

Abstract
A microglial response is part of the inflammatory processes in Alzheimer’s disease (AD). We have used APP23 transgenic mice overexpressing human amyloid precursor protein with the Swedish mutation to characterize this microglia response to amyloid deposits in aged mice. Analyses with MAC-1 and F4/80 antibodies as well as in vivo labeling with bromodeoxyuridine demonstrate that microglia in the plaque vicinity are in an activated state and that proliferation contributes to their accumulation at the plaque periphery. The amyloid-induced microglia activation may be mediated by scavenger receptor A, which is generally elevated, whereas the increased immunostaining of the receptor for advanced glycation end products is more restricted. Although components of the phagocytic machinery such as macrosialin and Fc receptors are increased in activated microglia, efficient clearance of amyloid is missing seemingly because of the lack of amyloid-bound autoantibodies. Similarly, although up-regulation of major histocompatibility complex class II (IA) points toward an intact antigen-presenting function of microglia, lack of T and B lymphocytes does not indicate a cell-mediated immune response in the brains of APP23 mice. The similar characteristics of microglia in the APP23 mice and in AD render the mouse model suitable to study the role of inflammatory processes during AD pathogenesis.
Inflammatory processes are thought to play a key role in the pathogenesis of Alzheimer’s disease (AD), as indicated by epidemiological studies with nonsteroidal anti-inflammatory drugs that markedly reduced the risk of AD. 1,2 Accordingly, in the brains of patients, microglia cells associated with amyloid plaques are activated. 2-5 In vitro studies further showed cytokine and neurotoxin release by Aβ-treated microglia cells. 6-11 These data argue in favor of an essential role of microglia cells in chronic inflammatory processes that may ultimately lead to neuronal degeneration as observed in AD.
To study the extent of inflammatory processes that accompany amyloid plaque formation, we used a transgenic mouse model, APP23, overexpressing the human β-amyloid precursor protein (APP) with the Swedish mutation. 12,13 Amyloid plaques in these mice are first observed at an age of 6 months and dramatically increase in size and number during aging. The mostly congophilic, dense-core Aβ deposits show many characteristics of human AD plaques such as enlarged dystrophic neurites and neuron loss. 14 Similar to AD, vascular amyloid is also present in aged APP23 animals. 15 Compact amyloid deposits are associated with microglia cells showing a characteristic activated morphology, 16 and with reactive astrocytes. 12 Studies from Frautschy and colleagues 17 have also demonstrated that microglia cells in another transgenic mouse line, Tg 2576, carrying human APP with the Swedish mutation, are activated, when located in close association with amyloid deposits.
In the present study, we immunohistochemically define the activation state of microglia in APP23 mice and, furthermore, identify mechanisms that may contribute to amyloid-associated microglia activation. In addition, we examine the expression of marker proteins for microglia phagocytosis and antigen presentation.
Materials and Methods
Animals
The generation of APP23 transgenic mice has previously been described. 12,13 These mice express the human APP751 cDNA with the Swedish double mutation under control of the neuron-specific mouse Thy-1 promoter fragment. APP23 mice, established on a B6D2 background, have been continuously back-crossed to C57BL/6J. Eighteen- to 23-month-old heterozygous mice from generations 6 and 7 were analyzed.
Tissue Preparation
Mice were anesthetized, decapitated and brains were removed, shock-frozen with liquid nitrogen, and stored in sealed plastic bags at −80°C. Sagittal sections were cut at 15 to 20 μm on a cryostat and mounted on Superfrost slides (Menzel-Gläser; Braunschweig, Germany). In addition, fresh-frozen sections from a mouse with a mechanical lesion to the frontal cortex were used. 18
Immunohistochemistry
Fresh-frozen, cryostat-cut tissue sections were either fixed in 1) acetone for 10 minutes at −20°C (for FA-11, F4/80, and 2.4G2 antibodies), or 2) 3% paraformaldehyde for 10 minutes on ice (for MAC-1, SRA [2F8], and NT11 antibodies), or 3) methanol:acetone (1:1) for 45 seconds at −20°C (IA antibody). Sections were then pretreated with H2O2 for 30 minutes and blocked with 2.5% bovine serum albumin/2% normal serum for 2 hours at room temperature. The tissue sections were incubated with the appropriate primary antibody (3.5 hours at room temperature or overnight at 4°C), followed by incubation with a secondary biotinylated antibody for 2 hours. Bound antibodies were visualized using the avidin-biotin-peroxidase method (Vectastain ABC Elite Kit; Vector Laboratories, Burlingame, CA) with diaminobenzidine (Boehringer Mannheim, Mannheim, Germany) or Vector Vip (Vector Laboratories) as the chromogens. Between all steps, tissue sections were rinsed with phosphate-buffered saline. Some sections were stained with alkaline phosphatase-conjugated secondary antibodies and further processed with naphthol phosphate (Sigma Chemical Co., St. Louis, MO). Finally, sections were counterstained in Mayer’s hemalum (Merck Darmstadt, Germany).
