

Abstract
We performed high-resolution allelotyping for loss of heterozygosity (LOH) analysis on microdissected samples from 45 primary breast cancers, 47 mammary preneoplastic epithelial foci, and 18 breast cancer cell lines, using a panel of 27 polymorphic chromosome 3p markers. Allele loss in some regions of chromosome 3p was detected in 39 of 45 (87%) primary breast tumors. The 3p21.3 region had the highest frequency of LOH (69%), followed by 3p22-24 (61%), 3p21.2-21.3 (58%), 3p25 (48%), 3p14.2 (45%), 3p14.3 (41%), and 3p12 (35%). Analysis of all of the data revealed at least nine discrete intervals showing frequent allele loss: D3S1511-D3S1284 (U2020/DUTT1 region centered on D3S1274 with a homozygous deletion), D3S1300-D3S1234 [fragile histidine triad (FHIT)/FRA3B region centered on D3S1300 with a homozygous deletion], D3S1076-D3S1573, D3S4624/Luca2.1-D3S4597/P1.5, D3S1478-D3S1029, D3S1029 (with a homozygous deletion), D3S1612-D3S1537, D3S1293-D3S1597, and D3S1597-telomere; it is more than likely that additional localized regions of LOH not examined in this study also exist on chromosome 3p. In multiple cases, there was discontinuous allele loss at several 3p sites in the same tumor. Twenty-one of 47 (45%) preneoplastic lesions demonstrated 3p LOH, including 12 of 13 (92%) ductal carcinoma in situ, 2 of 7 (29%) apocrine metaplasia, and 7 of 25 (28%) usual epithelial hyperplasia. The 3p21.3 region had the highest frequency of LOH in preneoplastic breast epithelium (36%), followed by 3p21.2-21.3 (20%), 3p14.2/FHIT region (11%), 3p25 (10%), and 3p22-24 (5%). In 39 3p loci showing LOH in both the tumor and accompanying preneoplasia, 34 (87%) showed loss of the same parental allele (P = 1.2 × 10−6, cumulative binomial test). In addition, when 21 preneoplastic samples showing LOH were compared to their accompanying cancers, 67% were clonally related, 20% were potentially clonally related but were divergent, and 13% were clonally unrelated. Overall this demonstrated the high likelihood of clonal relatedness of the preneoplastic foci to the tumors. We conclude that: chromosome 3p allele loss is a common event in breast carcinoma pathogenesis; involves multiple, localized sites that often show discontinuous LOH with intervening markers retaining heterozygosity; and is seen in early preneoplastic stages, which demonstrate clonal relatedness to the invasive cancer.
An estimated 182,800 new cases of invasive breast cancer (IBC) and 40,000 breast cancer-related deaths are expected to occur among women in the United States during 2000. 1 Epidemiological studies have identified several putative precursor lesions of IBC such as usual ductal hyperplasia (UEH), atypical ductal hyperplasia (ADH), and ductal carcinoma in situ (DCIS). 2 Many of the molecular abnormalities found in IBC are also seen in these precursor lesions, 3-5 supporting the notion that breast cancer, like other epithelial tumors, develops by a stepwise accumulation of genetic hits. Although DCIS shares most of the genetic alterations seen in IBC, it lacks the ability to invade and metastasize and is thus considered within the spectrum of preneoplastic breast disease. 5 The biology of other histological entities found in breasts with IBC, such as apocrine metaplasia (AM), is less well defined and most experts would be reticent to refer to these lesions as “preneoplastic.” However, as we will see, our molecular analysis shows that even these lesions, hitherto considered benign, do reveal clonal 3p genetic abnormalities similar to those found in the adjacent IBC. Therefore all non-IBC foci analyzed in this study [UEH, AM, DCIS, and intraductal papilloma (IDP)] will be henceforth arbitrarily referred to as “preneoplastic” foci.
Numerous putative tumor suppressor genes (TSGs) have been implicated in the multistage pathogenesis of breast cancers. Approximately 10 to 15% of breast cancers have a strong familial tendency, appearing in a younger subset of women than the general population. 6,7 Linkage studies in these families have led to the isolation of two breast cancer susceptibility genes—BRCA1 (for BReast CAncer 1) on chromosome 17q21 8 and BRCA2 on chromosome 13q12-13. 9 More than 20 other chromosomal regions have been implicated in breast cancer pathogenesis by virtue of exhibiting LOH, including 17p13 (the p53 locus), 11q23 (the ataxia telangiectasia gene, ATM, locus), 1p36, 4q32-qter, 8p12-21, and 16q12-25, underscoring the genetic diversity of IBC. 10-12
Loss of heterozygosity (LOH) involving polymorphic markers on the short arm of chromosome 3 is a frequently observed genetic alteration in a variety of human epithelial tumors. 13-15 This phenomenon was first detected in lung cancers by cytogenetic analysis showing 3p deletions, 16-18 and later confirmed by allele loss and/or comparative genome hybridization studies in breast, lung, bladder, cervix, and colon cancers. 13,14,19-21 So far, at least three distinct 3p regions have been identified as showing frequent allele losses in breast cancer including 3p24-26, 3p21.3-22, and 3p14.2, 22-25 implicating the existence of several different TSGs located in the 3p region. Homozygous deletions of chromosome 3p also have been reported, further defining TSG locations in breast cancers in the 3p12 U2020/DUTT1, 26 3p14.2 fragile histidine triad (FHIT)/FRA3B, 19 and 3p21.3 regions. 27 In a few cases, 3p alterations also have been detected in benign breast epithelium, by LOH analysis 28,29 or cytogenetic studies, 30,31 suggesting that these changes may occur in the development of breast cancer.
To further understand and clarify the role of chromosome 3p allele loss in the pathogenesis of breast cancer, and to more precisely identify TSG candidate targets for positional cloning efforts, we performed a detailed allelotyping analysis of the entire chromosome 3p arm using 27 microsatellite markers. We studied 45 primary breast tumors, which included 18 tumors used to establish breast cancer cell lines as well as the breast cancer cell lines themselves. Additionally we studied 47 preneoplastic epithelial lesions accompanying these tumors. These encompassed a variety of histologies such as UEH, AM, IDP, and DCIS. We found that a high proportion of primary breast cancers and preneoplastic lesions show 3p allele loss, which often occurs in a discontinuous pattern of involvement, implicating multiple discrete 3p loci. We have also found that the preneoplastic foci could be clonally related to the IBCs from the same individual. These findings confirm and significantly extend previous analysis of chromosome region 3p in breast cancer pathogenesis.
Materials and Methods
Archival Tumor Specimens
Paraffin-embedded materials from 45 surgically resected primary breast carcinomas (1992 to 1998) were retrieved from the surgical pathology archives of University of Texas Southwestern Medical Center, Dallas, TX, and screened for breast cancer histology and preneoplastic lesions by five pathologists on the panel (AM, IIW, RA, SM, and AFG). The clinicopathological features, extracted from the patient charts, are summarized in Table 1 ▶ . Serial 5-μm sections were cut from archival, formalin-fixed, paraffin-embedded tissue. All slides were stained with hematoxylin and eosin, and one of the slides was coverslipped. The coverslipped slide was used as a guide to localize regions of interest for microdissection of the other slides. In addition to the tumors, 47 preneoplastic epithelial foci were microdissected from 18 of those cases in which adequate noncancerous breast parenchyma was available. Classified by histology, these included 13 DCIS, 2 IDP, 7 AM, and 25 UEH.
Table 1.
