

Abstract
Familial British dementia (FBD), pathologically characterized by cerebral amyloid angiopathy (CAA), amyloid plaques, and neurofibrillary degeneration, is associated with a stop codon mutation in the BRI gene resulting in the production of an amyloidogenic fragment, amyloid-Bri (ABri). The aim of this study was to assess the distribution of ABri fibrillar and nonfibrillar lesions and their relationship to neurofibrillary pathology, astroglial and microglial response using immunohistochemistry, confocal microscopy, and immunoelectron microscopy in five cases of FBD. Abnormal tau was studied with immunoblotting. We present evidence that ABri is deposited throughout the central nervous system in blood vessels and parenchyma where both amyloid (fibrillar) and pre-amyloid (nonfibrillar) lesions are formed. Ultrastructurally amyloid lesions appear as bundles of fibrils recognized by an antibody raised against ABri, whereas Thioflavin S-negative diffuse deposits consist of amorphous electron-dense material with sparse, dispersed fibrils. In contrast to nonfibrillar lesions, fibrillar ABri is associated with a marked astrocytic and microglial response. Neurofibrillary tangles and neuropil threads occurring mainly in limbic structures, are found in areas affected by all types of ABri lesions whereas abnormal neurites are present around amyloid lesions. Immunoblotting for tau revealed a triplet electrophoretic migration pattern. Our observations confirm a close link between ABri deposition and neurodegeneration in FBD.
Familial British dementia (FBD) is the latest of the many terms introduced for an autosomal dominant, neurodegenerative disorder, first documented in affected members of a large British pedigree with a clinical presentation that is characterized by dementia, spastic tetraparesis, and cerebellar ataxia. 1-3 The neuropathological hallmarks include extensive cerebral amyloid angiopathy (CAA), cerebellar degeneration because of both severe CAA and parenchymal amyloid plaques, hippocampal amyloid plaques as well as neurofibrillary tangles (NFTs), and white matter degeneration similar to that seen in Binswanger’s disease. 2,4,5 In FBD the cytoskeletal pathology, most severe in the mediotemporal structures, has been found both immunohistochemically and ultrastructurally to be indistinguishable from that seen in Alzheimer’s disease (AD). 5 Although no fresh tissue samples were available for further studies, from these findings it was hypothesized that tau in FBD would have an electrophoretic migration pattern similar to that seen in AD. 5
Recently it has been demonstrated that a stop codon mutation of the gene BRI, located on chromosome 13, is associated with FBD. 6 The wild-type precursor protein (BriPP), which is a type-II, single-spanning transmembrane protein, is composed of 266 amino acids, whereas the mutated, extended precursor protein (ABriPP) has 277 amino acids. Cleavage of 34 amino acids at the C terminal end of ABriPP generates a 4-kd amyloidogenic fragment, ABri, which has been shown to be deposited as amyloid fibrils in leptomeningeal blood vessels. 6 The BRI gene is also associated with another familial cerebrovascular amyloidosis, familial Danish dementia, in which a decamer duplication results in the production of a 34-amino-acid long amyloidogenic peptide, ADan with a C-terminus different from that in FBD. 7
In this study of five cases of FBD we studied the topographical distribution and patterns of ABri deposition using immunohistochemistry with an antibody recognizing the C-terminus of ABri (Ab 338) alone and in combination with Thioflavin S. The distribution and relationship of NFTs neuropil threads (NTs) and abnormal neurites (ANs) to ABri deposits were investigated with the antibody AT8 alone and together with Ab 338. The astrocytic and microglial response to ABri deposits was studied using glial fibrillary acidic protein (GFAP) and CD68 antibodies, respectively, for double immunohistochemistry. Ultrastructural characteristics of amyloid and diffuse deposits were studied by immunoelectron microscopy using Ab 338. Western blots of brain homogenates from frozen hippocampal tissue were prepared to study the electrophoretic migration pattern of tau in one case.
Materials and Methods
Tissue Collection
Brain and spinal cord samples from five cases of FBD, five sporadic AD cases, and three age-matched controls were collected at post mortem. The pathological diagnosis of AD was made using standard criteria. 8 Cases 1–3 were from the original pedigree, 4 case 4 from a second, probably unrelated pedigree with identical clinical and pathological features, 3 and case 5 is from a recently identified third family with typical clinical presentation and mutation of the BRI gene in affected members. For immunohistochemistry tissue samples were fixed in 10% formalin in phosphate-buffered saline (PBS). For immunoelectron microscopy selected tissue samples from the hippocampal formation (cases 1 and 5) and temporal neocortex (case 5) were fixed in 3% buffered glutaraldehyde. For Western blot analysis of tau unfixed tissue samples from case 5 were taken at post mortem (post mortem delay 7 hours), frozen, and stored at −70°C.
Antibodies
A rabbit polyclonal antibody (Ab338) was raised to the C-terminal 10 amino acids of ABriPP and its specificity confirmed by absorption experiments, as previously described. 6 Pre-immune serum from the same animal was also used as a control. An anti-CD68 antibody (PG-M1; DAKO, Ely, UK) was used to demonstrate cells of the microglia/macrophage lineage and anti-GFAP antibody (DAKO) for astrocytes. For tau immunohistochemistry, the AT8 antibody (Innogenetics, Ghent, Belgium) was used, whereas for immunoblotting the rabbit, polyclonal antiserum TP70 9 the phosphorylation-dependent mouse monoclonal antibody PHF1 (a gift from Peter Davies, Albert Einstein College of Medicine, New York, NY) and the AT8, AT180, AT270, and AT100 (all Innogenetics) monoclonal antibodies were used.
Congo Red Staining
Congo red staining was performed using a standard protocol and viewed under polarized light. In selected areas including 1) the hippocampal formation with parahippocampus, 2) amygdala, 3) upper pons with the locus coeruleus, 4) medulla with the inferior olive, and 5) cerebellar cortex including white matter and the dentate nucleus serial, 15-μm sections were cut and alternate sections stained with Congo red or Ab 338.
Immunohistochemistry
Representative areas of formalin-fixed brain and spinal cord tissue were embedded in paraffin wax. Seven-μm sections were deparaffinized in xylene and rehydrated using graded alcohols. Ab 338 immunohistochemistry required pretreatment with 99% formic acid for 10 minutes. Endogenous peroxidase activity was blocked (0.3% H2O2 in methanol, 10 minutes) and nonspecific binding with 10% dried milk solution. Tissue sections were incubated with Ab 338 (1:2,000, 1 hour at room temperature) followed by biotinylated anti-rabbit IgG (1:200, 30 minutes; DAKO), and ABC complex (30 minutes; DAKO). Color was developed with diaminobenzidine/H2O2. For AT8 immunohistochemistry 7-μm sections were pretreated in a microwave oven in sodium citrate buffer for 20 minutes. After washes in PBS and 10% milk, sections were incubated with the AT8 antibody (1:600) overnight at 4°C followed by biotinylated anti-mouse antibody (1:200, 30 minutes; DAKO). The remaining steps were as described above.
For double-staining with AT8 and Ab 338, 15-μm tissue sections were first pretreated in formic acid as described above and then incubated with AT8 (1:600) for 1 hour at room temperature followed by steps as described above. Sections were then incubated with Ab 338 (1:2,000) overnight at 4°C followed by appropriate secondary antibody and color development.
The AD and normal control cases were stained in a similar manner. For negative controls Ab 338 was replaced with the pre-immune serum or the pre-absorbed Ab 338.
