

Abstract
Alveolar rhabdomyosarcoma (ARMS) is consistently associated with the characteristic translocations t(2;13)(q35;q14) and t(1;13)(p36;q14), which encode for the PAX3-FKHR and PAX7-FKHR fusion oncoproteins respectively. We have investigated the relationship between PAX3-FKHR expression and ARMS histogenesis in primary tumors and cell culture systems. In a blinded histological review of discrepant primary tumors in which there was PAX3-FKHR expression but embryonal histology, we found small areas of alveolar histology in 6 of 11 cases. This suggests that histology alone may under-represent the association between PAX3-FKHR and ARMS, and we investigated this link by examining the effect of ectopic PAX3-FKHR expression on RMS cells. Two cell lines, RD and HX170C, were stably transfected with a PAX3-FKHR expression construct. In cloned transfectants derived from both lines, PAX3-FKHR expression resulted in increased proliferative rate in vitro and promoted cell growth in the absence of added growth factors. Tumors that formed as xenografts in immunodeficient mice were faster growing, more locally invasive, and had a denser, more pleomorphic architecture than untransfected or empty vector transfected tumors. The characteristic clefts and alveolar spaces of ARMS, however, were not seen. In contrast, tumors grown as xenografts from individual clones derived from ARMS cell lines showed all of the classical morphological features of ARMS suggesting divergence in vivo from precursor cells propagated in culture.
Rhabdomyosarcomas can be broadly subdivided into embryonal and alveolar histological types, with spindle cell and botryoid variants of the former and a solid variant of the latter. 1-3 In recent years, molecular correlates with histology have been described. In particular, the translocation t(2;13)(q35;q14) and the rare variant translocation t(1;13)(p36;q14) 4,5 that result in the generation of the fusion proteins PAX3-FKHR and PAX7-FKHR, respectively, 6-8 are characteristic of alveolar rhabdomyosarcoma (ARMS). These two translocations involve the fusion of the DNA binding elements of the PAX transcription factors with the transactivation domain of the FKHR protein, and may be referred to collectively as FKHR disrupting translocations. Embryonal rhabdomyosarcoma (ERMS) cells express wild-type PAX3 that binds the same DNA targets as PAX3-FKHR but has weaker transcription activation. FKHR fusions have been reported to occur in about 80% of ARMS cases. Cytogenetic and comparative genomic hybridization studies have shown that ERMS tumors have a pattern of whole chromosome gains and losses whereas ARMS tumors have a high incidence of translocations and discrete amplicons, the significance of which, if any, has yet to be determined. 9 There are also molecular changes in common between the different histological subtypes, including disruption of the p53 pathway through mutation or mdm2 amplification, 10,11 and deregulation of imprinted genes at chromosome region 11p15.5. 12,13 Hence it is an attractive hypothesis that ERMS and ARMS represent distinct pathologies, rather than being part of a spectrum, and that the essential difference resides in the presence of the FKHR disrupting translocations. The implication is that both ERMS and ARMS are derived from cells committed to myogenic differentiation, but that PAX3-FKHR expression in ARMS results in the activation of an array of downstream transcriptional target genes that confer a distinct and more aggressive phenotype to ARMS tumors.
To test this hypothesis, we have extended a previous study of phenotype/genotype correlations in rhabdomyosarcoma tumors (Anderson et al, submitted) by focusing on cases where there is an apparent discrepancy between translocation status and histological type. A blinded histological review of these cases has confirmed the strong association between alveolar histology and FKHR disrupting translocations. We have ectopically expressed the fusion gene in two different PAX3 expressing RMS cell lines. We chose to transfect PAX3-expressing ERMS cells so that consistent changes in phenotype might be attributable to the contribution of the FKHR transactivation domain. To investigate the impact of PAX3-FKHR on tumor morphology we have grown the modified cells as tumor xenografts in immunodeficient mice. Moreover, by growing tumors derived from clones of native ARMS cells, we have demonstrated that the classical features of ARMS including cellular pleomorphism, varying degrees of morphological differentiation, and development of giant cells all represent divergence in vivo from the pluripotent cells propagated in culture.