Clinicopathological Features of 45 Patients with Breast Carcinoma
| Age | |
| Mean | 53.3 years |
| Median | 51 years |
| Range | 24–85 years |
| Histology | |
| Infiltrating ductal carcinoma | 36/45 (80%) |
| Infiltrating lobular carcinoma | 4/45 (9%) |
| Sarcomatoid carcinoma | 5/45 (11%) |
| Grade | |
| (modified Bloom-Richardson’s criteria) | |
| Moderate (grade II) | 20/45 (44%) |
| Poor (grade III) | 25/45 (56%) |
| Tumor size | |
| Mean | 4.7 cm |
| Median | 3 cm |
| Range | 1–16 cm |
| pT1 (<2 cm) | 15/45 (33%) |
| pT2 (2–5 cm) | 17/45 (38%) |
| pT3 (>5 cm) | 13/45 (29%) |
| Axillary node status | |
| pN0 | 15/45 (34%) |
| pN1 or more | 30/45 (66%) |
| Estrogen/Progesterone receptor | |
| ER+/PR+ | 14/45 (31%) |
| ER+/PR− | 3/45 (7%) |
| ER−/PR+ | 6/45 (13%) |
| ER−/PR− | 22/45 (49%) |
| Aneuploid | 27/39 (70%) |
Cell Line Specimens
Eighteen of the 45 primary tumors were successfully cultured and the corresponding tumor cell lines were also studied as previously described. 32,33 The clinicopathological features of these 18 cell lines have been previously reviewed. 32,33 All of the breast cancer cell lines as well as the BL lines were initiated by the authors or co-workers at the Hamon Center for Therapeutic Oncology Research, Dallas, TX (HCCxxxx lines). Cells were grown in RPMI 1640 medium supplemented with 5% fetal bovine serum and DNA was prepared by standard methods. As previously reported, there is a high degree of concordance in genetic abnormalities between the breast cancer cell lines and the corresponding primary tumors. 33 Because of the high frequency concordance in genetic abnormalities between the 18 breast cancer cell lines and primary tumors, the data on breast tumors is used in the analyses unless otherwise stated.
Microdissection and DNA Extraction
Microdissection from archival paraffin-embedded tissues was performed by laser capture microdissection (PixCell II apparatus; Arcturus Engineering, Inc, Mountain View, CA) 34 from multiple microslides of each sample. The number of microdissected nuclei was approximated from the diameter of the laser beam (30 to 60 μm) used for microdissection. DNA extraction was performed as previously described (Figure 1) ▶ . 35 Dissected lymphocytes from histologically uninvolved axillary lymph nodes or stromal cells from the breast were used as a source of constitutional DNA from each case. After DNA extraction, 5 μl of the proteinase K-digested samples, containing DNA from at least 100 nuclei, were used for each multiplex polymerase chain reaction (PCR) reaction. To prevent artifactual LOH from less than optimal quantities of DNA, 36 equivalent numbers of nuclei from tumors and preneoplastic lesions was used in each PCR multiplex.
Figure 1.
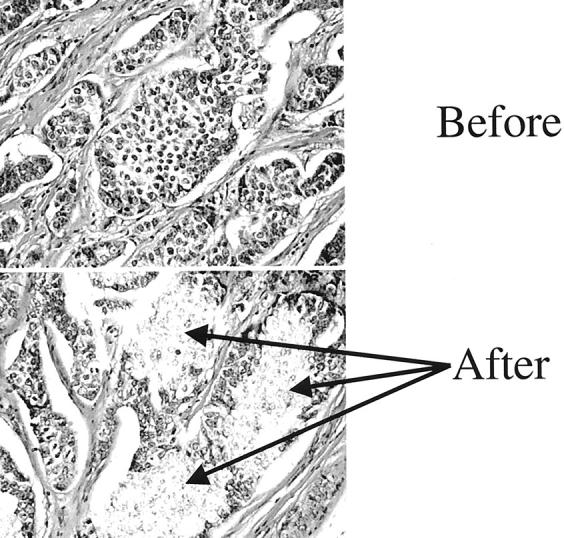
Laser capture microdissection of breast cancer. Before: Breast cancer before microdissection. After: Serial tissue section after laser capture, with selective isolation of neoplastic cells.
Polymorphic DNA Markers and PCR-LOH Analysis
To evaluate LOH, we used primers flanking dinucleotide and multinucleotide microsatellite repeat polymorphisms spanning the entire chromosome 3p arm (Figure 2) ▶ . Twenty-one of 27 polymorphic markers used were distributed as follows: 3p12 (D3S1511, D3S1274, D3S1284), 3p14.2 (D3S1300, D3S4103, D3S1234), 3p14.3 (D3S1766), 3p21.2-21.3 (ITIH-1, D3S1076, D3S1582, D3S1573), 3p21.3 (D3S1447, D3S1478, D3S1029), 3p22-24 (D3S1612, D3S1537, D3S2432, D3S1293), and 3p25 (D3S1111, D3S1597, D3S1351). Primer sequences were obtained from the Genome Database for all these markers. We recently reported 22 new polymorphic loci in the 3p21.3 region, using the genomic DNA sequence of a 630-kb (cosmid and P1 phage) clone contig 37 covering the 120-kb deletion overlap region at 3p21.3 27 and a novel computational system for the prediction of human polymorphisms. 38 A subset of six polymorphic markers from within this 600-kb region at 3p21.3 (D3S4597/P1.5, D3S4604/Luca19.1, D3S4614/Luca8.2, D3S4622/Luca4.1, D3S4623/Luca2.2, and D3S4624/Luca2.1) suitable for paraffin-embedded tissues were used in our study. 13
Figure 2.

Diagram of the short arm of chromosome 3 (3p) showing the 27 microsatellite markers used in the allelotyping analysis. Their order and approximate locations are derived from the Genome Database. Right: Regions characterized by homozygous deletions, including one at 3p12 (U2020 deletion) and the second at 3p21.3, in the 120-kb minimal deletion region that overlaps in four cell lines (HCC1500, GLC20, NCI-H740, and NCI-H1450). Left: A few of the recognized genes on chromosome 3p that have been implicated in tumor suppression.
For cell lines, PCR-LOH analysis was performed directly on genomic DNA as described, 39 whereas for microdissected samples a two-round PCR strategy was used as previously described. 35 LOH was scored by visual detection of complete absence of one allele in autoradiographs. Microsatellite alterations were identified by shift in the mobility of one tumor allele compared to the constitutional alleles, irrespective of the presence of LOH. All of the data were put into an Excel spreadsheet and analyzed with our own software constructed in Microsoft Visual Basic (available on request) designed to facilitate color formatting and visualization of the data as shown. The fractional allele loss (FAL) index, which is the measure of LOH for all chromosomal loci, was calculated for each individual sample as follows:
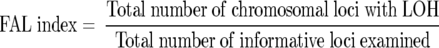 |
Ploidy and Immunohistochemical Analyses
Ploidy and immunohistochemical analyses were performed on archival tissue specimens, as previously described. 33,40 Immunohistochemical analyses was performed using primary mouse monoclonal antibodies for estrogen receptor (dilution 1:160; DAKO Corp., Carpinteria, CA), progesterone receptor (dilution 1:200; Zymed Laboratories Inc., South San Francisco, CA), Her-2/neu (AO485, dilution 1:14,400; DAKO), and Fhit (clone ZR44, dilution 1:200; Zymed). Immunostaining was performed using the Biotek automated immunostainer (Ventana, Tucson, AZ) using a standard avidin-biotin immunoperoxidase method. We have previously reported the scoring criteria for estrogen receptor, progesterone receptor, and Her-2/neu. 33,40 For Fhit, a three-tier-scoring scheme was used, as previously described. 41 Briefly, immunoreactivity was scored as 0 to 4+ for percentage of cells showing positive expression, and as 0 to 3+ for intensity of expression. A combined score was generated with 0 to 4 being designated “low,” 6 to 8 as “intermediate,” and 9 to 12 as “high” levels of expression.