Fluorescence Labeling for Confocal Microscopy
Tissue sections from the hippocampal formation, amygdala, cerebellum including dentate nucleus, cerebral cortex, and white matter were prepared using double-staining with Ab 338 and either GFAP or CD68. To determine the relationship between protein deposition and amyloid formation sections were double-stained with Ab 338 and Thioflavin S.
Twenty-μm sections were de-waxed and re-hydrated. Endogenous peroxidase activity was blocked as described above and sections were pretreated with trypsin (0.1%, 10 minutes at 37°C), then blocked with nonfat milk solution for 30 minutes. Sections were incubated with either GFAP (1:1,000, 1 hour at room temperature; DAKO) or CD68 (1:150, 1 hour at room temperature; DAKO), followed by the respective secondary antibodies (DAKO) and ABC complex. The reaction product was visualized using the tetramethylrhodamine signal amplification kit (NEN Life Science Products, Boston, MA). Incubation with the second antibody, Ab 338 (1:2,000) was performed (overnight, 4°C) without the usual formic acid pretreatment. Sections were then treated with biotinylated anti-rabbit antiserum (1:200) followed by the ABC complex. This reaction was visualized using the fluorescein signal amplification kit.
Double-staining with Ab 338 and Thioflavin S was performed by pretreating sections using 70% formic acid for 10 minutes and blocking in nonfat milk for 30 minutes. Ab 338 (1:2,000) was incubated overnight at 4°C followed by biotinylated anti-rabbit antibody (1:200) for 30 minutes, and finally ABC reagent for 30 minutes. Antibody binding was visualized using the tetramethylrhodamine signal amplification kit. Sections were counterstained in aqueous Thioflavin S (1.0%, 7 minutes; Sigma, Poole, UK) and differentiated with ethanol (70%, 5 minutes). The sections were then washed overnight in PBS.
Sections were viewed with a Leica TCS4D confocal microscope using a 3-channel scan head and argon/krypton laser.
Evaluation of 338 and AT8 Immunohistochemistry
The presence and frequency of Ab 338-positive plaques and CAA were evaluated in different anatomical regions using a semiquantitative approach. For CAA a four-tiered scoring system was devised such that score 0 was given for unaffected areas; ‘+’ if a proportion of the arterioles/small arteries were affected, but no or only an occasional capillary was stained; ‘+ +’ if the majority of arterioles as well as a minority of the capillaries were positive; and ‘+ + +’ for areas in which the majority of the arterioles and capillaries were Ab 338-positive.
For the quantitation of Ab 338-positive parenchymal deposits plaques were defined as Ab 338-, Congo red-, and Thioflavin S-positive structures with or without an associated blood vessel, whereas diffuse deposits were often ill-defined, Ab 338-positive, Congo red-negative structures, which were only weakly stained, if at all, with Thioflavin S. Using a ×10 objective plaques and diffuse deposits were scored separately in each case following a principle similar to that recommended by The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) for quantitating neuritic plaques in AD. 8 Score 0 described the absence of either plaques or diffuse deposits, score ‘+’ corresponded to sparse, score ‘+ +’ to moderate, and score ‘+ + +’ to frequent plaques or diffuse deposits. Variation between cases was recorded as shown in Tables 1 and 2 ▶ ▶ .
Table 1.
Distribution of the Different ABri Lesion Types and Tau Pathology in Familial British Dementia
| Type of lesion | ||||||
|---|---|---|---|---|---|---|
| Plaques, including perivascular | Diffuse deposits | CAA | NFT | AN | NT | |
| Olfactory bulb | ++ | +++ | ++ | + | + | + |
| Retina | + | + | ++ | 0 | 0 | 0 |
| Optic nerve | + | ++ | ++ | N/A | 0 | 0 |
| Optic chiasm | 0 | 0 | + | N/A | 0 | 0 |
| Optic tract | + | + | + | N/A | 0 | 0 |
| Hippocampus | +++ | +++ | +++ | +++ | +++ | +++ |
| Amygdala basal | +++ | +++ | +++ | +++ | +++ | +++ |
| Amygdala lateral | + | +++ | ++ | + | + | ++ |
| Uncus | ++ | +++ | ++ | ++ | ++ | +++ |
| Entorhinal cortex | + | +++ | ++ | +++ | + | +++ |
| Fusiform gyrus | + | +++ | ++ | +++ | 0 | +++ |
| 1st, 2nd, 3rd temporal gyri | + | + | ++ | + | 0 | + |
| Frontal cortex | 0–+ | + | ++ | 0 | 0 | 0 |
| Cingulum | + | + | ++ | + | 0 | + |
| Insular cortex | + | ++ | ++ | + | + | ++ |
| Parietal cortex | + | + | ++ | 0 | 0 | 0 |
| Occipital cortex | 0 | + | ++ | 0 | 0 | 0 |
| Cerebral white matter | + | ++ | ++–+++ | N/A | 0 | 0 |
| Hypothalamus | ++ | ++ | ++ | 0 | 0 | 0 |
| Striatum | 0 | 0 | 0 | 0 | 0 | 0 |
| Globus pallidus | 0 | 0 | 0 | 0 | 0 | 0 |
| Claustrum | + | ++ | ++ | + | 0 | + |
| Meynert nucleus | 0 | + | + | ++ | 0 | + |
| Thalamus | 0–+ | 0–+ | + | 0 | 0 | 0 |
| Subthalamus | + | + | ++ | 0 | 0 | 0 |
CAA, cerebral amyloid angiopathy; NFT, neurofibrillary tangle; AN, abnormal neurite; NT, neuropil thread; N/A, not applicable.
Table 2.
Distribution of the Different ABri Lesion Types and Tau Pathology in Familial British Dementia
| Type of lesion | ||||||
|---|---|---|---|---|---|---|
| Plaques, including perivascular | Diffuse deposits | CAA | NFT | AN | NT | |
| Tectum | 0 | ++ | ++ | + | 0 | 0 |
| Periaqueductal gray | + | ++ | + | + | 0 | + |
| Midbrain raphe | 0 | + | + | + | 0 | + |
| Substantia nigra | 0 | 0 | 0 | + | 0 | + |
| Red nucleus | ++ | ++ | +++ | 0 | 0 | 0 |
| Cerebral peduncle | + | ++ | ++ | N/A | 0 | 0 |
| Locus coeruleus | +++ | +++ | +++ | + | 0 | 0 |
| Pontine raphe | 0 | 0 | 0 | ++ | 0 | ++ |
| Pontine base | 0 | 0 | + | 0 | 0 | 0 |
| Inferior olive | +–++ | +–++ | ++ | 0 | 0 | 0 |
| Pyramid | 0 | +–++ | ++ | N/A | 0 | 0 |
| Cerebellar cortex | +++ | ++ | +++ | 0 | 0 | 0 |
| Cerebellar white matter | 0–+ | 0 | ++ | N/A | 0 | 0 |
| Dentate nucleus | +++ | + | +++ | 0 | 0 | 0 |
| Cervical cord LC | 0–+ | +++ | ++ | N/A | 0 | 0 |
| Cervical cord PC | 0–+ | +–++ | ++ | N/A | 0 | 0 |
| Cervical cord VC | 0–+ | 0–++ | +–++ | N/A | 0 | 0 |
| Cervical cord GM | + | +–++ | ++ | 0 | 0 | 0 |
| Thoracic LC | + | +++ | ++ | N/A | 0 | 0 |
| Thoracic PC | + | + | + | N/A | 0 | 0 |
| Thoracic VC | 0 | 0–+ | + | N/A | 0 | 0 |
| Thoracic GM | 0 | 0–+ | + | 0 | 0 | 0 |
| Lumbar LC | 0 | 0 | + | N/A | 0 | 0 |
| Lumbar PC | + | 0–+ | 0–+ | N/A | 0 | 0 |
| Lumbar VC | 0 | 0–+ | + | N/A | 0 | 0 |
| Lumbar GM | 0–+ | ++ | ++–+++ | 0 | 0 | 0 |
| Sacral LC | 0 | 0 | + | N/A | 0 | 0 |
| Sacral PC | 0 | 0 | ++ | N/A | 0 | 0 |
| Sacral VC | 0 | 0 | + | N/A | 0 | 0 |
| Sacral GM | 0–+ | ++ | ++ | 0 | 0 | 0 |
CAA, cerebral amyloid angiopathy; NFT, neurofibrillary tangle; AN, abnormal neurite; NT, neuropil thread; LC, lateral column; PC, posterior column; VC, ventral column; GM, gray matter; N/A, not applicable.