Antibodies
The following primary antibodies were used: rat monoclonal antibody MAC-1 (anti CR3, CD11b; diluted 1:1,000) (Serotec, Oxford, England); rabbit antiserum F4/80 against a macrophage/microglial-specific 160-kd protein (diluted 1:200; Serotec) as well as rat monoclonal antibodies against T cell markers CD4 and CD8 19 (diluted 1:50; kindly supplied by Drs. R. M. Zinkernagel and B. Odermatt, University of Zürich, Zürich, Switzerland); rat monoclonal FA-11 antibody against macrosialin 20 (CD68; diluted 1:100); monoclonal antibody to murine CD45R (B220, diluted 1:75; ImmunoKontact, Frankfurt/Main, Germany); monoclonal armenian hamster antibody to CD3ε (diluted 1:50; Pharmingen, San Diego, CA); monoclonal rat IA 21 (clone M5/114, dilution 1:50, gift from Dr. I. L. Campbell, The Scripps Research Institute, La Jolla, CA); monoclonal 2F8 rat antibody to scavenger receptor A (SRA) (diluted 1:100; gift of Dr. J. El Khoury, Massachusetts General Hospital, Boston, MA); rabbit anti-receptor for advanced glycation end products (RAGE) (dilution 1:10; kindly provided by Dr. Thomas Hughes, Novartis Pharmaceuticals Corp., Summit, NJ); monoclonal rat 2.4G2 against FcγII and III receptors (CD16/CD32); rabbit Aβ1-40 antiserum NT11 22 (dilution 1:1,000).
The following secondary antibodies were used: Biotinylated rabbit anti-rat IgG (dilution 1:200; Serotec), biotinylated goat anti-rat IgG (dilution 1:70; Southern Biotechnology Associates, Birmingham, AL); biotinylated mouse anti-hamster IgG (dilution 1:70; Pharmingen); biotinylated goat anti-rabbit IgG (dilution 1:200; Vector Laboratories); alkaline phosphatase-conjugated goat anti-rat IgG (dilution 1:75; Tago, Burlingame, CA); and alkaline phosphatase-conjugated donkey anti-goat IgG (dilution 1:75; Jackson ImmunoResearch Laboratories, West Grove, PA).
Immunohistochemical Analyses of Cell Proliferation
The 5′-bromodeoxyuridine (BrdU) incorporation method was used to study cell proliferation. Mice were injected daily by intraperitoneal infusion with the thymidine analogue BrdU (50 μg/g body weight; Sigma) for 5 consecutive days. Two hours after the last injection, mice were transcardially perfused with phosphate-buffered saline, followed by 4% paraformaldehyde. Brains were postfixed in paraformaldehyde, cryoprotected in 30% sucrose, frozen in isopentane at −20°C, and cut with a freezing-sliding microtome. Sections were incubated in 50% formamide/50% 2× standard saline citrate buffer at 65°C for 2 hours. After rinsing in 2× standard saline citrate, sections were incubated in 2 N HCl for 30 minutes at 37°C, and then rinsed in 0.1 mol/L borate buffer (pH 8.5) for 10 minutes as described by others. 23 Subsequently, free-floating sections were immunostained with an antibody to BrdU (MAS250c, dilution 1:1,000; Accurate Ltd., Westbury, NY) according to the above described protocol.
Results
Activated Microglia near Dense-Core Amyloid Plaques Proliferate
To define the activation state of microglia cells in the brains of APP23 mice, we performed immunohistochemical analyses with antibodies directed to several microglial proteins known to be up-regulated during activation. Figure 1A ▶ shows staining with the MAC-1 antibody detecting the macrophage/microglial-specific complement receptor 3. Intensively MAC-1-stained microglia were found in close association with amyloid plaques in all brain regions containing compact amyloid plaques such as the neocortex and the hippocampus. Microglia staining was weak or absent in nontransgenic, age-matched control mouse brain (Figure 1C) ▶ . When fixed under different conditions, however, fresh-frozen control and transgenic brain tissue devoid of amyloid plaques revealed positive, although less intense microglia immunoreactivity, consistent with previous reports. 12,16 High-power light microscopy revealed that MAC-1-positive microglial processes cover nearly the entire surface of the amyloid plaque (Figure 1B) ▶ . Moreover, MAC-1-positive microglia cell bodies occasionally formed darkly labeled clusters. Overall, immunostainings demonstrated that microglia with high expression levels of MAC-1 are located in the vicinity of virtually all compact amyloid plaques, indicating that these cells are not in a resting, but activated state. The fact that they are activated was verified by immunohistochemistry using the F4/80 antibody, which detects a macrophage/microglia-specific 160-kd glycoprotein 24 and clearly showed similar cell staining as MAC-1 (Figure 1, D and E) ▶ .
Figure 1.