Materials and Methods
Histological Assessment
In the review of primary tumors, a set of representative hematoxylin and eosin stained slides were marked with identifying numbers and reviewed individually by two pathologists (R.C. and S.G.) blinded to all clinical information. Tumors were assessed in accordance with modern diagnostic criteria. 14 Hematoxylin and eosin sections from xenografted tumors were assessed blind to cell line type by one pathologist (A.R.) and scored for morphological and ultrastructural features and local invasion. Staining for Ki67 15 and desmin 16 used antibodies from DAKO (Ely, UK) and standard techniques.
Cloning, Cell Lines, and Transfection
A PAX3-FKHR mixed murine/human cDNA, which uses the second ATG of PAX3, was a gift from J. Epstein (University of Pennsylvania, Philadelphia, PA). A six-amino-acid HA epitope tag was added to the N terminus by polymerase chain reaction (PCR) and the PCR product cloned into pBK-CMV (Stratagene) to give high level expression under the control of the CMV immediate early gene promoter. This construct is called pCMV-P3F. In an alternate construct, pMyoD-P3F, the 7-kb MyoD enhancer 17 was inserted upstream of PAX3-FKHR. The cell lines RH30, RH18, and RH36 (from Peter Houghton, Saint Jude’s Children’s Research Hospital, Memphis, TN), RMS, RD, and HX170C were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (FCS) (GIBCO-BRL). Transfection of RH30 and RMS cells was by electroporation using standard techniques. RD and HX170C cells were transfected using Fugene 6 reagent (Roche, Lewes, UK). Stable transfectants containing PAX3-FKHR or empty vector were selected in G418 and emergent colonies picked off with cloning loops.
Western Blots
10 6 cells were lysed in 50 μl of buffer containing 1% NP40, 150 mmol/L NaCl, 1 mmol/L Tris, and protease inhibitors. Lysates were resolved on 10% polyacrylamide gels and transferred to PDVF membranes, which were probed with an anti-HA mouse monoclonal antibody HA11 (BABCO, Berkley, CA).
Xenografting and Tumor Growth Assays
Animals were housed in accordance with Home Office regulations. Cell lines were grown as tumors in immunodeficient mice (RAG−/−, common γ chain−/−, or NOD-SCID). In tumor growth assays, 5 × 10 6 unirradiated cells were injected subcutaneously into the flank; up to four mice of the same strain were used per engraftment study depending on availability. Tumors were harvested from all of the mice in each experiment once one tumor had reached 15 mm in diameter. For analysis of development of lung metastases during the growth of subcutaneous tumor xenografts, one lung was fixed in formalin, stained in hematoxylin and eosin, and subjected to histological assessment. The other lung was homogenized and plated out on collagen-coated dishes containing DMEM and G418 to allow for colony formation from micrometastases.
In Vitro Proliferation Assays
10 5 cells were plated out per well of 30 mm plates and growth as monolayers was assessed after incubation in serum-free DMEM or DMEM containing 10% FCS. Cell numbers were assessed by trypan blue exclusion of trypsinized pooled cells. Cell culture supernatants were similarly examined for the presence of dead or dying cells Individual clones were plated out and counted in triplicate.
Results
Relationship between Presence of Translocations and Histological Type in Primary Tumors
In a separate study of 91 rhabdomyosarcoma primary tumors, reverse transcriptase-polymerase chain reaction (RT-PCR), fluorescence in situ hybridization (FISH), and cytogenetic analysis were used to determine the presence or absence of FKHR disrupting translocations (Anderson et al, submitted). In 21 of these cases there was a discrepancy between translocation status and histological diagnosis. In seven cases alveolar histology was not associated with a FKHR disrupting translocation/fusion gene and 14 cases showed non-alveolar histology in association with the presence of a translocation/fusion gene. We hypothesized that some of the fusion gene-positive but alveolar histology-negative cases might be reclassified when subjected to modern diagnostic criteria. Therefore, representative diagnostic slides from all available discrepant cases were reviewed by two pathologists blinded to all clinical and cytogenetic data (Figure 1) ▶ .
Figure 1.

Schema of blinded histological review.
Histological review identified three fusion-negative tumors with typical alveolar histology, confirming the existence of this phenomenon. We were interested in the group of 11 cases with FKHR disrupting translocation and embryonal histology according to the original pathological assessment. Review changed the diagnosis to that of ARMS in six cases by identifying small areas of classical alveolar histology. Four of these six had been diagnosed before 1989 when the diagnostic criteria of alveolar had been changed. Five further translocation-positive discrepant cases remained as embryonal following review. One of these “true discrepant” cases was of particular interest having PAX7-FKHR and botryoid histology although the majority of cells in the tumor sample were nonmalignant stromal cells. There were no distinctive morphological appearances in the four remaining discrepant cases, in which PAX3-FKHR was confirmed by RT-PCR and FISH (one case) or RT-PCR alone (three cases). Nine concordant centrally reviewed control cases were also reviewed blind and confirmed the pathologists’ adherence to modern diagnostic criteria.