The severity of NFT, NT, and AN pathologies was evaluated separately in a semiquantitative manner using a ×10 objective; score 0 was used if the pathological change was absent, ‘+’ was used if it was sparse, ‘+ +’ moderate, and ‘+ + +’ if the change was severe.
Immunoelectron Microscopy
Ultrathin sections were mounted on etched nickel grids, and treated with sodium metaperiodate solution (4.7% for 1 hour). Grids were washed in 0.5 mol/L Tris-buffered saline (TBS) (pH 7.2) containing 1% Triton X-100 and blocked with 0.5 mol/L TBS/bovine serum albumin (BSA) (pH 7.2) containing 0.05% sodium azide. Grids were then incubated in 10% normal goat serum in 0.5 mol/L TBS/BSA for 1 hour followed by Ab 338 (1:20,000) overnight at 4°C. Washes were performed with 0.5 mol/L TBS/BSA before incubation with goat anti-rabbit IgG gold conjugate (1:15) with a particle size of 20 nm (60 minutes at 37°C; Sigma). After further washes in 0.5 mol/L TBS/BSA the grids were postfixed in 2.5% glutaraldehyde in 0.05 mol/L sodium cacodylate buffer (10 minutes), and counterstained with uranyl acetate/lead citrate using a standard protocol.
Western Blotting
Insoluble tau was extracted from fresh, frozen samples of the hippocampus of case 5, as described previously. 10 In brief, 0.1 to 0.2 g of brain tissue was hand-homogenized in 50 mmol/L 2-[N-Morpholino]ethanesulphonic acid (MES) buffer, pH 6.5, containing 1 mol/L NaCl, 50 mmol/L imidazole, 0.1 mmol/L phenylmethyl sulfonyl fluoride, 20 mmol/L NaF, 10 mmol/L sodium pyrophosphate, and 25 mmol/L Naβ-glycerophosphate and the homogenate was centrifuged at 28,000 rpm for 30 minutes at 4°C. The supernatant was retained and re-centrifuged at 48,000 rpm for 60 minutes at 4°C. The pellet of the second centrifugation step was solubilized in Laemmli sample buffer and analyzed on sodium dodecyl sulfate-polyacrylamide gel electrophoresis using a discontinuous buffer method. 11 The resolved proteins were electroblotted (15 V, 30 minutes) onto nitrocellulose membranes (pore size, 0.45 μm; Schleicher & Schuell, Andermann, London, UK). Blocking was performed with TBS containing 0.2% Tween 20 and 3% dried skimmed milk (TBS-T-M) before incubation overnight with PHF-1 (1:2,000), TP70 (1:100,000), AT8, AT180, AT270, and AT100 (all 1:200) diluted in TBS-T-M. Blots were developed using either horseradish peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG (1:1,000) as appropriate followed by enhanced chemiluminescence detection as described by the manufacturer (Nycomed-Amersham, Little Chalfont, UK).
Results
Ab 338 Immunohistochemistry, Congo Red, and Thioflavin S Staining of Parenchymal Deposits
Amyloid plaques, which were both Congo red- and Thioflavin S-positive, were seen in many of the central nervous system areas. Ab 338 stained all previously described plaque types including the large and small plaques of the hippocampal formation and also the parenchymal amyloid plaques in the cerebellum and other structures (Figure 1, A ▶ –C). 2,5 Ab 338 and Thioflavin S double-staining showed that the staining patterns overlapped well in the majority of hippocampal plaques of all types and cerebellar amyloid plaques, although examples were found whose margins, determined with Ab 338, extended beyond those seen with Thioflavin S (Figure 2 ▶ , A–C). This was also seen in the cerebellum, particularly in the granular cell layer, where Ab 338 often stained structures more extensively and/or intensely than Thioflavin S. The same pattern was also apparent in some of the perivascular plaques (Figure 2 ▶ , D–F).
Figure 1.
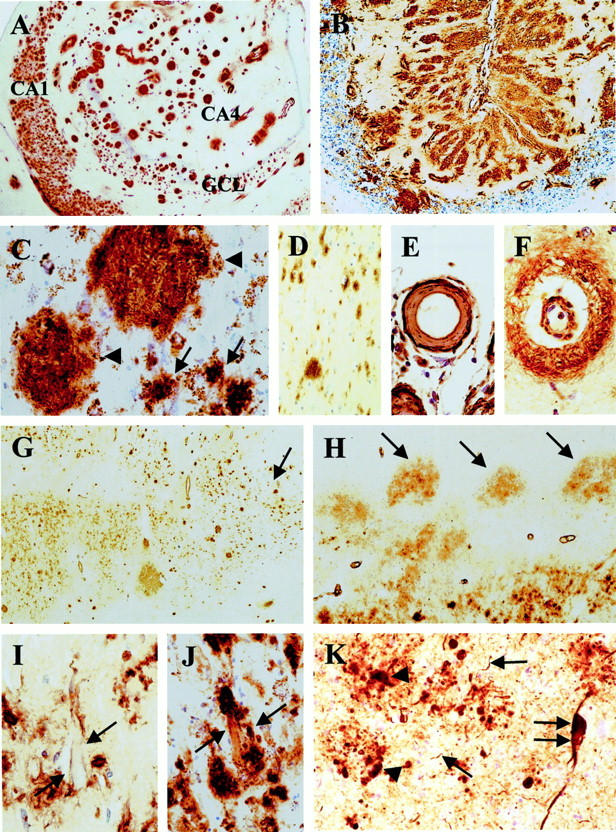
A: Numerous Ab 338-positive structures outlining the anatomical regions of the hippocampus. B: Extensive Ab 338-positive structures in the cerebellar cortex. C: Large (arrowhead) and small (arrow) plaques in the (hippocampus) CA2 subregion. D: Diffuse deposits in the fusiform gyrus. E: An affected leptomeningeal vessel showing ABri deposition with enhanced staining of the periphery. F: Small parenchymal vessel with staining of the vessel wall and perivascular parenchyma. G: The Ab 338 staining pattern of the entorhinal cortex showing a bi-laminar appearance becoming united toward the transenthorhinal cortex (arrow). H: Ab 338-positive diffuse deposits in the parvopyramidal cell clusters (arrow) of the pre- and parasubiculum. I: Membranous staining of a pyramidal neuron in CA1 (arrow). J: An extracellular neurofibrillary tangle labeled by Ab 338 partly incorporated into a small plaque (arrow). K: Tau immunohistochemistry in the basal nucleus of the amygdala showing NFT (double arrow), NTs (arrow), and ANs (arrowhead). Ab 338 immunohistochemistry (A–J) and AT8 immunohistochemistry (K). GCL, granular cell layer of the dentate fascia. Original magnifications: ×6 (A); ×8 (G and H); ×30 (B); ×70 (C and D); ×130 (E, F, and K); ×200 (I and J).