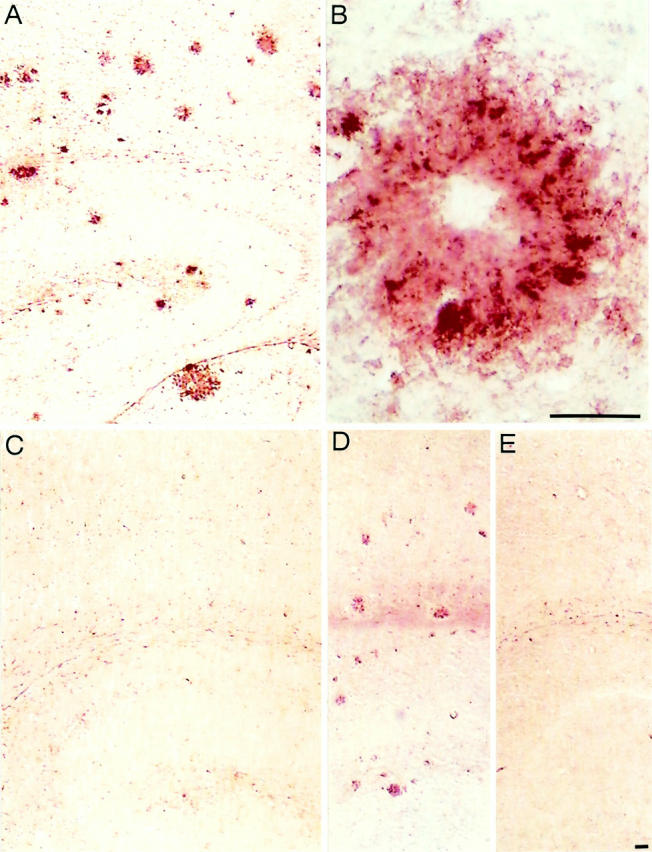
Amyloid plaque-associated microglia are activated. A: MAC-1-immunostained microglia cells associated with compact amyloid deposits in the neocortex and hippocampus of a 23-month-old APP23 transgenic mouse. Deposits of various sizes are visualized. B: Higher magnification of a single amyloid plaque showing several clusters of MAC-1-positive microglial cell bodies that frame the amyloid. Microglial processes cover nearly the entire amyloid, thereby revealing the morphology of this dense-core plaque. The center of the plaque (core) is not always stained. C: No MAC-1 immunostaining is obtained in equivalent brain regions of an age-matched control mouse under the labeling conditions used. D: F4/80 staining of sections containing neocortex and hippocampus of a 23-month-old APP23 mouse. The staining shows microglia cells closely associated with compact amyloid plaques. E: Lack of F4/80 reactivity in a corresponding brain section from a control mouse. Scale bar, 50 μm.
The high abundance of activated microglia near amyloid deposits led us to determine whether these cells proliferate. For this purpose, we performed in vivo labeling of APP23 mice with BrdU. BrdU-labeled cells were more numerous in the APP23 mouse neocortex as compared to age-matched wild-type brain. These cells were clustered around amyloid plaques and only a few BrdU-labeled cells were found away from the deposits (Figure 2, A and B) ▶ . Higher magnification shows the staining of cell bodies around a plaque (Figure 2C) ▶ . Double immunohistochemistry with antibodies directed to BrdU and MAC-1 revealed incorporation of this thymidine analogue into the DNA of microglia, indicating that these plaque-associated microglia cells are in a proliferative state (Figure 2D) ▶ .
Figure 2.
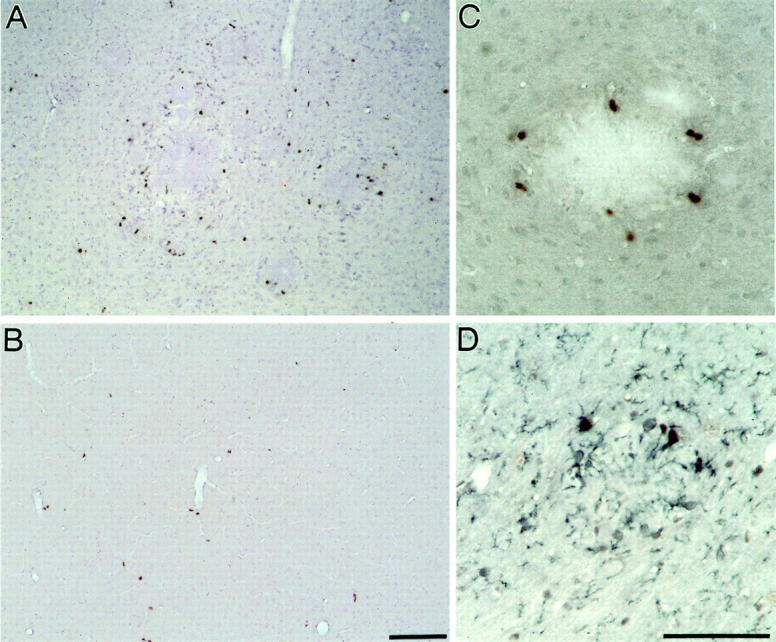
Activated microglia near amyloid plaques proliferate. A: BrdU-positive cells in the neocortex of a 12.5-month-old APP23 mouse are predominantly associated with amyloid plaques. B: BrdU labeling of a wild-type mouse of the same age shows only a few positive cells in the neocortex. C: Higher magnification shows BrdU labeled cells around an amyloid plaque. D: Double-labeling with BrdU (black nuclear staining) and the microglia marker MAC-1 (blue staining of plasma membrane processes) identifies the amyloid-associated BrdU-labeled cells as microglia. Scale bar, 50 μm.
Plaque-Associated Microglia Show Increased Levels of SRA
In vitro studies have shown that SRA is capable of mediating microglial adhesion to fibrillar Aβ resulting in their activation. 10,11,25 Using 2F8, an antibody directed to SRA that is expressed on mononuclear phagocytes, 11 we found increased immunoreactivity associated with virtually all compact amyloid plaques (Figure 3A) ▶ . Strongest labeling was observed at the outer edge of the plaques, at sites where microglial processes are in contact with the amyloid. In addition, SRA reactivity was found associated with vascular amyloid deposits (Figure 3B) ▶ . These results indicate that microglia may interact with fibrillar Aβ through SRA, and that this interaction may contribute to their activation and retention at these sites.
Figure 3.