Development of Alveolar Structures in Xenografted Tumors
In the light of the apparent strong correlation between translocation fusion gene expression and histological subtype, we sought further to investigate the development of alveolar rhabdomyosarcoma histology structures using xenografted tumors. The neoplasms were grown in immunodeficient mice and derived from cell lines with known translocation status as determined by RT-PCR (Table 1) ▶ . Between two and four mice were injected with 5 × 10 6 cells of each cell line, and histology of resultant tumors was reviewed blind to cell type of origin (Table 1) ▶ . There was a consistent histological appearance for tumors derived from each particular cell line. Three cell lines (RH30, RMS, RH18) showed very similar histological appearance with classical alveoli and deep clefts being prominent features. In RMS and RH30 this correlated with expression of PAX3-FKHR, whereas in RH18 this appearance was seen despite the absence of both PAX3-FKHR and PAX7-FKHR. These alveolar tumors also had marked cellular pleomorphism. Clefts, alveoli, and pleomorphism were absent from the embryonal tumors RD, HX170C, and RH36, all of which were negative for FKHR disrupting fusions by RT-PCR. In their wild-type form, cell lines RD and HX170C develop relatively slow growing tumors that do not invade into local structures and have a relatively monomorphic microscopic appearance. In the case of RD, the cells when grown as tumor xenografts, are spindle-shaped and resemble classical embryonal rhabdomyosarcoma, whereas HX170C are more rounded and denser and resemble the so-called solid alveolar rhabdomyosarcoma 1 (Figure 6g) ▶ . Interestingly, the tumor from which cell line HX170C was originally derived was a relapsed bladder rhabdomyosarcoma from a 5-year-old boy, which showed some classical alveolar features on relapse, although not in the diagnostic sample. 18 RH36 gave rise to tumors composed of uniform spindle cells.
Table 1.
Summary of Morphology of Xenografts
| Clonal or mixed population | Translocation status | No. of tumors | Tumor growth rate index (mg/day)* | Cells | Classical alveolar features | Giant cells | Necrosis | Local invasion | |
|---|---|---|---|---|---|---|---|---|---|
| RH30 | C | PAX3-FKHR | 4 | 95 | Pleomorphic | + | ++ | + | + |
| RMS | M | PAX3-FKHR | 3 | 175 | Round/pleomorphic | + | − | +/− | + |
| RH18 | M | No translocation | 4 | 1.4 | Round/pleomorphic | Mixed | ++ | − | − |
| RH36 | M | No translocation | 2 | 1.8 | Round | − | − | + | − |
| RD | M | No translocation | 4 | 1.8 | Spindle | − | + | +/− | − |
| RD/empty vector† | C | No translocation | 4 | 0.86 | Spindle | − | + | − | − |
| RD/PAX3-FKHR† | C | PAX3-FKHR | 6‡ | 9.06 | Spindle/pleomorphic | − | + | + | + |
| HX170C/empty vector† | C | No translocation | 1 | 0.2 | Round/loose | − | − | − | − |
| HX170C/PAX 3-FKHR† | C | PAX3-FKHR | 1 | 2.8 | Round/dense/fasciculated | − | − | + | + |
*Growth rate index is expressed as average tumor weight per day of growth.
†Transfected cells were all derived from single clones.
‡RD transfected with PAX3-FKHR 2 separate clones of cells were used to generate 3 tumors each.
Figure 6.