Statistical Analyses
The cumulative binomial test 42 was used to examine the likelihood that the occurrence of a particular event (eg, loss of the same parental allele in the IBC and an associated preneoplastic epithelial sample) would occur at a particular probability when observed in repeated trials. Fisher’s exact test was used to examine the probability of two events (eg, allele loss at 3p14.2 and loss of Fhit expression) occurring by chance alone. To increase the stringency of comparison for chance occurrence versus nonoccurrence, a probability value of P < 0.01 was regarded as statistically significant.
Results
The Majority of Primary Breast Cancers Demonstrate 3p Allele Loss
Thirty-nine of 45 (87%) primary breast cancers had LOH at one or more 3p loci (Figures 3 and 4) ▶ ▶ . The mean FAL index for the tumors was 0.4 (range, 0-1.0). The 3p21.3 region had the highest frequency of LOH (69%), followed by 3p22-24 (61%), 3p21.2-21.3 (58%), 3p25 (48%), 3p14.2 (45%), 3p14.3 (41%), and 3p12 (35%) (Table 2) ▶ . Fifty percent of the tumors showed LOH of at least one marker in the 3p21.3 630-kb region that encompasses the breast and small-cell lung cancer overlapping homozygous deletions. 27,40,43,44 Of note, most primary tumors had discontinuous LOH with regions of 3p allele loss (blue boxes) separated by intervening regions of retention of heterozygosity (green boxes) (Figure 3) ▶ . For purposes of discussion, we refer to the junctions between the blue and green boxes as “breakpoints.” These junctions could occur by either physical deletion and/or by mitotic recombination. We emphasize, our use of the term “breakpoint” does not imply a mechanism but only refers to the detection of juxtaposed regions of allele retention and allele loss (see Discussion).
Figure 3.

Chromosome 3p allelotyping analysis of 45 primary breast cancers using 27 microsatellite markers. Blue box, LOH; green box, heterozygous; gray box, marker tested but not informative (homozygous in the normal DNA); and hatched box, microsatellite alteration with or without LOH. The yellow box marks the D3S1511-D3S1284 interval overlapping with the U2020/DUTT1 homozygous deletion in 3p12, the red box marks the D3S1300-D3S1234 interval corresponding to the FHIT gene locus on 3p14.2, the pink box marks the D3S4624-D3S4597 interval that overlaps with the 630-kb homozygous deletion in the 3p21.3 region, and the orange box marks the D3S1293-D3S2432 interval at the RARβ locus. The data were analyzed with Visual Basic software in Excel spreadsheets and sorted horizontally as to descending order of the number of allele losses present in each primary tumor.
Figure 4.

Representative autoradiographs using polymorphic markers to demonstrate examples of chromosome 3p LOH in paired normal and tumor samples. In each example, the left lane is constitutional DNA (lymphocytes and/or stroma), whereas the right lane is microdissected tumor DNA. Arrowheads indicate the position of parental allele lost. The individual polymorphic marker is indicated above each autoradiograph and its location on 3p in the accompanying chromosome map.
Table 2.
Chromosome 3p Loss of Heterozygosity in Primary Breast Tumors and Preneoplastic Lesions
| Region | Primary tumor | Preneoplasia* |
|---|---|---|
| 3p25 | 20 /42 (48%) | 3 /31 (10%) |
| 3p22-24 | 27 /44 (61%) | 2 /38 (5%) |
| 3p21.3 | 31 /45 (69%) | 17 /47 (36%) |
| 3p21.2-21.3 | 25 /43 (58%) | 9 /46 (20%) |
| 3p14.3 | 13 /32 (41%) | NE† |
| 3p14.2 | 20 /44 (45%) | 5 /45 (11%) |
| 3p12 | 13 /37 (35%) | NE† |
| Any 3p | 39 /45 (87%) | 21 /47 (45%) |
* Thirteen polymorphic 3p markers were examined in preneoplasia and included: 3p14.2 (D3S1300, D3S4103), 3p21.2-21.3 (D3S1076, D3S1582, ITIH-1), 3p21.3 (D3S4624/Luca2.1, D3S4622/Luca 4.1, D3S4597/P1.5, D3S1447, D3S1478, D3S1029), 3p22-24 (D3S1612), and 3p25 (D3S1597).
†NE = not examined.
Analyses of the data from 45 primary tumors using 27 polymorphic markers revealed at least 9 discrete intervals of localized allele loss: D3S1511-D3S1284 (U2020/DUTT1 region centered on D3S1274), D3S1300-D3S1234 (FHIT/FRA3B region centered on D3S1300), D3S1076-D3S1573, D3S4624/Luca2.1-D3S4597/P1.5, D3S1478-D3S1029, D3S1029, D3S1612-D3S1537, D3S1293-D3S1597, and D3S1597-telomere (Figure 4 ▶ and Table 3 ▶ ). In view of the discontinuous pattern of LOH on chromosome 3p, it is more than likely that additional sites of localized losses also exist for polymorphic marker intervals not examined in this study.
Table 3.
Informativeness, Loss of Heterozygosity and Microsatellite Alterations at the 27 Chromosome 3p Polymorphic Markers in 45 Primary Breast Cancers

* Informativeness and loss of heterozygosity (LOH) are in percentages (%), microsatellite alterations (MA) are in numbers.
Double-headed arrows on the right delineate nine 3p intervals with frequent localized areas of allele loss (breakpoints).
Thirteen of 18 (72%) breast cell lines demonstrated one or more locus of LOH at chromosome 3p, with a mean FAL index of 0.5 (0 to 1.0). Of 282 informative loci, 259 (92%) showed either retention of heterozygosity or LOH in both cell lines and primary tumors. In all cases in which both the cell line and corresponding tumor showed LOH, the loss was in the identical parental allele. In 20 of 23 discordant events, a genetic alteration was found in the cell line but not in the primary tumor. These could represent a new genetic alteration occurring during tissue culture, or outgrowth of a clone of cells representing a subset of the primary tumor. The tumor cell lines also allowed detection of three homozygous deletions centered on D3S1274 (U2020/DUTT1 region), 31 D3S1300 (FRA3B/FHIT region) 19 and D3S1029 (Figure 5) ▶ . For breast cancer cell line HCC1500, an additional homozygous deletion of the 3p21.3 RASSFIA region with sequencing of the breakpoint junction has been reported. 27
Figure 5.