Figure 2.
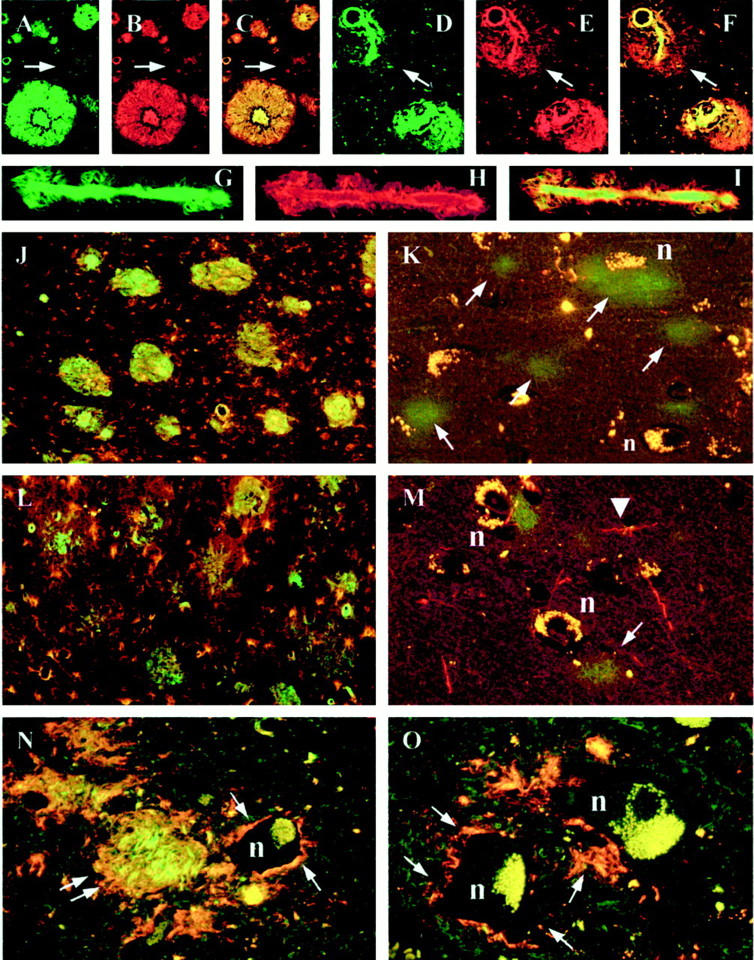
Confocal images. A, B, and C: There is a good overlap between Thioflavin S (green) and Ab 338 (red) staining of plaques as demonstrated in the combined image (C). However a small area is Thioflavin S-negative suggesting nonfibrillary protein (arrow). D, E, and F: Hippocampal blood vessel with perivascular Ab 338 staining, which is Thioflavin S-negative (arrow). G, H, and I: Hippocampal blood vessel with a good overlap between Thioflavin S and Ab 338 indicating fibrillar ABri. J and K: Ab 338 (green) and CD68 (red) double immunohistochemistry showing a marked microglial response around hippocampal amyloid plaques (J), but sparse reaction in the entorhinal cortex where the protein is predominantly nonfibrillar (K). L and M: Ab 338 (green) and GFAP (red) double immunohistochemistry showing a similar pattern of astrocytic reaction in the same areas; on M arrowhead points to an astrocytic process. N and O: Thioflavin S and Ab 338 double-staining shows a nonfibrillar perineuronal staining pattern (arrow) in the inferior olive (N), anterior horn of the sacral cord (O). There is an amyloid plaque in the inferior olive (double arrow). n = neuron. Original magnifications: ×20 (A–I), ×10 (J and L), and ×40 (K, M, N, and O).
Ab 338 immunohistochemistry, supplemented with Congo red and Thioflavin S methods combined with semiquantitative evaluation, showed little variation between cases (Tables 1 and 2) ▶ ▶ . We found not only that the hippocampus and cerebellum were most severely affected by amyloid plaques, but also that amyloid plaques were present in considerable numbers in structures such as the hypothalamus, amygdala, locus coeruleus, inferior olive, and the cerebellar dentate nucleus (Tables 1 and 2) ▶ ▶ (Figure 2 ▶ N). In contrast the striatum, globus pallidus, and substantia nigra, which also lacked CAA, were entirely unaffected by parenchymal deposits whereas the thalamic involvement varied between cases. Diffuse deposits, defined as Ab 338-positive and Congo red-negative structures, unstained or only weakly stained with Thioflavin S, were noted in varying numbers in other areas such as the entorhinal cortex, neocortex, tectum, periaqueductal gray, the medullary pyramids, and spinal cord gray and white matter (Figure 1, D, G, and H ▶ ; and Figure 2 ▶ , K and M). Both amyloid plaques, which were mostly of the perivascular type, and diffuse deposits were sparse in the neocortex where they appeared randomly distributed throughout the cortical laminae. In the cerebral cortices loosely arranged diffuse deposits were also seen to surround neurons, often with an accentuation of staining adjacent to the perikaryon. This phenomenon was found at several other locations, including in the pyramidal cell and granular cell layers of the hippocampus, around Purkinje cells, anterior horn cells of the cord, and inferior olivary neurons (Figure 2 ▶ , N and O). A delicate membranous staining pattern highlighting neuronal cell margins was noted most readily in hippocampal pyramidal cells (Figure 1I) ▶ . In the CA1 region and subiculum a number of less compact NFTs, not associated with visible neuronal nucleus or plasma membrane (extracellular tangles, 12,13 ), were stained with Ab 338 and a few such structures seemed to have been incorporated into amyloid plaques (Figure 1J) ▶ .
Between the hippocampus and third temporal gyrus Ab 338-positive deposits affected all allocortical areas and also the isocortical fusiform gyrus. An anatomically determined sharp transition between two staining patterns was noted at the junction between the subiculum and presubiculum; the subiculum contained a significant number of Ab 338-, Congo red-, and Thioflavin S-positive amyloid plaques, whereas the presubiculum and the parasubiculum had an ill-defined diffuse staining pattern. The size and shape of the Ab 338 positivity closely matched the anatomical boundaries of the pre- and parasubicular parvopyramidal cell islands (Figure 1H) ▶ . In the entorhinal cortex the deposits were predominantly of the diffuse type with a striking bilaminar distribution. There was an inner wide band, occupying the deeper cortical laminae, with an ill-defined, confluent staining pattern and a narrower outer band composed of often better-circumscribed deposits (Figure 1G) ▶ . The outer Ab 338-positive band closely overlapped with the Pre-α neuronal clusters and, with the Pre-α neurons themselves, 14 gradually descended in the transenthorhinal cortex to form a single band of well-defined Ab 338-positive, mainly diffuse deposits in the deeper laminae of the fusiform gyrus.
In many cerebral and cerebellar areas and also in the spinal cord Ab338 stained subpial deposits, which were variably positive on Congo red or Thioflavin S preparations. Ab 338 also stained subependymal areas around the ventricles and cerebral aqueduct in a diffuse pattern. Clearly defined fibrillar staining was also seen associated with the basolateral aspect of the ependyma.
The cerebral, cerebellar, and spinal cord white matter showed mostly diffuse deposits, although perivascular amyloid plaques were also noted (Tables 1 and 2) ▶ ▶ .
No staining of vascular amyloid, plaques or diffuse deposits of the FBD cases was observed in controls using the pre-immune serum, the Ab 338 pre-absorbed with synthetic peptide or when the primary antibody was omitted. Immunohistochemical staining with Ab 338 of brain tissues from individuals with AD or normal controls were also negative (results not shown).