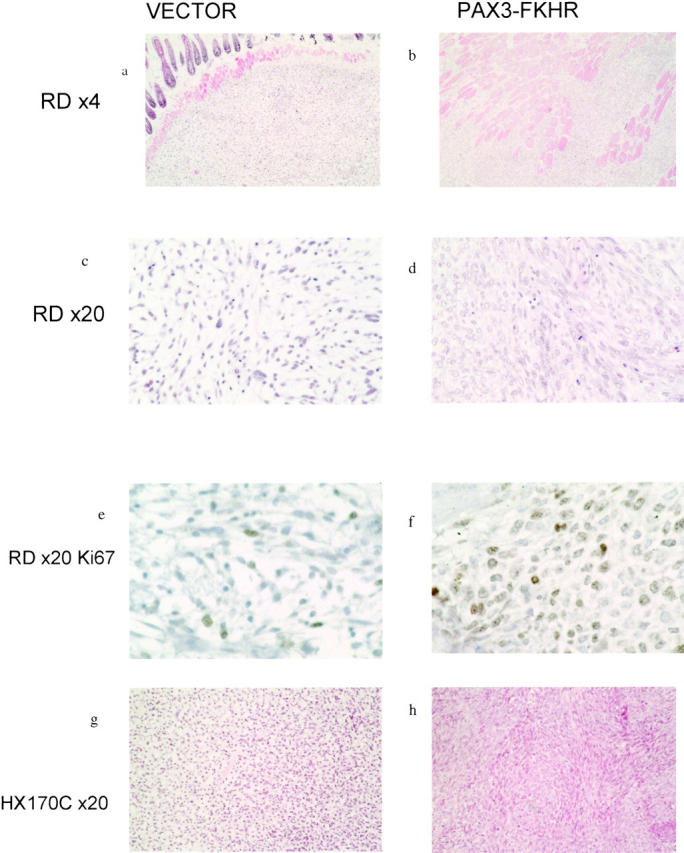
Characteristic histological appearances of tumors grown as xenografts from clones of cell stably transfected with either empty vector or PAX3-FKHR. Cell lines and magnification on left. Cells were transfected with vector (a, c, e, g) or PAX3-FKHR (b, d, f, h). In b, there is evidence of increased local invasion. Individual muscle fibers are separated by infiltrating tumor cells. In contrast, in a the tumor has a well circumscribed edge and there is no evidence of significant splitting of individual fibers. d shows the increase in cellularity, nuclear size, and cellular pleomorphism in the PAX3-FKHR transfected tumor and f confirms the increased proliferation rate. g and h show a similar increase in cellularity and the grouping of cells into fascicular bundles that result from the expression of PAX3-FKHR in HX170C cells.
Xenografted Tumors Demonstrate Differentiation from Stem Cells Propagated in Culture
We were interested in the cause of the cellular heterogeneity (cellular pleomorphism, region-specific differences in architecture, and the existence of giant cells) seen in rhabdomyosarcoma primary tumors and xenografted tumors derived from cell lines. For example, are the alveolar areas in an ARMS tumor genetically distinct as a result of genetic heterogeneity maintained during passage in culture? To test this we made use of subclones of cell lines derived following transfection with pBK-CMV. Xenografted tumors derived from different subclones of ARMS cell line RH30 and ERMS cell line RD were histologically indistinguishable from the tumors derived from the original cell population. Specifically, three different RH30 subclones all gave rise to tumors showing a high degree of cellular pleomorphism, and showed the presence of giant cells and alveolar areas intermixed with areas of more uniform morphology (Figure 2A) ▶ . Tumor size and local invasion were not significantly different between subclones. A single subclone of ERMS cell line RD was used to generate four different tumors, all of which had a uniform appearance of loose uniform spindle cells intermixed with occasional giant cells. Therefore, all of the features that we had previously seen were present in tumors grown up from single cells.
Figure 2.
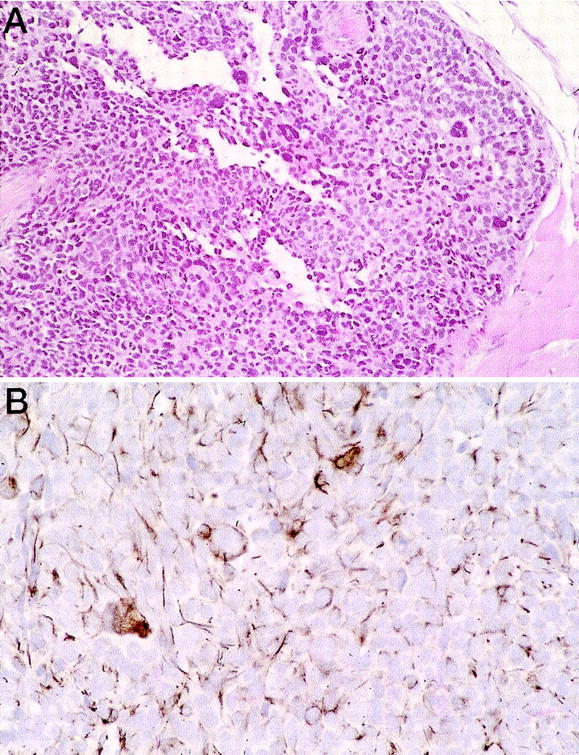
Tumors grown from individual clones derived from cell lines. A: Tumor from a clone of cell line RH30 showing giant cells, clefts, and alveolar histology. B: Desmin staining of a clone derived from RD showing focal desmin positivity and staining of giant cells.