Scavenger receptor A may contribute to microglia activation. A: Immunostaining of a single neocortical amyloid plaque with the 2F8 antibody directed to SRA shows strongest reactivity at the outer edge of the deposit, where microglial processes cover the amyloid. The weaker staining throughout the plaque may be related to a lower density of microglial processes. The contours of cell bodies surrounding the plaques are also visualized. B: Microglial 2F8 reactivity at a blood vessel containing vascular amyloid. C: RAGE immunoreactivity of neocortical plaque-associated cells, revealing a strong, but very limited staining (the contours of the plaque are not well visible at this magnification). RAGE reactivity was only detected at a minority of the amyloid plaques. D: RAGE staining at the vascular endothelium shows the contours of the vessel. Scale bar, 50 μm.
The RAGE has also been demonstrated to mediate interaction of microglia with Aβ in vitro, thereby resulting in their activation. 26 RAGE has additionally been found on endothelial cells and affected neurons in the brains of AD patients. 26 In APP23 mice, RAGE immunohistochemistry revealed only very few, but strongly stained spots in the plaque vicinity (Figure 3C) ▶ and at the vascular endothelium (Figure 3D) ▶ . Moreover, RAGE staining was present only at a small percentage of all amyloid plaques. Although the staining did not allow us to unequivocally localize the immunoreactivity to microglia cells, it was apparent that at best a subpopulation of activated microglia was RAGE-positive. Therefore, a general contribution of RAGE to microglial activation in the vicinity of amyloid plaques in the APP23 mice seems to be unlikely.
Components for Phagocytosis Such as Macrosialin and Fc Receptors Are Elevated in Plaque-Associated Microglia
To further assess the functional potential of activated microglial cells, we used the antibody FA-11, which is directed to the lysosomal membrane protein macrosialin, 20,27 the murine homologue of CD68. Increased immunoreactivity of this protein is found in cells that accumulate lysosomal vacuoles, which represent a typical feature of phagocytic cells. 28 As visualized by the strong staining, plaque-associated microglia in APP23 mice accumulate macrosialin (Figure 4A) ▶ . Sections from control mice or from brain regions devoid of amyloid plaques show only weak labeling, reflecting a low macrosialin expression in all resting microglia cells (Figure 4C) ▶ . At higher magnification, plaque-associated microglial cell bodies are visible (Figure 4B) ▶ . Staining of microglia processes is also evident, possibly because of the appearance of lysosomal structures in microglial processes or because of an expression of a small fraction of macrosialin at the cell surface. 27,29 The elevation of macrosialin hints at a phagocytic capability of microglia cells in the vicinity of plaques.
Figure 4.
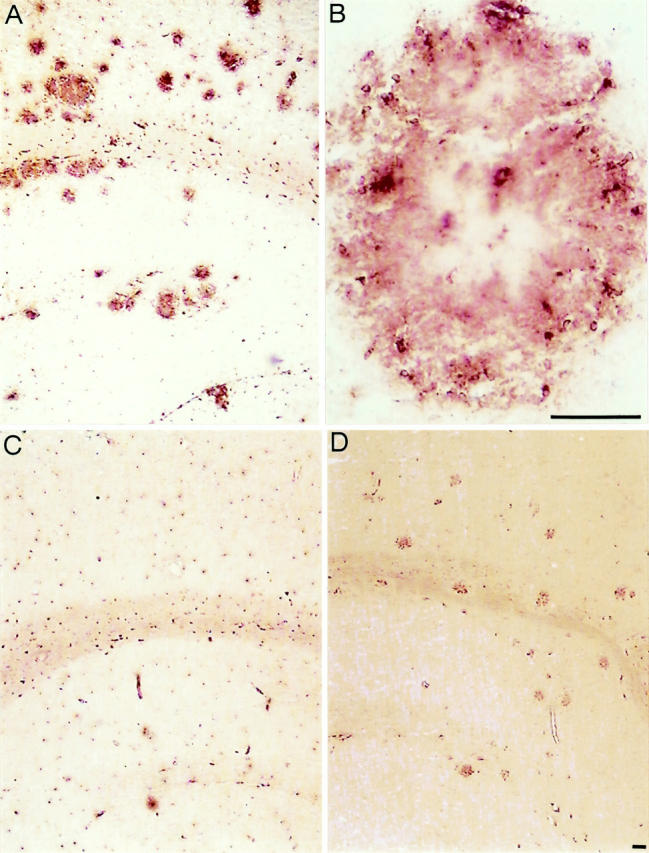
Activated microglia are capable to phagocytose. A: FA-11 (macrosialin) immunostaining of a section through the neocortex and hippocampus of a 23-month-old APP23 transgenic mouse shows strong macrosialin-positive amyloid plaque-associated microglia cells. B: A higher magnification of a single amyloid plaque visualizes macrosialin-positive microglia cell bodies that are surrounding the amyloid. Weaker labeling is found at the amyloid, thereby revealing the contours of the plaque. C: Microglia in the neocortex and hippocampus of a control mouse show a basal macrosialin staining in agreement with published data. 27 D: 2.4G2 staining of a section through the neocortex and hippocampus demonstrates an up-regulation of Fc receptors FcγRII and FcγRIII on activated microglia cells at amyloid plaques. Scale bar, 50 μm.
To further study this observation we stained brain sections of APP23 mice with antibodies directed to FcγRII and FcγRIII that bind to IgG-opsonized particles. 30,31 Both Fc receptors have been reported to be up-regulated on phagocytic cells. 32 In APP23 mice we also found an up-regulation of these receptors on microglia in the plaque vicinity (Figure 4D) ▶ . However, when performing immunohistochemical analyses with antibodies directed to mouse IgG, we did not obtain any evidence of autoantibodies bound to the amyloid plaques (data not shown). In addition, antibody titers against Aβ were not detectable in the sera of APP23 mice (data not shown). These results indicate some phagocytic capability of plaque-associated microglia, but they do not support Fc receptor-mediated phagocytosis of amyloid by microglia.