Ab 338 Immunohistochemistry, Congo Red, and Thioflavin S Staining of Blood Vessels
All affected blood vessels stained with Congo red and Thioflavin S were also Ab 338-positive. Furthermore on preparations double-stained with Ab 338 and Thioflavin S there was an overall good overlap between the different staining patterns, although a smaller proportion of affected blood vessels, especially those with perivascular plaques were more extensively stained with Ab 338 (Figure 2 ▶ , D–F, G–I). Many of the leptomeningeal and parenchymal small arteries, arterioles, and capillaries were stained throughout the central nervous system, although the severity of CAA varied considerably in the different areas; the striatum, globus pallidus, and substantia nigra were entirely unaffected (grade, 0), in the thalamus and pontine base angiopathy was of moderate severity (grade, + to ++), whereas in other areas such as the hippocampus and cerebellar cortex it was severe (grade, + + +). In the cerebellum there was a difference in the staining pattern of the blood vessels within the white matter in that the blood vessels of the deep white matter, with the exception of those found in the vicinity of the dentate nucleus, were more rarely stained with Ab 338 than those of the white matter in the cerebellar folia.
The majority of the Ab 338-positive blood vessels were arterial, but a proportion of venous channels was also affected. These latter were more frequent in the leptomeninges, but were also found in the parenchyma. Although small arteries with a diameter of up to 1 mm were stained with the Ab 338, the majority of the affected blood vessels were smaller than 300 μm. The larger vessels were usually less affected in that Ab 338 primarily stained the abluminal aspect of the media and the adventitia. In contrast, in many of the small arteries and arterioles Ab 338 positivity was seen throughout the full thickness of the wall, often with severe disruption of its normal structure and sometimes occlusion of the lumen. An accentuation of the staining in the outer perimeter of some small arteries and arterioles, was noted (Figure 1E) ▶ .
Longitudinal sections of affected arterioles and capillaries showed that Ab 338 often stained vessel walls in a linear manner although the full length was not always affected. Other staining patterns included the presence of globular deposits along vessel walls or fine Ab 338-positive spicules radiating from the walls of affected arterioles and capillaries (Figure 2 ▶ , G–I). In all regions the glia limitans around many affected vessels was stained with an extension into the adjacent parenchyma, often giving rise to plaque-like structures, previously described as ‘perivascular plaques’ (Figure 1F) ▶ . 2
There was substantial vascular amyloid in many retinal vessels and, similar to the subpial deposits, there were small amyloid plaques and diffuse deposits along the retinal inner limiting membrane. No gross decrease in cell numbers could be discerned in any of the retinal layers and the photoreceptor outer segments and retinal pigmented epithelial cells appeared identical to age-matched controls. There was an increase in GFAP expression in Müller’s glial cells, which has been previously documented in aging rat retina. 15
AT8 Immunohistochemistry and Its Correlation with Ab 338 Immunohistochemistry
Severe involvement by NFTs, and NTs of mediotemporal and other limbic structures, including the hippocampus, subiculum, entorhinal and transenthorhinal cortices, amygdala, uncus, cingulum, and insular cortex was readily noted (Figure 1K) ▶ . Double AT8 and Ab 338 immunohistochemistry also revealed that the amyloid plaques, occurring in these structures, tended to be surrounded with ANs, many of which were fine and thread-like and were often incorporated within the large hippocampal amyloid plaques in a radial pattern. ANs clustering around an occasional blood vessel with amyloid deposition were also seen in the hippocampus, amygdala, and temporal neocortex. There was a strikingly similar distribution of AT8-positive structures and Ab 338-positive amyloid plaques within the amygdala in that both pathologies affected the basal nucleus more severely than the lateral nucleus (Figure 1K) ▶ . Occasional NFTs and NTs were seen in the parvopyramidal clusters of the pre- and parasubiculum. In the entorhinal cortex, which was rich in nonfibrillar ABri and contained only relatively few amyloid plaques, the Pre-α and Pri-α neurons were severely affected, containing frequent NFTs and NTs, which because of their large numbers, produced darkly stained bands in both cell layers. The Pre-β and Pre-γ neurons also contained moderate numbers of NFTs. In the fusiform gyrus, which contained predominantly nonfibrillar diffuse plaques, there were numerous NFTs and NTs. These gradually decreased in number toward the neighboring third temporal gyrus, which, similar to the first and second temporal gyri contained only sparse NFTs and NTs. Other neocortices, except the cingulum and insular cortex, were unaffected. The hypothalamus, claustrum, midbrain tectum, periaqueductal gray, and locus coeruleus contained sparse and the pontine raphé moderate numbers of NFTs. The cerebellum and spinal cord were unaffected by tau pathology (Tables 1 and 2) ▶ ▶ .
Correlation between Ab 338, CD68, and GFAP Staining of Different Lesion Types
Tissue sections double-stained with Ab 338 and CD68 and examined by confocal microscopy showed a good co-localization between cells of microglial/macrophage lineage and both amyloid plaques and vascular amyloid. However such cells were few in areas in which diffuse deposits were predominant (Figure 2 ▶ , J and K). A similar relationship between GFAP-positive astrocytes and AB 338-positive structures was seen on preparations double-labeled with Ab 338 and GFAP (Figure 2 ▶ , L and M).
Immunoelectron Microscopy
Ultrathin sections from the hippocampus and temporal neocortex, labeled with Ab 338 using the immunogold method, were examined. Tissue preservation was such that blood vessels, neurons, astrocytes, and both neuronal and glial processes could be identified. In the hippocampus frequent amyloid plaques were present and many small blood vessels had amyloid fibrils deposited within their walls thus confirming the findings of light microscopy. Amyloid plaques varied in size and were composed of short fibrils of ∼10 nm diameter, which were randomly distributed but varied in density, some plaques being composed of loosely arranged fibrils whereas others were more compact. The amyloid fibrils were consistently heavily labeled by Ab 338. Glial processes containing intermediate filaments were often seen within plaques (Figure 3, A and B) ▶ .
Figure 3.
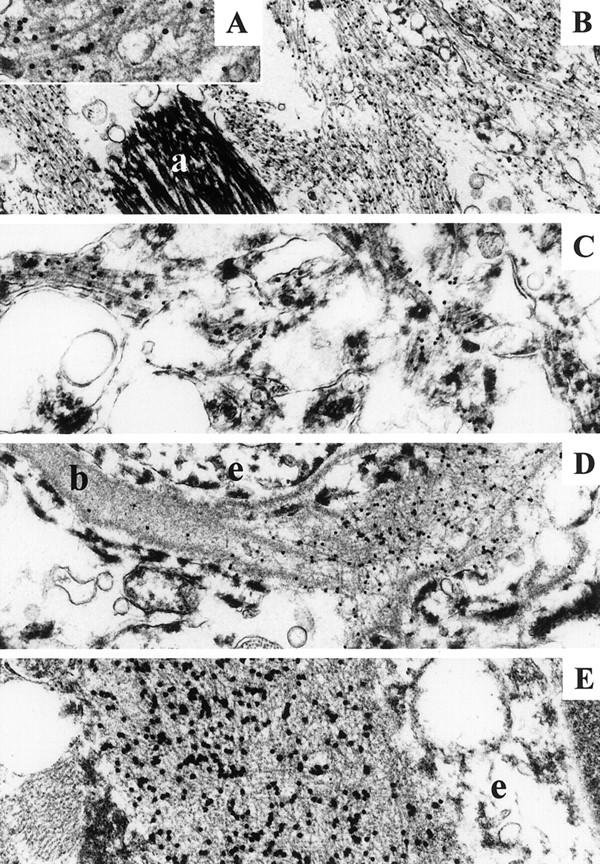
Ab 338 immunoelectron microscopy. B: Bundles of labeled fibrils in a hippocampal plaque. An astrocytic process (a) is unlabeled. Insert A showing labeled fibrils with high magnification. C: Amorphous electron-dense material together with sparse fibrils is decorated with Ab 338 in a Thioflavin S-negative area of temporal neocortex. D: Small vessel showing labeling of the basal lamina (b), which is focally disrupted by amyloid fibrils. E: A vessel showing extensive deposition of labeled fibrils in the basal lamina. D and E: Endothelial cells (e) are unlabeled. Original magnifications: ×16,000 (A); ×6,600 (B); ×10,000 (C); ×8,300 (D); and ×13,000 (E).