Chromosome 3p allelotyping analysis of 18 breast cancer cell lines using 27 microsatellite markers. Blue box, LOH; green box with “H”, homozygous deletion; green box, heterozygous; gray box, marker tested but not informative (homozygous in the normal DNA); and hatched box, microsatellite alteration with or without LOH. The yellow box marks the D3S1511-D3S1284 interval overlapping with the U2020/DUTT1 homozygous deletion in 3p12, the red box marks the D3S1300-D3S1234 interval corresponding to the FHIT gene locus on 3p14.2, the pink box marks the D3S4624-D3S4597 interval that overlaps with the 630-kb homozygous deletion in the 3p21.3 region, and the orange box marks the D3S1293-D3S2432 interval at the RARβ locus. The data were analyzed with Visual Basic software in Excel spreadsheets and sorted horizontally as to descending order of the number of allele losses present in each tumor line. The tumor cell lines are indicated above. Please note that for HCC1500, in addition to the homozygous deletion of D3S1029, we previously have reported on another interstitial homozygous deletion in the 630-kb region with sequencing of the breakpoint junction. 27
Relative Frequency of LOH at 3p in Preneoplastic Breast Lesions Parallels Invasive Tumors, Shows Allele-Specific Mutation, and Is Often Clonally Related to the Invasive Tumor
Forty-seven preneoplastic lesions in resection specimens of 18 breast cancers were analyzed for allele losses on chromosome 3p, with one to eight separate epithelial foci examined in any given case. Because the amount of tissue amenable to microdissection from preneoplastic foci is often small, we examined only 13 of the 27 polymorphic markers on chromosome 3p that we could readily amplify using multiplex technology (Figures 6 and 7) ▶ ▶ . These included the five chromosomal regions 3p14.2 (D3S1300, D3S4103), 3p21.2-21.3 (D3S1076, D3S1582, ITIH-1), 3p21.3 (D3S4624/Luca2.1, D3S4622/Luca 4.1, D3S4597/P1.5, D3S1447, D3S1478, D3S1029), 3p22-24 (D3S1612), and 3p25 (D3S1597). Twenty-one of 47 (45%) preneoplastic lesions had 3p LOH, with a mean FAL index of 0.21 (range, 0 to 1.0). LOH of at least one 3p locus was seen in 12 of 13 (92%) DCIS, 2 of 7 (29%) AM, and 7 of 25 (28%) UEH. Thus, LOH on chromosome 3p was seen in some of the earliest stages of preneoplastic breast epithelium, including AM and UEH. AM is usually not associated with an increased risk of subsequent breast cancer, unlike the other components of proliferative breast disease such as UEH or ADH. LOH in AM adjacent to cancers is a recently observed phenomenon, 45 and may reflect a better ability from microdissection to enrich the cells of interest for allelotyping.
Figure 6.

Chromosome 3p allelotyping analysis of 47 preneoplastic mammary epithelial foci and corresponding primary tumor (18 cases) using 13 microsatellite markers. Blue box, LOH; green box, heterozygous; gray box, marker tested but not informative (homozygous in the normal DNA); and hatched box, microsatellite alteration with or without LOH. The cases and their associated preneoplastic lesions are sorted in increasing order by the case number assigned to the corresponding tumors (as given in Figure 3 ▶ ). In each individual epithelial focus analyzed, the case number and corresponding histology are indicated above the columns. T, tumor.
Figure 7.

Representative autoradiographs using polymorphic markers to demonstrate examples of chromosome 3p LOH in preneoplastic mammary epithelium. In each example, the first lane to the left is constitutional DNA (lymphocytes and/or stroma), and the next lane to the right is microdissected tumor DNA. Subsequent lane(s) represent one or more examples of preneoplastic epithelium, which are indicated above the corresponding lane. Asterisks indicate the position of parental allele lost. The individual polymorphic marker is indicated below each autoradiograph.
The relative frequency of LOH in preneoplastic epithelium usually paralleled those seen in primary tumors at the various chromosome 3p regions tested. The 3p21.3 region had the highest frequency of LOH in preneoplastic breast epithelium (36%). Less frequent LOH were also detected in 3p21.2-21.3 (20%), 3p14.2/FHIT region (11%), 3p25 (10%), and 3p22-24 (5%) (Table 2) ▶ . Interestingly, two of two AM and four of seven UEH with 3p LOH had loss at the D3S1029 locus, a marker that is also altered in a high proportion of IBCs. Other 3p21.3 polymorphic loci tested (D3S1447, D3S1478, D3S4624Luca 2.1, D3S4622/Luca 4.1, and D3S4597/P1.5) also demonstrated LOH in one or more UEH foci. By contrast, some regions such as 3p14.2 (D3S1300 and D3S4103) and 3p25 (D3S1597) usually showed LOH at the stage of DCIS or invasive cancer. This pattern is identical to the sequence observed in the stages of histological progression in bronchial epithelium, 13 where 3p21.3 LOH is seen in normal or mildly abnormal epithelium, whereas 3p14.2 or 3p25 LOH is usually seen at the stage of dysplasia or CIS. Alternatively, the higher frequency of LOH at 3p21.3 could be a methodological bias introduced by testing for a higher number of markers (n = 6) than either 3p14.2 (n = 2) or 3p25 (n = 1). In either case, a conceivable argument can be made that LOH at 3p21.3 represents one of the earliest changes in preneoplastic epithelium.
Thirty-nine of 45 (87%) informative loci showed LOH in both the tumor and its corresponding preneoplastic lesion, whereas in 6 of 45 (13%) loci only the preneoplastic epithelium showed LOH. Of the 39 informative loci demonstrating LOH in both tumor and preneoplasia, 34 (87%) showed loss of the same parental allele, a phenomenon previously described by us as “allele-specific mutation.” 46 Using the cumulative binomial test, the probability of this occurring by chance alone is 1.2 × 10−6, making it highly unlikely that allele-specific mutation at chromosome 3p is a random event. Although the mechanism of allele-specific mutation remains unknown, this phenomenon has also been demonstrated with other tumors such as lung carcinomas and their associated preneoplastic foci. 13 Several possibilities have been suggested such as seeding of multiple sites in epithelium at risk with a common progenitor clone, 47 inherited (eg, polymorphic) differences in allele susceptibility to loss, 48 and potential epigenetic differences in alleles that arise during development (eg, methylation) that might predispose one to preferential loss.
We further examined the possibility of whether any of the preneoplastic lesions could be clonally related to the corresponding invasive tumor, which may explain the phenomenon of allele-specific mutation. We defined three categories of lesions with respect to the genetic alterations in the tumor: clonally related (preneoplasia showing identical genetic losses as the tumor, with or without additional LOH in the latter); clonally divergent (preneoplasia and tumor showing both identical and discordant genetic losses); and clonally unrelated (preneoplasia and tumor having completely discordant LOH). This definition was made with the understanding that the categories are not absolute, and reflects the results of molecular analysis involving 13 polymorphic loci on a single chromosome arm. In other words, a preneoplastic lesion that appears clonally unrelated in our analysis may actually represent an example of clonal divergence if other chromosomal loci were also examined. We found that of the 21 of 45 preneoplastic lesions demonstrating LOH, 14 (67%) were clonally related to the invasive tumor, 5 (20%) were clonally divergent, and only 2 were clonally unrelated. Thus, the majority of preneoplastic epithelial foci (19 of 21) could potentially have emerged from the same clone as the corresponding breast tumor, with or without clonal divergence during multistage progression.
LOH in Individual 3p Regions Correlates with other Clinicopathological Parameters
There were no statistically significant associations between 3p LOH and numerous clinicopathological variables, including histological grade of tumor, presence or absence of axillary lymph node metastasis, estrogen and progesterone receptor expression, and DNA ploidy. However when the comparisons were made for the individual 3p regions, there was a statistically significant correlation between loss of progesterone receptor expression and LOH at 3p25 (P = 0.006) and poor histological grade and LOH at 3p22-24 (P = 0.009). Loss of Fhit expression by immunohistochemistry was significantly correlated with LOH at the 3p14.2 FRA3B/FHIT locus (P = 0.004). Because of the small numbers of cases, most of which had limited follow-up, we did not correlate LOH at 3p with survival.