A Subpopulation of Activated Microglial Cells Is Major Histocompatibility Complex (MHC) Class II-Positive
Phagocytosis and intracellular processing of external proteins are required for their presentation as antigenic peptides in association with MHC class II molecules. Given the increase of phagocytic markers in APP23 mice, we addressed the question whether the antigen-presenting machinery in microglia is intact. We examined expression levels of MHC class II molecules using an antibody directed to the murine MHC class II molecule IA. Strong reactivity was observed in association with the amyloid plaques, although only a small portion of plaque-associated microglia appeared to be reactive (Figure 5A) ▶ . To study the spatial relationship of these MHC class II-positive microglia cells and amyloid plaques in more detail, we performed double-labeling experiments with antibodies directed against Aβ and IA. As shown in Figure 5B ▶ , MHC class II protein reactivity was found within as well as very close to the compact amyloid plaques. Additionally, we observed IA reactivity associated with blood vessels containing vascular amyloid but not with diffuse amyloid deposits. To confirm that up-regulated IA was found exclusively on microglia cells, we used double staining with MAC-1 and IA (data not shown). These results indicate that the antigen-presenting machinery in amyloid plaque-associated microglia is intact.
Figure 5.
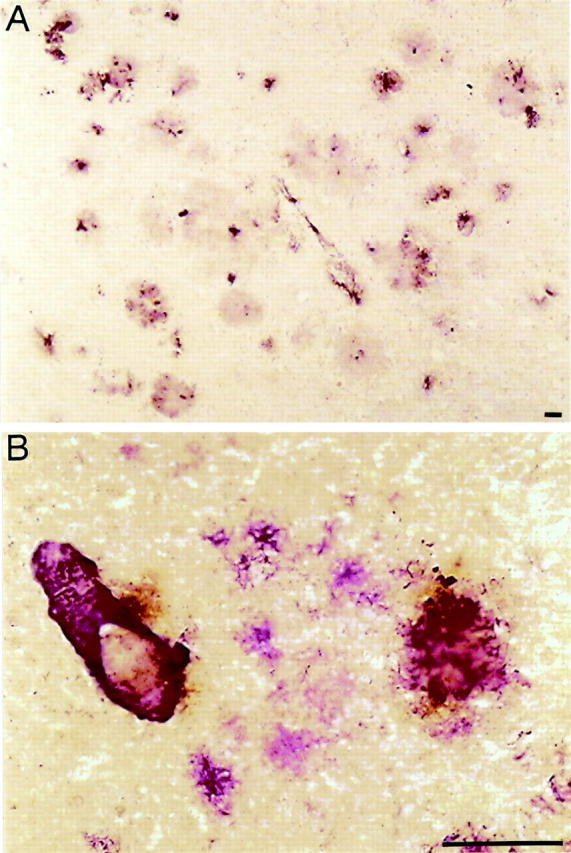
A subpopulation of activated microglia is MHC class II-positive. A: IA (MHC class II protein)-positive microglia are closely associated with amyloid plaques as well as capillary endothelium, as shown in a neocortex section of a 23-month-old APP23 transgenic mouse. B: Double-labeling of amyloid (purple reaction product) and IA (brown reaction product) reveals MHC class II protein reactivity within and around the compact amyloid deposits. Although MHC class II staining is also visible at a blood vessel and an amyloid plaque, it was not detected at diffuse, nonfibrillary amyloid as present in the center of the picture. Scale bar, 50 μm.
No Evidence for Lymphocyte Recruitment in Brain
The increase in MHC class II reactivity at amyloid plaques in the APP23 transgenic mouse brain led us to examine a potential recruitment of lymphocytes in the inflammatory processes. We, therefore, searched for T cells that could be activated by antigenic peptides, which are associated with MHC class II molecules. Using an antibody against CD3ε, a component associated with the T cell receptor, we did not obtain any evidence for T cells in the cerebral cortex of APP23 transgenic mice (Figure 6A) ▶ including the plaque regions where MHC class II-positive microglia have been found. To demonstrate the antibody reactivity, staining of T lymphocytes, located in the splenic white pulp, was used as a positive control (Figure 6B) ▶ . Further immunohistochemical staining was performed with an anti-CD4 antibody, directed against T helper cells to verify the lack of T cells in aged transgenic mouse brain. Analyses of the cerebral cortex of APP23 mice containing numerous amyloid deposits repeatedly did not provide any evidence for the presence of T cells (Figure 6C) ▶ . T cells in the white pulp of the spleen, however, showed a positive immunoreactivity when stained with the same antibody (Figure 6D) ▶ . The same result was obtained with CD8 antibodies, which recognize cytotoxic T cells that are activated by antigen associated with MHC class I molecules (data not shown). For direct comparison we also stained post mortem brain tissue from AD patients with a human T cell-specific CD3ε antibody and detected a few single positive cells in the capillaries of the cerebral cortex (data not shown). Only one of these T cells appeared to be located at the outer endothelium of the capillary. Neither the lack of detectable T lymphocytes in the cerebral cortex of APP23 transgenic mice nor the incidental detection of single T lymphocytes in the cerebral cortex of AD patients support a function of activated microglia as antigen-presenting cells.
Figure 6.