Regions of the temporal neocortex in which Ab 338-positive, but Thioflavin S-negative diffuse deposits had been identified in semithin sections were selected for immunoelectron microscopy. No amyloid plaques were identified ultrastructurally; however, extracellular immunogold labeling was seen associated with amorphous electron-dense material that was sometimes accompanied by small numbers of loosely arranged fibrils resembling the amyloid fibrils of plaques (Figure 3C) ▶ . Such electron-dense material was not found in association with hippocampal amyloid plaques.
Small blood vessels showed labeling of the basal lamina by Ab338. Several vessels had normal segments of basal lamina adjacent to areas in which a low level of labeling was present in its central part. The basal lamina then often became expanded into regions with a greater labeling density in which amyloid fibrils became apparent. In some vessels the intensity of labeling was greatest on the abluminal aspect of the basal lamina reflecting the accentuation of Ab 338 staining seen around the outer perimeter of some vessels using light microscopy. The accumulation of amyloid fibrils within the basal lamina produced increased expansion and eventual disruption of the abluminal surface whereas the luminal aspect of the basal lamina remained intact (Figure 3, D and E) ▶ .
NFTs composed of paired helical filaments were frequently found in both neuronal cell bodies and processes but these were unlabeled by Ab338.
No labeling was found using the pre-immunization rabbit serum or when the primary antibody was omitted. The antibody that had been previously absorbed using the immunizing peptide showed some residual labeling of amyloid fibrils but this was greatly reduced in comparison with untreated Ab338 (not shown).
Tau Immunoblotting
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting analysis of the insoluble tau extracted from the control case, with the polyclonal tau antiserum TP70 that recognizes all forms of tau protein, corresponded to the normal tau pattern of post mortem adult human brain because it consisted of 6 to 8 bands. The AD case showed the characteristic triplet with T54, T59, and T64 tau species and a minor T71 band when probed with all of the tau antibodies. In the FBD case, tau comprised a triplet pattern, which was indistinguishable from the pattern seen in the AD case (Figure 4) ▶ .
Figure 4.

Tau immunoblots in AD (lane 1) and FBD (lane 2) cases using the PHF1 antibody.
Discussion
In this study of five cases with FBD we showed that Ab 338 stained a large number of lesions of different morphological types in the central nervous system including the retina. The lesions stained included blood vessels affected by amyloid deposition, amyloid plaques, and also diffuse, nonfibrillar, parenchymal ABri deposits. Using the same antibody for immunoelectron microscopic examination we confirmed that bundles of amyloid fibrils in both plaques and blood vessel walls were labeled. In areas with diffuse parenchymal deposits, nonfluorescent with Thioflavin S, Ab 338 decorated amorphous, electron-dense extracellular material, which was interspersed with sparse fibrils, but not with bundles of fibrils. As such, tinctorial, optical, and ultrastructural appearances are thought to represent protein deposits in a nonfibrillar configuration, or pre-amyloid, 16,17 the presence of such lesions demonstrates that ABri deposits in both fibrillar and nonfibrillar forms throughout the central nervous system in FBD. It can be argued that the immunoreactivity detected by Ab 338 does not represent solely the presence of ABri because the specific epitope (TVKKNIIEEN) is shared by ABri, its precursor ABriPP and potential C-terminal fragments generated by the proteolytic processing of either species. However, preliminary mass spectrometry studies of the material extracted from amyloid and pre-amyloid lesions in a case of FBD indicates that more than 95% of the protein species are composed of full-length ABri peptide (J Ghiso, unpublished data).
This immunohistochemical study confirmed findings of previous pathological examinations, including ours, showing that in FBD the hippocampus and cerebellum are among the anatomical areas most severely affected by amyloid deposition in both blood vessels and parenchymal plaques. 2,5,18,19 Furthermore we were also able to show not only that ABri is widely deposited throughout the central nervous system, but also that the sites affected include other anatomically well-defined structures such as amygdala, hypothalamic nuclei, inferior olive, the dentate nucleus, and the parvopyramidal cell clusters of the pre- and parasubiculum; which may be affected by Aβ deposition in AD. 20-22
The hippocampal amyloid plaques had rarely been found to be associated with surrounding ANs when studied with silver impregnation techniques. 2 However, in addition to tau-positive dystrophic neurites, a rich network of fine thread-like processes, permeating the large amyloid plaques was readily detectable when the AT8 antibody, recognizing phosphorylated serine 202/threonine 205 epitopes of tau, 23-25 was used. 5 In addition to demonstrating the predominant involvement of limbic structures by neurofibrillary pathology in FBD, we also showed that AT8-positive neurites are associated with amyloid plaques and occasionally with vascular amyloid rather than with nonfibrillar diffuse deposits. In contrast, NFTs and NTs occur in association with both amyloid plaques and diffuse deposits. A number of NFTs with appearances of extracellular tangles 12,13 were stained with Ab 338 in the hippocampus and subiculum, which are among the structures with the highest parenchymal ABri load. It has been shown that in AD Aβ deposition within extracellular tangles, taking place in the vicinity of senile plaques, is a secondary phenomenon 26 and we suggest that a similar mechanism is likely in FBD.
The predominantly limbic distribution of the tau pathology together with mild involvement of the temporal cortex would suggest that the NFT pathology corresponds to stage IV in the system recommended by Braak and Braak 27 for staging NFT pathology in AD. In FBD the severity of the limbic involvement exceeds that expected in stage IV of AD and the presence of NFTs in structures such as the granular cell layer of the dentate fascia 5 and substantia nigra suggests that in FBD the progression of NFT pathology may be different from that seen in AD. We have confirmed our previous finding that NFTs in FBD are composed ultrastructurally of paired helical filaments 5 and showed, for the first time that insoluble tau has a triplet electrophoretic migration pattern on Western blots using a panel of phosphorylation-dependent and -independent antibodies, one of which (AT100) has been described as PHF tau-specific. 28
Although the mechanism of NFT formation in FBD remains to be investigated, the close topographic association of both fibrillar and nonfibrillar ABri with neurofibrillary pathology in limbic structures may support the notion that NFTs are principally because of ABri-mediated neurotoxicity. This hypothesis is supported by our finding of co-localization of reactive astrocytes and microglia with ABri in β-sheet conformation, which is strikingly similar to the findings in AD, in which a relationship between the presence of these cell types and the neurodegenerative events have been suggested. 29-31 Despite the close association between neurofibrillary pathology and ABri deposition in limbic structures, the absence of apparent neurodegeneration in some other well-defined protein-bearing regions such as the dentate nucleus and inferior olive, points to the importance of other biological variables in this process.