Giant cells were consistently apparent in tumors derived from three of the six cell lines but were not seen in cells propagated in culture. We were interested in whether giant cells represented an early stage of myogenic differentiation (i.e., cell fusion and synthesis of myotubes). We therefore applied an antibody against desmin (an intermediate filament expressed during skeletal muscle development) to sections from xenografted tumors derived from cell line subclones of RH30. Desmin positivity was highly non-uniform with a mixture of strongly staining cells and cells with no desmin positivity (Figure 2B) ▶ . Giant cells were usually strongly positive for desmin immunostaining.
Expression of PAX3-FKHR in Embryonal Rhabdomyosarcoma Cells Induces Histological and Phenotypic Changes in Vitro and in Vivo
The strong correlation between the presence of FKHR-disrupting translocations and alveolar histology led us to hypothesize that expression of PAX-FKHR fusion oncoproteins is directly responsible for histological alveolar structures, as well as the more aggressive tumor phenotype associated with ARMS. To test this we ectopically expressed PAX3-FKHR into embryonal rhabdomyosarcoma cells RD and HX170C and monitored changes in cell growth both in culture and as xenografts in immunodeficient mice. We demonstrated low level expression of PAX3 in both cell lines by Northern blot (not shown). We grew up clones of RD and HX170C transfected with pCMV-P3F, pMyoD-P3F, or an empty vector (pBK-CMV) control. Multiple clones derived from RD were obtained although HX170C, which has very low transfection efficiency, yielded only single clones transfected with pCMV-P3F or pBK-CMV respectively. Expression of PAX3-FKHR was confirmed by demonstration of the presence of the HA epitope by immunoblot and immunofluorescence. No significant difference in levels of expression was seen between individual clones used in subsequent experiments (Figure 3) ▶ .
Figure 3.

Representative Western blot showing expression of the HA epitope on tagged PAX3-FKHR in different clones of RD cells transfected with pCMV-P3F. a: Coomassie blue-stained gel made in parallel with blotted gel to demonstrate equivalence of loading. b: Band at about 120 kd corresponding to the expected size of PAX3-FKHR. C5 through C15 are different clones whereas “mixed” represents a mixed population of RD cells transfected with pCMV-P3F. L is protein standard ladder, WT is untransfected RD, and POS is a positive control cell line that highly expresses an HA tagged protein.
In in vitro growth assays, different clones of RD cells transfected with PAX3-FKHR grew significantly faster in the absence of added serum than empty vector transfected clones. However, in the presence of 10% serum, the proliferative advantage was not seen. Transfection with pCMVP3F and pMyoDP3F enabled RD cells to remain viable and continue to proliferate in culture in serum-free medium for two weeks. The vector-only transfected RD cells had all died by this time (Figure 4d) ▶ . HX170C cells transfected with pCMVP3F had a higher growth rate both in the presence and absence of added serum (Figure 4e) ▶ . No cell death, as determined by trypan blue uptake, was seen in wild-type or transfected cells during up to 96 hours growth in serum- free medium or 10% serum.
Figure 4.

a-e: Comparative proliferation assays. 100,000 cells of RD or 500,000 cells of HX170C were plated on day 1 and viable cell number determined after differing incubation times by trypan blue exclusion. Vector equals empty vector transfected cells, M and CMV refer to PAX3-FKHR expression construct with MyoD and CMV immediate early promoters respectively, and c refers to individual colony numbers.