Discussion
LOH analysis provides information on genetic alterations in tumors and their preneoplastic lesions, as well as for the purposes of localizing sites of potential TSGs, that can be subsequently isolated by positional cloning efforts. We were able to refine the extent of genetic alterations at chromosome 3p in breast cancer pathogenesis by a combination of microdissection of tumors and preneoplastic lesions and use of multiple polymorphic markers for the LOH analysis. With the use of microdissected material, the scoring of LOH in our series was unambiguous. For example, Sato and colleagues 25 reported LOH at 3p in 47% of 219 tumors analyzed by restriction fragment length polymorphism, whereas Matsumoto and colleagues 22 reported a frequency of 52% 3p LOH in 196 tumors analyzed by conventional allelotyping; both studies had used nonenriched frozen tumor tissue, which may explain the lower frequency of 3p LOH than in the current study (87%), where microdissected material was used. Additionally, Matsumoto and colleagues 22 were able to define only two noncontiguous regions of frequent LOH in their analysis (3p24.3-25.1 and 3p14.2-21.2), as compared to the current study, where we found multiple regions of LOH that are frequently, and often independently, lost in breast carcinomas. Analysis of our 3p allelotyping data pooled together from all of the specimens showed the LOH patterns were complex with at least nine discrete intervals and many chromosome 3p breakpoints (discontinuous regions of LOH). These results are nearly identical to those found in lung cancer. 13 It is not certain however whether all or some of these localized regions harbor TSGs; an alternative explanation could be that these regions represent areas of increased fragility that undergo LOH under the influence of circumstances that promote chromosome breakage.
Because we have been interested in isolating potential TSGs in the critical 3p21.3 region, an attempt was made to determine whether high-resolution allelotyping could further narrow the areas of interest for positional cloning. We 49 and Dammann and colleagues 50 have recently characterized an isoform of a human RAS effector homologue (termed RASSF1A) located in the previously described 3p21.3 120-kb minimal homozygous deletion region in breast and lung cancers, that undergoes epigenetic inactivation by promoter methylation in 100% of small-cell lung cancers, more than a third of non-small-cell lung cancers and 49% of primary breast cancers. In addition, we showed loss of expression of RASSF1A in breast cancer cell lines and primary breast tumors, with retention in normal breast epithelium. 49 Because the 3p21.3 region shows LOH of the other parental allele in the majority of lung 13 carcinomas, RASSF1A fulfills Knudson’s two-hit hypothesis of a recessive TSG, even in the absence of demonstrable mutations. Twenty-two of 39 breast carcinomas (56%) with 3p LOH had breakpoints in the 3p21.3 region, and 14 of the 22 breakpoints (64%) were consistent with the presence of TSG(s) in the 120-kb minimal deletion region containing RASSF1A. The presence of frequent LOH and breakpoints in breast cancers within the 3p21.3 region is not surprising, considering that as many as 19 putative TSGs are currently being evaluated in this region for their potential role in carcinogenesis. 51
The polymorphic marker D3S1029, also located within the 3p21.3 region, showed allele losses in 50% of breast carcinomas, including several examples of localized allele losses, and was also homozygously deleted in the breast cancer cell line HCC1500. The HCC1500 homozygous deletion at D3S1029 is noncontiguous and discrete from the previously described 120-kb 3p21.3 deletion in this same tumor, 27 and indicates a new 3p21.3 area for a potential TSG that may be inactivated in breast tumorigenesis. Kholodnyuk and colleagues 52 have previously reported that the D3S1029 marker is located within a 7-cM common eliminated region within SCID mouse-derived tumors from human-mouse microcell hybrids. In a subsequent analysis, the authors localized the common eliminated region containing D3S1029 to a 1.6-cM interval that was telomeric to the 3p21.3 120-kb minimal deletion region in breast and small-cell lung cancers. 53 In summary, our allelotyping data emphasizes that there are several potential TSGs in the 3p21.3 region, some of which have been cloned and are under active investigation, 51 whereas others (particularly at the D3S1029 locus) merit further study. The completion of the human genome effort should aid in this search particularly in combination with new informatics approaches for gene discovery and analysis. 38,51
Besides the 3p21.3 region, we detected a high frequency of LOH in three regions previously known to harbor putative TSGs. The 3p12 region bordered by the markers D3S1511-D3S1284, and centered on the HCC38 D3S1274 homozygous deletion, overlaps with the previously reported 8-Mb U2020 small-cell lung cancer cell line deletion. 26 Recently, a new candidate TSG, DUTT1, has been cloned residing in the U2020/HCC38 3p12 deletion region at D3S1274. 26 However, its tumor suppressing activity and protein expression patterns in tumors are unknown. The 3p14.2 region, bordered by the markers D3S1300-D3S1234, is the locus of one of the most extensively studied TSGs on chromosome 3p—the fragile histidine triad (FHIT) gene located in the aphidicolin-sensitive FRA3B fragile site at 3p14.2. 15,54,55 Abnormalities of the FHIT gene have been detected in breast cancer cell lines, primary tumors, and preneoplastic lesions at both the genomic and expression levels. 19,22,56,57 Several studies have shown loss of expression of Fhit protein in 50 to 75% of breast cancers, 56-58 and loss of Fhit expression has been shown to correlate with established clinicopathological parameters such as increased proliferation and larger tumor size. 57 We detected LOH at 3p14.2 in 45% of IBCs, and found a statistically significant correlation between LOH at 3p14.2 and down-regulation of Fhit expression (P = 0.004). Finally, the 3p25 region, defined by the interval D3S1111-D3S1351, showed LOH in 48% of tumors, and has been previously implicated as a site of frequent deletions in IBCs. 22,23 LOH in this region has been reported to have a significantly adverse effect on postoperative survival. 59 It is not clear which gene is the target of inactivation in these deletions, because the best characterized TSG at this locus, the Von Hippel Lindau (VHL) gene, 60 is mutated virtually exclusively in renal cell carcinomas. It may be possible that the VHL gene or another putative TSG also undergoes inactivation by the increasingly apparent epigenetic pathway of promoter methylation rather than mutation, but this remains to be determined.
In addition to the four regions of frequent LOH on 3p discussed above, we also found two novel regions that have not been well described in the context of IBCs, but which were found to be deleted in a high proportion of our microdissected tumors. The first is the 3p22-24 region, defined by the markers D3S1612-D3S1293, which showed LOH in 61% of IBCs. One candidate TSG in this region—the retinoic acid receptor-β2 (RAR-β2) gene located at 3p24—undergoes inactivation by allele loss or promoter methylation in virtually all small-cell lung cancers, the majority of non-small-cell lung cancers, 61 and up to 40% of breast cancers. 62 The second region lies within the centromeric 3p21.2-21.3 region, defined by the interval D3S1076-D3S1573, which showed LOH in 58% of IBCs. A significant proportion of both small-cell and non-small-cell lung carcinomas, as well as their preneoplastic lesions demonstrate LOH at 3p21.2-21.3. 13 The probe D3S2 that is reported to be homozygously deleted in a primary breast cancer also localizes to this region, 63 and human-mouse microcell hybrids have demonstrated this region to be a common eliminated region in SCID-derived tumors. 52 Currently, no definitive TSG(s) has been identified in this region.
Although it is tempting to speculate that these discrete areas may harbor one or more TSG(s) critical in carcinogenesis, an alternative argument that needs to be addressed is whether these discontinuous breaks merely represent mutational predisposition. 64 It has been shown in cohorts of hereditary breast cancer patients with germline BRCA2 mutations that LOH at 3p14.2 (ie, the FHIT/FRA3B locus) are more frequent than in sporadic tumors. 58 This has been used to foster the argument that the increased frequency of LOH in these patients arise not from selection but from a generalized defect of DNA damage repair caused by BRCA2 mutations. Similarly, recent reports of whole genome allelotyping in breast cancers has shown that tumors with defective double-strand break repair have a higher frequency of double-strand break-induced chromosomal instability. 11 The double-strand break-induced chromosome breakage is considered central to the pathway for genomic deletion or mitotic recombination leading to LOH. 65,66 In the context of lung carcinomas, it has been convincingly demonstrated that chromosome 3p undergoes frequent discontinuous LOH, 13 and the stimulus for double-strand break is presumed to be oxidative damage from environmental causes, most importantly cigarette smoking. 67 For breast epithelium, it is possible that the reproductive hormone estrogen and/or its metabolites function as carcinogenic stimuli, 68,69 by induction of oxidative damage and depurination, potentially resulting in clustered abasic sites on both DNA strands, and subsequent formation of double-strand breaks. 70 Additionally, the recent demonstration of tumor-specific promoter methylation in several putative TSGs located within these frequent breakpoint regions, 50,61 and the absence of promoter methylation in corresponding normal tissue also suggests a causal role in tumor promotion or tumor initiation for chromosome 3p LOH. Thus, although mutational predisposition from defective double-strand break-repair combined with exposure to oxidative damage may turn out to be the common underlying pathway by which discontinuous fragile regions in the human genome (such as on chromosome 3p) undergo damage, it does not by itself detract from the downstream consequences of losing critical portions of DNA that harbor growth-suppressing elements.