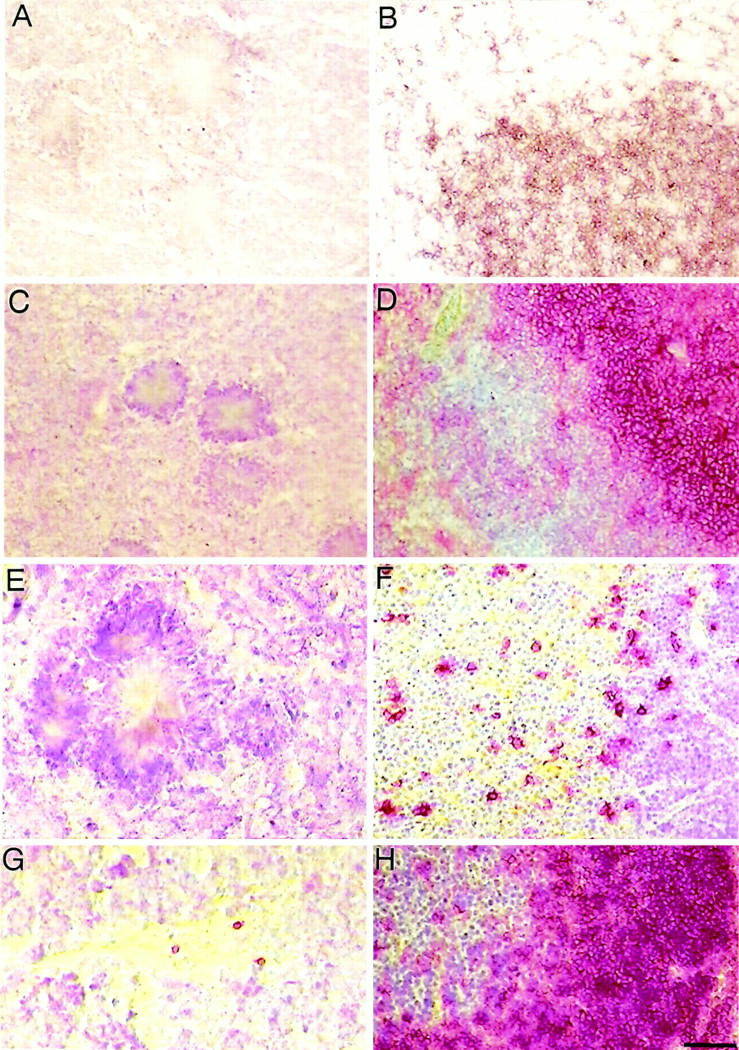
No detectable lymphocyte recruitment during inflammatory processes in the brains of APP23 mice. A: The CD3ε staining of a neocortex section with three compact amyloid plaques does not provide any evidence of T lymphocytes in aged APP23 mice. B: As a positive control the specific reactivity of CD3ε antibodies with T cells is shown in a staining of the splenic white pulp of a 6-week-old control mouse. C: CD4 staining did not identify T helper cells in the neocortex of APP23 transgenic animals. D: The reactivity of the CD4 antibodies was shown by labeling CD4-positive T helper cells (in red color) that are located at high density in a splenic white pulp section. Surrounding cells, mostly B cells, were counterstained with Mayer’s hemalaun (blue color). E: CD45R (B220) staining of a neocortex section showing a single compact amyloid plaque does not provide evidence for B lymphocytes. F: A control section stained with CD45R reveals B cell staining (red reaction product), located primarily in the medulla of the thymus of a 6-week-old control mouse. G: CD45R staining of a mouse brain section near a mechanical frontal cortex lesion that is infiltrated by three individual B lymphocytes demonstrates single cell detection. H: Control section showing part of a murine splenic white pulp packed with CD45R-positive B lymphocytes. Scale bar, 50 μm.
In addition to T lymphocytes, we also studied B lymphocytes in murine as well as human brain tissue. However, staining with anti-CD45R (B220), a typical B lineage marker, did not reveal B cells in the cerebral cortex of aged APP23 mice (Figure 6E) ▶ . Analyses of neocortical tissue from AD patients also did not provide any evidence of B lymphocytes (data not shown). Sections from several control tissues demonstrated many B cells in the white pulp of a murine spleen (Figure 6H) ▶ . The detection of single B lymphocytes near a mechanical brain lesion of the frontal cortex 18 (Figure 6G) ▶ as well as in the medulla of a murine thymus (Figure 6F) ▶ served as additional positive controls.
Discussion
Amyloid plaque pathology in the AD brain is accompanied by an accumulation of cells and proteins known to be mediators of inflammation. In particular, microglia cells are considered to play a role in chronic inflammation and possibly neurodegeneration in AD brains. 4,33 In a recent study we have described an APP transgenic mouse line, APP23, and demonstrated abundant microglia reactivity in the vicinity of compact amyloid plaques in the brains of these animals. 12,16 Using another APP transgenic mouse, Tg2576, an increase in microglia number and size around compact amyloid plaques has also been described. 17 These observations are very similar to findings in the brains of AD patients, where microglia cells have been shown to be located within or near amyloid plaques. 34
To better understand the microglial response, we evaluated their activation state in the brains of APP23 transgenic mice. The observation that both the microglial/macrophage-specific protein MAC-1 and a 160-kd glycoprotein recognized by the F4/80 antibody are increased, demonstrates that microglia cells associated with compact amyloid plaques are in an activated state. Some of the activated cells proliferate, as shown by in vivo labeling of APP23 mice with BrdU. This finding suggests that proliferation of resident or invading microglia/monocytes contributes to the high abundance of microglia cells near amyloid plaques.
