubMed] [Google Scholar]
- 44.Korn MW, Olde Weghuis DEM, Suijkerbuijk RF, Schmidt U, Otto T, Du Manoir S, Geurts van Kessel A, Seeber S, Becher R: Detection of chromosomal DNA gains and losses in testicular germ cell tumors by comparative genomic hybridization. Genes Chromosom Cancer 1996, 17:78-87 [DOI] [PubMed] [Google Scholar]
- 45.Mostert MC, Verkerk AJMH, Van de Pol M, Heighway J, Marynen P, Rosenberg C, Geurts van Kessel A, van Echten J, Oosterhuis JW, Looijenga LHJ: Identification of the crucial region of 12p overrepresentation in testicular germ cell tumors of adolescents and adults. Oncogene 1998, 16:2617-2627 [DOI] [PubMed] [Google Scholar]
- 46.Mostofi FK, Sesterhenn IA: Histological Typing of Testis Tumours. 1998. Springer, Berlin
- 47.Pugh RCB: Combined tumours. Pugh RCB eds. Pathology of the Testis. 1976, :pp 245-258 Blackwell, Oxford [Google Scholar]
- 48.Mosselman S, Looijenga LHJ, Gillis AJM, Van Rooijen MA, Kraft HJ, Van Zoelen EJJ, Oosterhuis JW: Aberrant platelet-derived growth factor α-receptor transcript as a diagnostic marker for early human germ cell tumors of the adult testis. Proc Natl Acad Sci USA 1996, 93:2884-2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin RW, Iwaki T, Kitamoto T, Tateishi J: Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab Invest 1991, 64:693-702 [PubMed] [Google Scholar]
- 50.Castedo SMMJ, De Jong B, Oosterhuis JW, Seruca R, Idenburg VJS, Dam A, Te Meerman GJ, Schraffordt Koops H, Sleijfer DT: Chromosomal changes in human primary testicular nonseminomatous germ cell tumors. Cancer Res 1989, 49:5696-5701 [PubMed] [Google Scholar]
- 51.Castedo SMMJ, De Jong B, Oosterhuis JW, Seruca R, Te Meerman GJ, Dam A, Schraffordt Koops H: Cytogenetic analysis of ten human seminomas (two of them lacking the i(12p)). Cancer Res 1989, 49:439-443 [PubMed] [Google Scholar]
- 52.Mitelman F: ISCN An International System for Human Cytogenetic Nomenclature. 1995. S. Karger, Basel
- 53.Rosenberg C, Mostert MC, Bakker Schut T, Van de Pol M, Van Echten-Arends J, De Jong B, Raap T, Tanke H, Oosterhuis JW, Looijenga LHJ: Chromosomal constitution of human spermatocytic seminomas: comparative genomic hybridization supported by conventional and interphase cytogenetics. Genes Chromosom Cancer 1998, 23:286-291 [PubMed] [Google Scholar]
- 54.Maniatis T, Fritsch EF, Sambrook J: Isolation of high molecular-weight, eukaryotic DNA from cells grown in tissue culture. Molecular Cloning. 1982, :pp 280-281 Cold Spring Harbor Laboratory, New York [Google Scholar]
- 55.Rosenberg C, Van Gijlswijk RP, Vos CBJ, Wiegant J, Cornelisse CJ, Tanke HJ, Raap AK: Comparative genomic hybridization with lissamine- and fluorescein-labelled nucleotides. Cytometry 1998, 32:337-341 [PubMed] [Google Scholar]
- 56.Rosenberg C, Bakker Schut T, Mostert MC, Tanke HJ, Raap AK, Oosterhuis JW, Looijenga LHJ: Chromosomal gains and losses in testicular germ cell tumors of adolescents and adults investigated by a modified CGH approach. Lab Invest 1999, 79:1447-1451 [PubMed] [Google Scholar]
- 57.Benhattar J, Losi L, Chaubert P, Givel JC, Costa J: Prognostic significance of K-ras mutations in colorectal carcinoma. Gastroenterology 1993, 104:1044-1048 [DOI] [PubMed] [Google Scholar]
- 58.Kahn SM, Jiang W, Culbertson TA, Weinstein IB, Williams GM, Tomita N, Ronai Z: Rapid and sensitive nonradioactive detection of mutant K-ras genes via “enriched” PCR amplification. Oncogene 1991, 6:1079-1083 [PubMed] [Google Scholar]
- 59.Van Echten-Arends J, Oosterhuis JW, Looijenga LHJ, Wiersma J, Te Meerman G, Schraffordt Koops H, Sleijfer DT, De Jong B: No recurrent structural abnormalities in germ cell tumors of the adult testis apart from i(12p). Genes Chromosom Cancer 1995, 14:133-144 [DOI] [PubMed] [Google Scholar]
- 60.Ford JH: Translocations of chromosome 12. II. A comparison of the distribution of sites of spontaneous and induced breakages. Hum Genet 1981, 58:279-281 [DOI] [PubMed] [Google Scholar]
- 61.Oosterhuis JW, Gillis AJM, Looijenga LHJ: In Vitro Survival, RAS Mutations, Apoptosis and Activation of the SAPK-Pathway in Human Seminoma Cells. 1997:pp 51-57 John Libbey & Company Ltd., Edited by I Appleyard. London
- 62.Rao PH, Houldsworth J, Palanisamy N, Murty VV, Reuter VE, Motzer RJ, Bosl GJ, Chaganti RS: Chromosomal amplification is associated with cisplatin resistance of human male germ cell tumors. Cancer Res 1998, 58:4260-4263 [PubMed] [Google Scholar]
- 63.Andrews PW, Casper J, Damjanov I, Duggan-Keen M, Giwercman A, Hata J-i, Von Keitz A, Looijenga LHJ, Oosterhuis JW, Pera M, Sawada M, Schmoll H-J, Skakkebæk NE, Van Putten W, Stern P: A comparative analysis of cell surface antigens expressed by cell lines derived from human germ cell tumors. Int J Cancer 1996, 66:806-816 [DOI] [PubMed] [Google Scholar]
- 64.Fenton RG, Hixon JA, Wright PW, Brooks AD, Sayers TJ: Inhibition of fas (CD95) expression and fas-mediated apoptosis by oncogenic ras. Cancer Res 1998, 58:3391-3400 [PubMed] [Google Scholar]
- 65.Peli J, Schroter M, Rudaz C, Hahne M, Meyer C, Reichmann E, Tschopp J: Oncogenic Ras inhibits Fas ligand-mediated apoptosis by downregulating the expression of Fas. EMBO J 1999, 18:1824-1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK: The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci USA 1998, 95:14950-14955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME: Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96:857-868 [DOI] [PubMed] [Google Scholar]
- 68.Vorechovsky I, Mazanec K: Is isochromosome i(12p) present in gonadal precancerous tissue? Neoplasma 1989, 36:697-700 [PubMed] [Google Scholar]
- 69.Vos A, Oosterhuis JW, De Jong B, Buist J, Schraffordt Koops H: Cytogenetics of carcinoma in situ of the testis. Cancer Genet Cytogenet 1990, 46:75-81 [DOI] [PubMed] [Google Scholar]
- 70.Looijenga LHJ, Rosenberg C, Van Gurp RJHLM, Geelen E, Van Echten-Arends J, De Jong B, Mostert MC, Oosterhuis JW: Comparative genomic hybridization of microdissected samples from different stages in the development of a seminoma and nonseminoma. J Pathol 2000, 191:187-192 [DOI] [PubMed] [Google Scholar]