In tumor xenograft experiments, mice were injected with 5 × 10 6 cells at an early passage following cloning thereby minimizing genetic variation within each clone. Tumors derived from clones of ERMS cell lines stably transfected with pCMVP3F grew faster than empty vector-transfected tumors (Figure 5) ▶ . Tumors derived from pCMVP3F-transfected RD cells showed the development of a high degree of cellular pleomorphism, higher proliferative rate (as determined by Ki 67 immunoreactivity) and greater local tissue invasion than were observed in either wild-type RD or a clone of vector only-transfected RD (Figure 6 ▶ and Table 1 ▶ ). Ectopic expression of PAX3-FKHR in HX170C resulted in a dramatic increase in cellularity, increased invasion, and increased cellular pleomorphism (Figure 6, g and h) ▶ .
Figure 5.

Mean tumor weight following subcutaneous injection in NOD-SCID mice of RD or HX170C rhabdomyosarcoma cells transfected with empty vector (vec) or pCMV-P3F. RD-P3f-13 and RD-P3f-14 refer to the same pCMV-P3F clones as used in the in vitro proliferation assays. Tumors were grown for four weeks. There were four mice per group in RD experiments and one mouse per group in HX170c experiments.
There was no detection of microscopic or macroscopic lung metastases with either wild-type or transfected RD and HX170C cells although, in parallel experiments, the ARMS cell line RH30 did result in macroscopic lung metastases over the same time course (data not shown).
Discussion
We have sought to test the hypothesis that ARMS is a distinct pathological entity from ERMS with a more aggressive phenotype, and that the difference can be accounted for by the expression of the FKHR-disrupting fusion proteins in ARMS tumors. In tumor xenografts and in vitro proliferation assays we have shown that ectopic expression of PAX3-FKHR in PAX3-expressing ERMS cells results in more rapid, growth factor autonomous growth, cellular pleomorphism, and enhanced tissue invasion.
We have previously shown that PAX3-FKHR-expressing tumors occur in older children and are more likely to show regional lymph node spread or distant metastases resulting in significantly worse overall and event-free survival. PAX7-FKHR-expressing tumors are relatively rare but appear to have a less severe phenotype (Anderson et al, submitted and 19 ). Moreover, PAX7-FKHR and PAX3-FKHR expression have also been closely identified with the distinctive alveolar histological subtype. Of great molecular interest, therefore, are tumors with discrepancy between FKHR fusion genes and histological type. For example, cell line RH18 originally derived from an alveolar rhabdomyosarcoma is negative for PAX3-FKHR and PAX7-FKHR by RT-PCR but maintains a classical alveolar appearance when grown as a tumor xenograft. A possible explanation for this cell line and the alveolar fusion-negative tumors is the presence of alternative translocations involving PAX3/7 or deregulation of PAX gene transcription by mechanisms other than fusion with the FKHR transactivation domain. Less well recognized is the phenomenon of tumors expressing a FKHR disrupting fusion protein but with no classical alveolar morphology. We had previously identified 14 such cases using molecular techniques and relying on histological reports. We were able to reanalyze 11 of these cases with two pathologists answering the question of whether the tumors fulfilled the modern criteria of alveolar, and blinded to all clinical and cytogenetic information. Six cases were re-defined as alveolar following blinded review, demonstrating the change in diagnostic criteria with time (only minimal amounts of alveolar histology in an otherwise embryonal tumor are now required to secure the diagnosis) and the importance of centralized review of histology. Moreover, the finding of small amounts of alveolar architecture in an otherwise undifferentiated rhabdomyosarcoma suggests PAX3-FKHR might result in the development of morphological and phenotypic features of ARMS if ectopically expressed in embryonal rhabdomyosarcoma.