Ab 338-positive congophilic angiopathy of variable severity was widespread and was only absent in the striatum, globus pallidus, and substantia nigra. In FBD the overall patterns of amyloid deposition in cerebral blood vessels are similar to those seen in congophilic angiopathies related to other amyloid peptides, in particular Aβ, in which arteries and arterioles are more frequently affected than veins. 32
The origin and mechanism of deposition of ABri in plaques and blood vessel walls is not known. One possibility is that ABri is produced by cellular components of the central nervous system itself. In support of such a hypothesis is the finding that BriPP mRNA is expressed at high levels in a number of areas of normal human brain including the hippocampus, amygdala, and cerebellum, 6 which we have shown, are those areas most affected by fibrillar and nonfibrillar ABri deposition. Furthermore furin, which may be involved in cleaving both the wild-type and mutant precursor proteins, releasing a 23-amino acid long C-terminal peptide and the amyloidogenic ABri from BriPP and ABriPP, respectively, 33 is also widely expressed in both neurons and glial cells in the central nervous system. 34 ABri is deposited in areas composed of particular neuronal groups such as the locus coeruleus, inferior olive, cerebellar dentate, or the parvopyramidal neuronal clusters of the pre- and parasubiculum and often shows a striking perineuronal distribution in areas of diffuse plaques and an association with neuronal plasma membranes. These morphological findings are similar to those that linked Aβ production with neurons, 20,22,35,36 and raise the possibility that neurons may be one of the cell types involved in cerebral ABri production/deposition. Both pathological and experimental evidence now exists to suggest that neuronal expression of an amyloidogenic protein can result in both parenchymal deposits and CAA. Aβ depositing as vascular amyloid may be of cerebral cortical origin and transported along periarterial interstitial fluid drainage pathways in both human AD 32 and in a transgenic mouse model of AD (APP23) that overexpresses mutant human amyloid precursor protein driven by the neuronal Thy-1 promoter. In this transgenic model of AD there are both amyloid plaques and significant deposition of Aβ in the cerebral vasculature. 37 In FBD the widespread deposition of ABri in small cerebral arteries and arterioles is also consistent with a nonneuronal, vascular smooth muscle cell, pericytic or even systemic origin of the ABri peptide. Preliminary data indicate that soluble ABri is present in the circulation as has been found for Aβ in AD 38,39 and some vascular amyloid has been found in blood vessels in organs (J Ghiso, unpublished data). The significance of these findings in relation to deposition of ABri in the central nervous system remains to be investigated.
FBD with CAA, parenchymal amyloid plaques, and diffuse deposits as well as neurofibrillary degeneration mimics important morphological features of AD, although there are distinct features in both diseases. Limbic structures are severely affected by both parenchymal amyloid deposition and neurofibrillary degeneration in both conditions, although in AD neocortical involvement is much more prominent than in FBD. The cytoskeletal pathology in FBD, immunohistochemically, ultrastructurally, and biochemically, is indistinguishable from that found in AD. 10,40 The morphology of limbic plaques shows some similarities with AD as the large plaques of FBD noticeably resemble the cotton wool plaques seen in variant AD with spastic paraparesis associated with PS1Δ9 41 or PS1 exon 4 DelIM mutation. 42 However, there are differences between the two lesions as the cotton wool plaques are mainly Congo red-negative; whereas the large hippocampal plaques of FBD are Congo red- and Thioflavin S-positive. CAA resulting in white matter degeneration is not a unique feature of FBD as this can be associated with a similar pathological picture in AD. 43,44 Therefore the study of FBD, including future development of different transgenic animal models, may result in a better understanding of the link between amyloid deposition and neurodegeneration.
Acknowledgments
We thank Professor Margaret Esiri for providing material from case 3; and Mr. Steve Durr and members of the Audiovisual Department, Institute of Neurology, for providing help with the illustrations.
Footnotes
Address reprint requests to T. Revesz, M.D., FRCPath, Department of Neuropathology, Institute of Neurology, Queen Square, London WC1N 3BG, United Kingdom. E-mail: t.revesz@ion.ucl.ac.uk.
Supported by National Institutes of Health grants AG05891 and AG08721 and a grant from the Alzheimer Association. T. R. was recipient of a Wellcome Trust Travel Award, and G. G. is supported by a grant from the Wellcome Trust.
References
- 1.Worster-Drought C, Hill TR, McMenemey WH: Familial presenile dementia with spastic paralysis. J Neurol Psychopathol 1933, 14:27-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plant GT, Revesz T, Barnard RO, Harding AE, Gautier-Smith PC: Familial cerebral amyloid angiopathy with nonneuritic amyloid plaque formation. Brain 1990, 113:721-747 [DOI] [PubMed] [Google Scholar]
- 3.Mead S, James-Galton M, Revesz T, Doshi RB, Harwood G, Pan EL, Ghiso J, Frangione B, Plant G: Familial British dementia with amyloid angiopathy: early clinical, neuropsychological and imaging findings. Brain 2000, 123:975-991 [DOI] [PubMed] [Google Scholar]
- 4.Worster-Drought C, Greenfield JG, McMenemey WH: A form of familial presenile dementia with spastic paralysis (including the pathological examination of a case). Brain 1940, 63:237-254 [Google Scholar]
- 5.Revesz T, Holton JL, Doshi B, Anderton BH, Scaravilli F, Plant GT: Cytoskeletal pathology in familial cerebral amyloid angiopathy (British type) with non-neuritic amyloid plaque formation. Acta Neuropathol (Berl) 1999, 97:170–176 [DOI] [PubMed]
- 6.Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J: A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 1999, 399:776-781 [DOI] [PubMed] [Google Scholar]
- 7.Vidal R, Revesz T, Rostagno A, Kim E, Holton JL, Bek T, Bojsen-Moller M, Braendgaard H, Plant G, Ghiso J, Frangione B: A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci USA 2000, 97:4920-4925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirra SS, Heyman A, Mckeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, Vanbelle G, Berg L: The consortium to establish a registry for Alzheimer’s disease (CERAD). 2. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41:479-486 [DOI] [PubMed] [Google Scholar]
- 9.Brion JP, Couck AM, Robertson J, Loviny TL, Anderton BH: Neurofilament monoclonal antibodies RT97 and 8D8 recognize different modified epitopes in paired helical filament-tau in Alzheimer’s disease. J Neurochem 1993, 60:1372-1382 [DOI] [PubMed] [Google Scholar]
- 10.Hanger DP, Brion JP, Gallo JM, Cairns NJ, Luthert PJ, Anderton BH: Tau in Alzheimer’s disease and Down’s syndrome is insoluble and abnormally phosphorylated. Biochem J 1991, 275:99-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laemmli U: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227:680-685 [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi H, Morimatsu M, Hirai S, Takahashi K: Alzheimer’s neurofibrillary tangles are penetrated by astroglial processes and appear eosinophilic in their final stages. Acta Neuropathol (Berl) 1987, 72:214–217 [DOI] [PubMed]