In conclusion, on the basis of high-resolution 3p allelotyping in microdissected breast cancers and their preneoplastic foci, we were able to demonstrate that there are at least nine discrete intervals of discontinuous LOH located along the entire short arm of chromosome 3p. LOH on chromosome 3p is not only frequent in IBCs, but also in the earliest preneoplastic epithelial lesions such as UEH and AM, besides DCIS. In the majority of instances, there is a demonstrable clonal relatedness between the tumor and the preneoplastic epithelium, suggesting that chromosome 3p allele loss is a nonrandom event that probably plays an important role in the multistep pathogenesis of breast cancers. It is anticipated that the current study will facilitate positional cloning of additional TSGs important in carcinogenesis, whereas ongoing functional studies will better elucidate the mechanism of inactivation of the various candidate TSGs on chromosome 3p.
Acknowledgments
We thank Luc Girard and Gina Mele for their contribution toward the completion of this work.
Footnotes
Address reprint requests to John D. Minna, M.D., Hamon Center for Therapeutic Oncology Research, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, Texas 75390. E-mail: john.minna@utsouthwestern.edu.
Supported by the National Institutes of Health (CA71618), the Early Detection Research Network, the G. Harold and Leila Y. Mathers Charitable Foundation, the Cancer Research Foundation of North Texas, and the Susan G. Komen Foundation. C. W. was supported by a fellowship from the Damon Runyon-Walter Winchell Cancer Research Fund.
References
- 1.American Cancer Society’s Breast Cancer Resource (http: //www.cancer.org)
- 2.Dupont WD, Page DL: Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med 1985, 312:146-151 [DOI] [PubMed] [Google Scholar]
- 3.O’Connell P, Pekkel V, Fuqua S, Osborne CK, Allred DC: Molecular genetic studies of early breast cancer evolution. Breast Cancer Res Treat 1994, 32:5-12 [DOI] [PubMed] [Google Scholar]
- 4.Radford DM, Fair KL, Phillips NJ, Ritter JH, Steinbrueck T, Holt MS, Donis-Keller H: Allelotyping of ductal carcinoma in situ of the breast: deletion of loci on 8p, 13q, 16q, 17p and 17q. Cancer Res 1995, 55:3399-3405 [PubMed] [Google Scholar]
- 5.O’Connell P, Pekkel V, Fuqua SA, Osborne CK, Clark GM, Allred DC: Analysis of loss of heterozygosity in 399 premalignant breast lesions at 15 genetic loci. J Natl Cancer Inst 1998, 90:697-703 [DOI] [PubMed] [Google Scholar]
- 6.Slattery ML, Kerber RA: A comprehensive evaluation of family history and breast cancer risk. The Utah Population Database. JAMA 1993, 270:1563-1568 [PubMed] [Google Scholar]
- 7.Easton DF, Ford D, Bishop DT: Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am J Hum Genet 1995, 56:265-271 [PMC free article] [PubMed] [Google Scholar]
- 8.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al: A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266:66-71 [DOI] [PubMed] [Google Scholar]
- 9.Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D, et al: Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12–13. Science 1994, 265:2088-2090 [DOI] [PubMed] [Google Scholar]
- 10.Osborne RJ, Hamshere MG: A genome-wide map showing common regions of loss of heterozygosity/allelic imbalance in breast cancer. Cancer Res 2000, 60:3706-3712 [PubMed] [Google Scholar]
- 11.Shen CY, Yu JC, Lo YL, Kuo CH, Yue CT, Jou YS, Huang CS, Lung JC, Wu CW: Genome-wide search for loss of heterozygosity using laser capture microdissected tissue of breast carcinoma: an implication for mutator phenotype and breast cancer pathogenesis. Cancer Res 2000, 60:3884-3892 [PubMed] [Google Scholar]
- 12.Forozan F, Mahlamaki EH, Monni O, Chen Y, Veldman R, Jiang Y, Gooden GC, Ethier SP, Kallioniemi A, Kallioniemi OP: Comparative genomic hybridization analysis of 38 breast cancer cell lines: a basis for interpreting complementary DNA microarray data. Cancer Res 2000, 60:4519-4525 [PubMed] [Google Scholar]
- 13.Wistuba II, Behrens C, Virmani AK, Mele G, Milchgrub S, Girard L, Fondon JW, III, Garner HR, McKay B, Latif F, Lerman MI, Lam S, Gazdar AF, Minna JD: High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res 2000, 60:1949-1960 [PubMed] [Google Scholar]
- 14.Wistuba II, Montellano FD, Milchgrub S, Virmani AK, Behrens C, Chen H, Ahmadian M, Nowak JA, Muller C, Minna JD, Gazdar AF: Deletions of chromosome 3p are frequent and early events in the pathogenesis of uterine cervical carcinoma. Cancer Res 1997, 57:3154-3158 [PubMed] [Google Scholar]
- 15.Boldog F, Gemmill RM, West J, Robinson M, Robinson L, Li E, Roche J, Todd S, Waggoner B, Lundstrom R, Jacobson J, Mullokandov MR, Klinger H, Drabkin HA: Chromosome 3p14 homozygous deletions and sequence analysis of FRA3B. Hum Mol Genet 1997, 6:193-203 [DOI] [PubMed] [Google Scholar]
- 16.Whang-Peng J, Bunn PA, Jr, Kao-Shan CS, Lee EC, Carney DN, Gazdar A, Minna JD: A nonrandom chromosomal abnormality, del 3p(14-23), in human small cell lung cancer (SCLC). Cancer Genet Cytogenet 1982, 6:119-134 [DOI] [PubMed] [Google Scholar]
- 17.Whang-Peng J, Kao-Shan CS, Lee EC, Bunn PA, Carney DN, Gazdar AF, Minna JD: Specific chromosome defect associated with human small-cell lung cancer; deletion 3p(14–23). Science 1982, 215:181-182 [DOI] [PubMed] [Google Scholar]
- 18.Pandis N, Bardi G, Mitelman F, Heim S: Deletion of the short arm of chromosome 3 in breast tumors. Genes Chromosom Cancer 1997, 18:241-245 [PubMed] [Google Scholar]
- 19.Ahmadian M, Wistuba II, Fong KM, Behrens C, Kodagoda DR, Saboorian MH, Shay J, Tomlinson GE, Blum J, Minna JD, Gazdar AF: Analysis of the FHIT gene and FRA3B region in sporadic breast cancer, preneoplastic lesions, and familial breast cancer probands. Cancer Res 1997, 57:3664-3668 [PubMed] [Google Scholar]
- 20.Li M, Zhang ZF, Reuter VE, Cordon-Cardo C: Chromosome 3 allelic losses and microsatellite alterations in transitional cell carcinoma of the urinary bladder. Am J Pathol 1996, 149:229-235 [PMC free article] [PubMed] [Google Scholar]
- 21.Thiagalingam S, Lisitsyn NA, Hamaguchi M, Wigler MH, Willson JK, Markowitz SD, Leach FS, Kinzler KW, Vogelstein B: Evaluation of the FHIT gene in colorectal cancers. Cancer Res 1996, 56:2936-2939 [PubMed] [Google Scholar]