Because diffuse amyloid deposits do not show microglia reactivity, 16 it is very likely that only fibrillar Aβ interacts with the microglia cells to induce their activation. Candidate receptors are SRA and RAGE, which were shown in vitro to mediate adhesion to Aβ fibrils and activation of microglia cells. 10,11,26 An up-regulation of SRA, at sites where microglial processes interact with Aβ, indicates that this receptor may be responsible for activation and accumulation of microglia at the amyloid plaques. A previous study has shown that lack of SRA does not alter the plaque formation itself in APP transgenic mice but the effect on microglia activation as well as other aspects of pathology have not been fully investigated. 35 RAGE was also found to be up-regulated on some plaque-associated cells in APP23 mice. Because only a subpopulation of cells showed this up-regulation, RAGE does not seem to be a strong candidate for a general mediator of microglia activation at plaques in the brains of APP23 mice. In addition, cytokines or chemokines may contribute to sustained microglia activation as they were shown to be present in glia cells around amyloid plaques of APP transgenic mice. 36
Evidence for a phagocytic capability of microglia cells in APP23 mice comes from an increase of macrosialin, the mouse homologue of CD68. Elevated levels of CD68, primarily a lysosomal membrane protein, indicate that cells are phagocytically active and digest internalized material because they accumulate lysosomal vacuoles. 28 Consistent with these results, we have previously demonstrated microglia with typical phagocytic morphology in APP23 mice using electron microscopy. 16 Interestingly, however, these microglia were in close contact with the dystrophic terminals around the amyloid plaques, whereas those in intimate contact with the amyloid fibrils did not appear phagocytic. Our finding of Fcγ receptor up-regulation further suggests that microglia in the APP23 mouse model are generally capable to phagocytose IgG-opsonized particles. 32 However, because we found neither autoantibodies bound to amyloid plaques, nor antibody producing B lymphocytes in the brain or Aβ antibody titers in the sera, our results indicate that Fc receptor-mediated phagocytosis does not occur to a large extent in the brains of APP23 mice. Efficient (likely Fc receptor-mediated) phagocytosis may be induced under specific circumstances as recently shown by Schenk and colleagues, 37 who demonstrated clearance of amyloid deposits in a human APP transgenic mouse line, PDAPP, after immunization with Aβ42 peptide. It is tempting to speculate that microglial cells may clear the amyloid and that binding of already up-regulated microglial Fc receptors to the Aβ antibody-amyloid ligands plays an important role in this process.
A subpopulation of plaque-associated, activated microglia cells is characterized by an increase in MHC class II protein and may be capable of presenting plaque-derived antigen, thereby activating T lymphocytes. However, an intensive search did not reveal any T lymphocytes in plaque-containing brain regions of aged APP23 mice, rendering this possibility unlikely. Because we also did not obtain any evidence for B lymphocytes, a contribution of cell-mediated immune responses to the inflammatory processes and to the plaque pathology seems unlikely in APP23 mice.
Taken together, the data presented in this study demonstrate that microglia in the plaque vicinity are in an activated state. Proliferation as well as interaction of the SRA with Aβ seem to contribute to microglia accumulation and activation at the periphery of amyloid plaques. Clearance of amyloid deposits seems to be suppressed for reasons that are not related to the phagocytic capability of microglia. Although the antigen-presenting function via MHC class II seems to be intact, the lack of T and B lymphocytes argues against cell-mediated immune responses in the brains of APP23 mice. Because of similar functional characteristics of microglia in APP23 mice compared to Alzheimer patients, these mice serve as a suitable model to further investigate the effect of inflammatory processes on AD pathology.
Acknowledgments
We thank Drs. Alphonse Probst (Institute for Pathology, University of Basel) and Paolo Paganetti (Novartis Pharma AG) for their review of the manuscript; and Dorothee Abramowski (Novartis Pharma AG) for helpful discussions during this project.
Footnotes
Address reprint requests to Dr. Matthias Staufenbiel, Novartis Pharma AG, WSJ-386.8.06, CH-4002 Basel, Switzerland. E-mail: matthias.staufenbiel@pharma.novartis.com.
Supported by grants 3130-44526.95 and 3130-56753.99 from the Swiss National Foundation.