- 13.Bondareff W, Harrington C, Wischik CM, Hauser DL, Roth M: Immunohistochemical staging of neurofibrillary degeneration in Alzheimer’s disease. J Neuropathol Exp Neurol 1994, 53:158-164 [DOI] [PubMed] [Google Scholar]
- 14.Braak H, Braak E: On areas of transition between entorhinal allocortex and temporal isocortex in the human-brain: normal morphology and lamina-specific pathology in Alzheimer’s disease. Acta Neuropathol (Berl) 1985, 68:325–332 [DOI] [PubMed]
- 15.Bjorklund H, Eriksdotter-Nilsson M, Dahl D, Rose G, Hoffer B, Olson L: Image analysis of GFA-positive astrocytes from adolescence to senescence. Exp Brain Res 1985, 58:163-170 [DOI] [PubMed] [Google Scholar]
- 16.Verga L, Frangione B, Tagliavini F, Giaccone G, Migheli A, Bugiani O: Alzheimer patients and Down patients: cerebral preamyloid deposits differ ultrastructurally and histochemically from the amyloid of senile plaques. Neurosci Lett 1989, 105:294-299 [DOI] [PubMed] [Google Scholar]
- 17.Giaccone G, Verga L, Bugiani O, Frangione B, Serban D, Prusiner SB, Farlow MR, Ghetti B, Tagliavini F: Prion protein preamyloid and amyloid deposits in Gerstmann-Straussler-Scheinker disease, Indiana kindred. Proc Natl Acad Sci USA 1992, 89:9349-9353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffiths RA, Mortimer TF, Oppenheimer DR, Spalding JM: Congophilic angiopathy of the brain: a clinical and pathological report on two siblings. J Neurol Neurosurg Psychiatry 1982, 45:396-408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love S, Duchen LW: Familial cerebellar ataxia with cerebrovascular amyloid. J Neurol Neurosurg Psychiatry 1982, 45:271-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalus P, Braak H, Braak E, Bohl J: The presubicular region in Alzheimer’s disease: topography of amyloid deposits and neurofibrillary changes. Brain Res 1989, 494:198-203 [DOI] [PubMed] [Google Scholar]
- 21.Ogomori K, Kitamoto T, Tateishi J, Sato Y, Suetsugu M, Abe M: Beta-protein amyloid is widely distributed in the central nervous system of patients with Alzheimer’s disease. Am J Pathol 1989, 134:243-251 [PMC free article] [PubMed] [Google Scholar]
- 22.Wisniewski HM, Sadowski M, Jakubowska-Sadowska K, Tarnawski M, Wegiel J: Diffuse, lake-like amyloid-beta deposits in the parvopyramidal layer of the presubiculum in Alzheimer disease. J Neuropathol Exp Neurol 1998, 57:674-683 [DOI] [PubMed] [Google Scholar]
- 23.Goedert M, Jakes R, Crowther RA, Six J, Lubke U, Vandermeeren M, Cras P, Trojanowski JQ, Lee VMY: The abnormal phosphorylation of tau-protein at ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci USA 1993, 90:5066-5070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su JH, Cummings BJ, Cotman CW: Early phosphorylation of tau in Alzheimer’s disease occurs at Ser-202 and is preferentially located within neurites. Neuroreport 1994, 5:2358-2362 [DOI] [PubMed] [Google Scholar]
- 25.Schmidt ML, Didario AG, Lee VMY, Trojanowski JQ: An extensive network of PHF-τ-rich dystrophic neurites permeates neocortex and nearly all neuritic and diffuse amyloid plaques in Alzheimer disease. FEBS Lett 1994, 344:69-73 [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi H, Nakazato Y, Shoji M, Okamoto K, Ihara Y, Morimatsu M, Hirai S: Secondary deposition of beta amyloid within extracellular neurofibrillary tangles in Alzheimer-type dementia. Am J Pathol 1991, 138:699-705 [PMC free article] [PubMed] [Google Scholar]
- 27.Braak H, Braak E: Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991, 82:239–259 [DOI] [PubMed]
- 28.ZhengFischhofer QY, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E: Sequential phosphorylation of Tau by glycogen synthase kinase-3 beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem 1998, 252:542-552 [DOI] [PubMed] [Google Scholar]
- 29.el Hachimi KH, Verga L, Giaccone G, Tagliavini F, Frangione B, Bugiani O, Foncin JF: Relationship between non-fibrillary amyloid precursors and cell processes in the cortical neuropil of Alzheimer patients. Neurosci Lett 1991, 129:119-122 [DOI] [PubMed] [Google Scholar]
- 30.Pike CJ, Cummings BJ, Cotman CW: Early association of reactive astrocytes with senile plaques in Alzheimer’s disease. Exp Neurol 1995, 132:172-179 [DOI] [PubMed] [Google Scholar]
- 31.Selkoe DJ: Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999, 399:A23-A31 [DOI] [PubMed] [Google Scholar]
- 32.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE: Cerebral amyloid angiopathy—amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 1998, 153:725-733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SH, Wang R, Gordon DJ, Bass J, Steiner DF, Lynn DG, Thinakaran G, Meredith SC, Sisodia SS: Furin mediates enhanced production of fibrillogenic ABri peptides in familial British dementia. Nat Neurosci 1999, 2:984-988 [DOI] [PubMed] [Google Scholar]
- 34.Day R, Schafer MK, Cullinan WE, Watson SJ, Chretien M, Seidah NG: Region specific expression of furin mRNA in the rat brain. Neurosci Lett 1993, 149:27-30 [DOI] [PubMed] [Google Scholar]
- 35.Probst A, Langui D, Ipsen S, Robakis N, Ulrich J: Deposition of beta/A4 protein along neuronal plasma membranes in diffuse senile plaques. Acta Neuropathol (Berl) 1991, 83:21–29 [DOI] [PubMed]
- 36.Mann DM, Jones D, South PW, Snowden JS, Neary D: Deposition of amyloid beta protein in non-Alzheimer dementias: evidence for a neuronal origin of parenchymal deposits of beta protein in neurodegenerative disease. Acta Neuropathol (Berl) 1992, 83:415–419 [DOI] [PubMed]
- 37.Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M: Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 1999, 96:14088-14093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B: Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 1992, 258:126-129 [DOI] [PubMed] [Google Scholar]
- 39.Ghiso J, Calero M, Matsubara E, Governale S, Chuba J, Beavis R, Wisniewski T, Frangione B: Alzheimer’s soluble amyloid beta is a normal component of human urine. FEBS Lett 1997, 408:105-108 [DOI] [PubMed] [Google Scholar]
- 40.Spillantini MG, Goedert M: Tau protein pathology in neurodegenerative diseases. Trends Neurosci 1998, 21:428-433 [DOI] [PubMed] [Google Scholar]
- 41.Crook R, Verkkoniemi A, PerezTur J, Mehta N, Baker M, Houlden H, Farrer M, Hutton M, Lincoln S, Hardy J, Gwinn K, Somer M, Paetau A, Kalimo H, Ylikoski R, Poyhonen M, Kucera S, Haltia M: A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med 1998, 4:452-455 [DOI] [PubMed] [Google Scholar]
- 42.Houlden H, Baker M, McGowan E, Lewis P, Hutton M, Crook R, Wood NW, Kumar-Singh S, Geddes JF, Swash M, Scaravilli F, Holton JL, Lashley T, Tomita T, Hashimoto T, Verkkoniemi A, Kalimo H, Somer M, Paetau A, Martin JJ, Van Broeckhoven C, Golde T, Hardy J, Haltia M, Revesz T: Variant Alzheimer’s disease with spastic paraparesis and ‘cotton wool’ plaques is caused by PS-1 mutations which lead to exceptionally high Aβ concentrations. Ann Neurol 2000, 68:806-808 [PubMed] [Google Scholar]
- 43.Gray F, Dubas F, Roullet E, Escourolle R: Leukoencephalopathy in diffuse hemorrhagic cerebral amyloid angiopathy. Ann Neurol 1985, 18:54-59 [DOI] [PubMed] [Google Scholar]
- 44.Vinters HV, Secor DL, Pardridge WM, Gray F: Immunohistochemical study of cerebral amyloid angiopathy. III. Widespread Alzheimer A4 peptide in cerebral microvessel walls colocalizes with gamma trace in patients with leukoencephalopathy. Ann Neurol 1990, 28:34-42 [DOI] [PubMed] [Google Scholar]