- 22.Matsumoto S, Kasumi F, Sakamoto G, Onda M, Nakamura Y, Emi M: Detailed deletion mapping of chromosome arm 3p in breast cancers: a 2-cM region on 3p14.3–21.1 and a 5-cM region on 3p24.3–25.1 commonly deleted in tumors. Genes Chromosom Cancer 1997, 20:268-274 [PubMed] [Google Scholar]
- 23.Chen LC, Matsumura K, Deng G, Kurisu W, Ljung BM, Lerman MI, Waldman FM, Smith HS: Deletion of two separate regions on chromosome 3p in breast cancers. Cancer Res 1994, 54:3021-3024 [PubMed] [Google Scholar]
- 24.Driouch K, Briffod M, Bieche I, Champeme MH, Lidereau R: Location of several putative genes possibly involved in human breast cancer progression. Cancer Res 1998, 58:2081-2086 [PubMed] [Google Scholar]
- 25.Sato T, Akiyama F, Sakamoto G, Kasumi F, Nakamura Y: Accumulation of genetic alterations and progression of primary breast cancer. Cancer Res 1991, 51:5794-5799 [PubMed] [Google Scholar]
- 26.Sundaresan V, Chung G, Heppell-Parton A, Xiong J, Grundy C, Roberts I, James L, Cahn A, Bench A, Douglas J, Minna J, Sekido Y, Lerman M, Latif F, Bergh J, Li H, Lowe N, Ogilvie D, Rabbitts P: Homozygous deletions at 3p12 in breast and lung cancer. Oncogene 1998, 17:1723-1729 [DOI] [PubMed] [Google Scholar]
- 27.Sekido Y, Ahmadian M, Wistuba II, Latif F, Bader S, Wei MH, Duh FM, Gazdar AF, Lerman MI, Minna JD: Cloning of a breast cancer homozygous deletion junction narrows the region of search for a 3p21.3 tumor suppressor gene. Oncogene 1998, 16:3151-3157 [DOI] [PubMed] [Google Scholar]
- 28.Euhus DM, Maitra A, Wistuba II, Alberts A, Albores-Saavedra J, Gazdar AF: Loss of heterozygosity at 3p in benign lesions preceding invasive breast cancer. J Surg Res 1999, 83:13-18 [DOI] [PubMed] [Google Scholar]
- 29.Deng G, Lu Y, Zlotnikov G, Thor AD, Smith HS: Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science 1996, 274:2057-2059 [DOI] [PubMed] [Google Scholar]
- 30.Dietrich CU, Pandis N, Teixeira MR, Bardi G, Gerdes AM, Andersen JA, Heim S: Chromosome abnormalities in benign hyperproliferative disorders of epithelial and stromal breast tissue. Int J Cancer 1995, 60:49-53 [DOI] [PubMed] [Google Scholar]
- 31.Petersson C, Pandis N, Rizou H, Mertens F, Dietrich CU, Adeyinka A, Idvall I, Bondeson L, Georgiou G, Ingvar C, Heim S, Mitelman F: Karyotypic abnormalities in fibroadenomas of the breast. Int J Cancer 1997, 70:282-286 [DOI] [PubMed] [Google Scholar]
- 32.Wistuba II, Behrens C, Milchgrub S, Syed S, Ahmadian M, Virmani AK, Kurvari V, Cunningham TH, Ashfaq R, Minna JD, Gazdar AF: Comparison of features of human breast cancer cell lines and their corresponding tumors. Clin Can Res 1998, 4:2931-2938 [PubMed] [Google Scholar]
- 33.Gazdar AF, Kurvari V, Virmani A, Gollahon L, Sakaguchi M, Westerfield M, Kodagoda D, Stasny V, Cunningham HT, Wistuba II, Tomlinson G, Tonk V, Ashfaq R, Leitch AM, Minna JD, Shay JW: Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int J Cancer 1998, 78:766-774 [DOI] [PubMed] [Google Scholar]
- 34.Bonner RF, Emmert-Buck M, Cole K, Pohida T, Chuaqui R, Goldstein S, Liotta LA: Laser capture microdissection: molecular analysis of tissue. Science 1997, 278:1481-1483 [DOI] [PubMed] [Google Scholar]
- 35.Wistuba II, Behrens C, Milchgrub S, Virmani AK, Jagirdar J, Thomas B, Ioachim HL, Litzky LA, Brambilla EM, Minna JD, Gazdar AF: Comparison of molecular changes in lung cancers in HIV-positive and HIV-indeterminate subjects. JAMA 1998, 279:1554-1559 [DOI] [PubMed] [Google Scholar]
- 36.Euhus DM, Maitra A, Wistuba II, Ashfaq R, Alberts A, Gibbons D, Gazdar AF: Use of archival fine-needle aspirates for the allelotyping of tumors. Cancer 1999, 87:372-379 [DOI] [PubMed] [Google Scholar]
- 37.Wei MH, Latif F, Bader S, Kashuba V, Chen JY, Duh FM, Sekido Y, Lee CC, Geil L, Kuzmin I, Zabarovsky E, Klein G, Zbar B, Minna JD, Lerman MI: Construction of a 600-kilobase cosmid clone contig and generation of a transcriptional map surrounding the lung cancer tumor suppressor gene (TSG) locus on human chromosome 3p21.3: progress toward the isolation of a lung cancer TSG. Cancer Res 1996, 56:1487-1492 [PubMed] [Google Scholar]
- 38.Fondon JW, III, Mele GM, Brezinschek RI, Cummings D, Pande A, Wren J, O’Brien KM, Kupfer KC, Wei MH, Lerman M, Minna JD, Garner HR: Computerized polymorphic marker identification: experimental validation and a predicted human polymorphism catalog. Proc Natl Acad Sci USA 1998, 95:7514-7519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Virmani AK, Fong KM, Kodagoda D, McIntire D, Hung J, Tonk V, Minna JD, Gazdar AF: Allelotyping demonstrates common and distinct patterns of chromosomal loss in human lung cancer types. Genes Chromosom Cancer 1998, 21:308-319 [DOI] [PubMed] [Google Scholar]
- 40.Daly MC, Xiang RH, Buchhagen D, Hensel CH, Garcia DK, Killary AM, Minna JD, Naylor SL: A homozygous deletion on chromosome 3 in a small cell lung cancer cell line correlates with a region of tumor suppressor activity. Oncogene 1993, 8:1721-1729 [PubMed] [Google Scholar]
- 41.Hao XP, Willis JE, Pretlow TG, Rao JS, MacLennan GT, Talbot IC, Pretlow TP: Loss of fragile histidine triad expression in colorectal carcinomas and premalignant lesions. Cancer Res 2000, 60:18-21 [PubMed] [Google Scholar]
- 42.Siegel S: Nonparametric Statistics for the Behavioral Sciences. 1956:pp 36-42 McGraw-Hill International Book Co., New York
- 43.Roche J, Boldog F, Robinson M, Robinson L, Varella-Garcia M, Swanton M, Waggoner B, Fishel R, Franklin W, Gemmill R, Drabkin H: Distinct 3p21.3 deletions in lung cancer and identification of a new human semaphorin. Oncogene 1996, 12:1289-1297 [PubMed] [Google Scholar]
- 44.Kok K, van den Berg A, Veldhuis PM, van der Veen AY, Franke M, Schoenmakers EF, Hulsbeek MM, van der Hout AH, de Leij L, van de Ven W, Buys C: A homozygous deletion in a small cell lung cancer cell line involving a 3p21 region with a marked instability in yeast artificial chromosomes. Cancer Res 1994, 54:4183-4187 [PubMed] [Google Scholar]