References
- 1.Stewart WF, Kawas C, Corrada M, Meter EJ: Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48:626-632 [DOI] [PubMed] [Google Scholar]
- 2.McGeer PL, Schulzer M, McGeer EG: Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996, 47:425-432 [DOI] [PubMed] [Google Scholar]
- 3.Mackenzie IR, Hao C, Munoz DG: Role of microglia in senile plaque formation. Neurobiol Aging 1995, 16:797-804 [DOI] [PubMed] [Google Scholar]
- 4.McGeer PL, McGeer EG: The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Rev 1995, 21:195-218 [DOI] [PubMed] [Google Scholar]
- 5.Eikelenboom P, Zhan SS, Van Gool WA, Allsop D: Inflammatory mechanisms in Alzheimer’s disease. Trends Pharmacol Sci 1994, 15:447-450 [DOI] [PubMed] [Google Scholar]
- 6.Klegeris A, Walker DG, McGeer PL: Activation of macrophages by Alzheimer beta amyloid peptide. Biochem Biophys Res Comm 1994, 199:984-991 [DOI] [PubMed] [Google Scholar]
- 7.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F: Activation of microglial cells by β-amyloid protein and interferon-γ. Nature 1995, 374:647-650 [DOI] [PubMed] [Google Scholar]
- 8.Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo LM, Roher AE: Specific domains of β-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci 1996, 16:6021-6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald DR, Brunden KR, Landreth GE: Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci 1997, 17:2284-2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD: Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature 1996, 382:716-719 [DOI] [PubMed] [Google Scholar]
- 11.El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC: Microglia, scavenger receptors, and the pathogenesis of Alzheimer’s disease. Neurobiol Aging 1998, 19:S81-S84 [DOI] [PubMed] [Google Scholar]
- 12.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti P, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B: Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 1997, 94:13287-13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staufenbiel M, Sommer B: Transgenic animal models in the development of therapeutic strategies for Alzheimer’s disease. Haass C eds. The Molecular Biology of Alzheimer’s Disease: Genes and Mechanisms Involved in Amyloid Generation. 1998, :pp 309-326 Harwood Academic Publishers, Amsterdam [Google Scholar]
- 14.Calhoun ME, Wiederhold K-H, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M: Neuron loss in APP transgenic mice. Nature 1998, 395:755-756 [DOI] [PubMed] [Google Scholar]
- 15.Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold K-H, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M: Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 1999, 96:14088-14093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M: Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol 1999, 154:1673-1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM: Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 1998, 152:307-317 [PMC free article] [PubMed] [Google Scholar]
- 18.Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH: Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci 1999, 11:3648-3658 [DOI] [PubMed] [Google Scholar]
- 19.Binder D, van den Broek MF, Kagi D, Bluethmann H, Fehr J, Hengartner H, Zinkernagel RM: Aplastic anemia rescued by exhaustion of cytokine-secreting CD8+ T cells in persistent infection with lymphocytic choriomeningitis virus. J Exp Med 1998, 187:1903-1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith MJ, Koch GCE: Differential expression of murine macrophage surface glycoprotein antigens in intracellular membranes. J Cell Sci 1987, 87:113-119 [DOI] [PubMed] [Google Scholar]
- 21.Stalder AK, Carson MJ, Pagenstecher A, Asensio VC, Kincaid C, Benedict M, Powell HC, Masliah E, Campbell IL: Late-onset chronic inflammatory encephalopathy in immune-competent and severe combined immune-deficient (scid) mice with astrocyte-targeted expression of tumor necrosis factor. Am J Pathol 1998, 153:767-783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paganetti PA, Lis M, Klafki H-W, Staufenbiel M: Amyloid precursor protein truncated at any of the gamma-secretase sites is not cleaved to beta-amyloid. J. Neurosci Res 1996, 46:283-293 [DOI] [PubMed] [Google Scholar]
- 23.Kuhn HG, Dickinson-Anson H, Gage FH: Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neuroscience 1996, 16:2027-2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Austyn JM, Gordon S: F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol 1981, 11:805-815 [DOI] [PubMed] [Google Scholar]
- 25.Paresce DM, Gosh RN, Maxfield FR: Microglial cells internalize aggregates of Alzheimer’s disease amyloid beta protein via a scavenger receptor. Neuron 1996, 17:553-565 [DOI] [PubMed] [Google Scholar]
- 26.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM: RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382:685-691 [DOI] [PubMed] [Google Scholar]
- 27.Verbeek MM, Otte-Holler I, Wesseling P: A lysosomal marker for activated microglial cells involved in Alzheimer classic senile plaques. Acta Neuropathol 1995, 90:493-503 [DOI] [PubMed] [Google Scholar]
- 28.Walker DG: Inflammatory markers in chronic neurodegenerative disorders with emphasis on Alzheimer’s disease. Wood PL eds. Neuroinflammation, Mechanisms and Management. 1998, :pp 61-90 Humana Press, Inc., Totowa [Google Scholar]
- 29.McKnight AJ, Gordon S: Membrane molecules as differentiation antigens of murine macrophages. Adv Immunol 1998, 68:289-291 [DOI] [PubMed] [Google Scholar]
- 30.Ravetch JV, Luster AD, Weinshank R, Kochan J, Pavlovec A, Portnoy DA, Hulmes J, Pan Y-CE, Unkeless JC: Structural heterogeneity and functional domains of murine immunoglobulin G Fc receptors. Science 1986, 234:718-725 [DOI] [PubMed] [Google Scholar]
- 31.Ravetch JV, Kinet J-P: Fc receptors. Annu Rev Immunol 1991, 9:457-492 [DOI] [PubMed] [Google Scholar]
- 32.Weinshank RL, Luster AD, Ravetch JV: Function and regulation of a murine macrophage-specific IgG Fc receptor, FcγRα. J Exp Med 1988, 167:1909-1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogers J, Webster S, Lue LF, Brachova L, Civin WH, Emmerling M, Shivers B, Walker D, McGeer P: Inflammation and Alzheimer’s disease pathogenesis. Neurobiol Aging 1996, 17:681-686 [DOI] [PubMed] [Google Scholar]
- 34.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG: Microglia in degenerative neurological disease. Glia 1993, 7:84-92 [DOI] [PubMed] [Google Scholar]
- 35.Huang F, Buttini M, Wyss-Coray T, McConlogue L, Kodama T, Pitas RE, Mucke L: Elimination of the class A scavenger receptor does not affect amyloid plaque formation or neurodegeneration in transgenic mice expressing human amyloid protein precursors. Am J Pathol 1999, 155:1741-1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR: Evidence for glial-mediated inflammation in aged APPsw transgenic mice. Neurobiol Aging 1999, 20:581-589 [DOI] [PubMed] [Google Scholar]
- 37.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P: Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400:173-177 [DOI] [PubMed] [Google Scholar]