- 45.Washington C, Dalbegue F, Abreo F, Taubenberger JK, Lichy JH: Loss of heterozygosity in fibrocystic change of the breast: genetic relationship between benign proliferative lesions and associated carcinomas. Am J Pathol 2000, 157:323-329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hung J, Kishimoto Y, Sugio K, Virmani A, McIntire DD, Minna JD, Gazdar AF: Allele-specific chromosome 3p deletions occur at an early stage in the pathogenesis of lung carcinoma. JAMA 1995, 273:558-563 [PubMed] [Google Scholar]
- 47.Franklin WA, Gazdar AF, Haney J, Wistuba II, La Rosa FG, Kennedy T, Ritchey DM, Miller YE: Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J Clin Invest 1997, 100:2133-2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wistuba II, Tomlinson GE, Behrens C, Virmani A, Geradts J, Blum JL, Minna JD, Gazdar AF: Two identical triplet sisters carrying a germline BRCA1 gene mutation acquire very similar breast cancer somatic mutations at multiple other sites throughout the genome. Genes Chromosom Cancer 2000, 28:359-369 [DOI] [PubMed] [Google Scholar]
- 49.Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI, Zabarousky E, White M, Minna JD: Epigenetic inactivation of RASSFIA in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst 2001, 93:691-699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP: Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 2000, 25:315-319 [DOI] [PubMed] [Google Scholar]
- 51.Lerman MI, Minna, JD for the International Lung Cancer Chromosome 3p21:3 Tumor Suppressor Gene Consortium: The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. Cancer Res 2000, 60:6116–6133 [PubMed]
- 52.Kholodnyuk I, Kost-Alimova M, Kashuba V, Gizatulin R, Szeles A, Stanbridge EJ, Zabarovsky ER, Klein G, Imreh S: A 3p21.3 region is preferentially eliminated from human chromosome 3/mouse microcell hybrids during tumor growth in SCID mice. Genes Chromosom Cancer 1997, 18:200-211 [PubMed] [Google Scholar]
- 53.Szeles A, Yang Y, Sandlund AM, Kholodnyuk I, Kiss H, Kost-Alimova M, Zabarovsky ER, Stanbridge E, Klein G, Imreh S: Human/mouse microcell hybrid based elimination test reduces the putative tumor suppressor region at 3p21.3 to 1.6 cM. Genes Chromosom Cancer 1997, 20:329-336 [PubMed] [Google Scholar]
- 54.Ong ST, Fong KM, Bader SA, Minna JD, Le Beau MM, McKeithan TW, Rassool FV: Precise localization of the FHIT gene to the common fragile site at 3p14.2 (FRA3B) and characterization of homozygous deletions within FRA3B that affect FHIT transcription in tumor cell lines. Genes Chromosom Cancer 1997, 20:16-23 [DOI] [PubMed] [Google Scholar]
- 55.Sard L, Accornero P, Tornielli S, Delia D, Bunone G, Campiglio M, Colombo MP, Gramegna M, Croce CM, Pierotti MA, Sozzi G: The tumor-suppressor gene FHIT is involved in the regulation of apoptosis and in cell cycle control. Proc Natl Acad Sci USA 1999, 96:8489-8492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gatalica Z, Lele SM, Rampy BA, Norris BA: The expression of Fhit protein is related inversely to disease progression in patients with breast carcinoma. Cancer 2000, 88:1378-1383 [PubMed] [Google Scholar]
- 57.Campiglio M, Pekarsky Y, Menard S, Tagliabue E, Pilotti S, Croce CM: FHIT loss of function in human primary breast cancer correlates with advanced stage of the disease. Cancer Res 1999, 59:3866-3869 [PubMed] [Google Scholar]
- 58.Ingvarsson S, Agnarsson BA, Sigbjornsdottir BI, Kononen J, Kallioniemi OP, Barkardottir RB, Kovatich AJ, Schwarting R, Hauck WW, Huebner K, McCue PA: Reduced Fhit expression in sporadic and BRCA2-linked breast carcinomas. Cancer Res 1999, 59:2682-2689 [PubMed] [Google Scholar]
- 59.Matsumoto S, Minobe K, Utada Y, Furukawa K, Onda M, Sakamoto G, Kasumi F, Nakamura Y, Emi M: Loss of heterozygosity at 3p24–p25 as a prognostic factor in breast cancer. Cancer Lett 2000, 152:63-69 [DOI] [PubMed] [Google Scholar]
- 60.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, et al: Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260:1317-1320 [DOI] [PubMed] [Google Scholar]
- 61.Virmani AK, Rathi A, Zochbauer-Muller S, Sacchi N, Fukuyama Y, Bryant D, Maitra A, Heda S, Fong KM, Thunnissen F, Minna JD, Gazdar AF: Promoter methylation and silencing of the retinoic acid receptor-beta gene in lung carcinomas. J Natl Cancer Inst 2000, 92:1303-1307 [DOI] [PubMed] [Google Scholar]
- 62.Widschwendter M, Berger J, Hermann M, Muller HM, Amberger A, Zeschnigk M, Widschwendter A, Abendstein B, Zeimet AG, Daxenbichler G, Marth C: Methylation and silencing of the retinoic acid receptor-beta2 gene in breast cancer. J Natl Cancer Inst 2000, 92:826-832 [DOI] [PubMed] [Google Scholar]
- 63.Buchhagen DL, Qiu L, Etkind P: Homozygous deletion, rearrangement and hypermethylation implicate chromosome region 3p14.3-3p21.3 in sporadic breast-cancer development. Int J Cancer 1994, 57:473-479 [DOI] [PubMed] [Google Scholar]
- 64.Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature 1998, 396:643-649 [DOI] [PubMed] [Google Scholar]
- 65.Windle B, Draper BW, Yin YX, O’Gorman S, Wahl GM: A central role for chromosome breakage in gene amplification, deletion formation, and amplicon integration. Genes Dev 1991, 5:160-174 [DOI] [PubMed] [Google Scholar]
- 66.Moynahan ME, Jasin M: Loss of heterozygosity induced by a chromosomal double-strand break. Proc Natl Acad Sci USA 1997, 94:8988-8993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turker MS, Gage BM, Rose JA, Elroy D, Ponomareva ON, Stambrook PJ, Tischfield JA: A novel signature mutation for oxidative damage resembles a mutational pattern found commonly in human cancers. Cancer Res 1999, 59:1837-1839 [PubMed] [Google Scholar]
- 68.Huang CS, Chern HD, Chang KJ, Cheng CW, Hsu SM, Shen CY: Breast cancer risk associated with genotype polymorphism of the estrogen-metabolizing genes CYP17, CYP1A1, and COMT: a multigenic study on cancer susceptibility. Cancer Res 1999, 59:4870-4875 [PubMed] [Google Scholar]
- 69.Cavalieri EL, Stack DE, Devanesan PD, Todorovic R, Dwivedy I, Higginbotham S, Johansson SL, Patil KD, Gross ML, Gooden JK, Ramanathan R, Cerny RL, Rogan EG: Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc Natl Acad Sci USA 1997, 94:10937-10942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harrison L, Hatahet Z, Wallace SS: In vitro repair of synthetic ionizing radiation-induced multiply damaged DNA sites. J Mol Biol 1999, 290:667-684 [DOI] [PubMed] [Google Scholar]