To test this and to analyze further the relationship between PAX3-FKHR, alveolar histology, and tumor phenotype, we have studied the effect of ectopic PAX3-FKHR expression in two ERMS cell lines RD and HX170C. We focused on the more common translocation t(2;13)(q36;+q14), encoding PAX3-FKHR, because it is more unequivocally associated with a more malignant phenotype. The only difference between ERMS cells stably transfected with the PAX3-FKHR expression construct and empty vector is the expression of PAX3-FKHR, and changes in growth phenotype and morphology can therefore be directly attributed to PAX3-FKHR. We have controlled for differences in level of expression of PAX3-FKHR by using two different promoters and several randomly selected clones of cell line RD. Ectopic expression of PAX3-FKHR in RD and HX170C cells resulted in increased tumor growth rate, in vitro proliferation, and Ki67 immunostaining of tumors. Increased in vitro proliferation was particularly evident in the absence of added serum, suggesting that increased production of growth factors by PAX3-FKHR may be responsible. It is unlikely that loss of contact inhibition was misinterpreted as increased proliferation rate, as the in vitro growth assays were performed at low density and both wild-type RD and HX170C cells continue to proliferate when confluent (not shown). Similarly, lack of cell death was not responsible for the growth advantage. Increased tumor growth rate in the presence of PAX3-FKHR is associated with a greater cellular density and invasion into local muscle. It is unlikely that the absence of tissue invasion in the empty vector-transfected cells is due to the smaller tumor size because, when wild-type RD or HX170C were grown to equivalent or greater sizes than the PAX3-FKHR-transfected tumors in comparative growth assays, muscle invasion was not seen. We therefore favor the hypothesis that PAX3-FKHR adds additional properties to rhabdomyosarcoma cells to allow invasion into local muscle. In metastasis assays, however, we did not find that local muscle infiltration translated into seeding of distant lung deposits. This may have been because the time course of the experiments was too short due to relatively rapid growth of RD cells as tumors, or that PAX3-FKHR regulated, metastatogenic factors in human rhabdomyosarcoma cells did not function in a murine tissue environment. Metastases at other sites may have been present.
Because ERMS cells express wild-type PAX3, the changes in phenotype that we have demonstrated may well reflect the contribution of the FKHR transactivation domain fused to PAX3. Moreover, alveolar rhabdomyosarcoma cells typically express both PAX3 and PAX3-FKHR, so the cellular systems we have generated may well be more reflective of normal ARMS histogenesis than studies using PAX3-FKHR-transfected myoblasts. 20 Transient transfection studies in heterologous cells have demonstrated that PAX3-FKHR is a more potent transcriptional activator than PAX3 and one method of tumorigenesis may be enhanced levels of transcription of PAX3 target genes. 21,22 However, there is evidence that PAX3-FKHR can also transcriptionally regulate genes that are not targets of wild-type PAX3. 23 The anti-apoptotic gene Bcl-xL is a transcriptional target of both PAX3 and PAX3-FKHR suggesting that the enhanced tumorigenicity of PAX3-FKHR may be a result of inhibition of apoptosis. 24,25 PAX3 and PAX3-FKHR are known to be important anti-apoptotic survival factors in both developing myoblasts and rhabdomyosarcoma cells. 26,27 Recently, inhibition of transcription of PAX3 targets through expression of a KRAB-PAX3 repressor in ARMS cells has been shown to result in inhibition of tumor growth and marked apoptosis. 25 However, as both wild-type PAX3 and PAX3-FKHR targets were repressed in that study, it is impossible to attribute the effects specifically to PAX3-FKHR and our data suggest that PAX3-FKHR imparts a proliferative advantage independent from inhibition of cell death. Another possible mechanism of action of PAX3-FKHR is dominant negative interaction with undisrupted FKHR which is expressed in rhabdomyosarcoma cells. FKHR is downstream of AKT and plays a pro-apoptotic role in 293T cells, so disruption of normal apoptotic pathways through inhibition of FKHR transcriptional activity may be a mechanism of enhanced tumorigenesis by PAX3-FKHR.
Although both cell lines studied undergo similar morphological and phenotypic changes following ectopic PAX3-FKHR expression, the background histology of the untransfected cells, neither of which express of FKHR fusion gene, are distinct. Tumor xenografts of RD are classical ERMS with loose spindle cells whereas HX170C tumors are more solid and were originally classified as solid variant alveolar. HX170C was derived from the bladder of a 5-year-old boy, a site and age at which histology is almost invariably embryonal. We therefore consider it unlikely that this cell line represents one of the alveolar FKHR fusion negative tumors. The change to a more aggressive morphology and growth following PAX3-FKHR expression supports this. Hence we have shown in two separate PAX3 expressing RMS cell lines that PAX3-FKHR expression regulated by two different promoters does not lead to formation of alveoli and clefts but does resulting dramatic increase in cell density, pleomorphism, proliferation rate, and tissue invasion. This provides direct evidence that PAX3-FKHR, and the genes that it regulates, is responsible for the more malignant behavior of ARMS compared with ERMS.
Finally, the tumors grown from different subclones of the original cell lines have identical morphology and include alveolar spaces, deep clefts, giant cells, and variable desmin staining. This cellular heterogeneity was not seen in the same cells cultured in vitro as monolayers. The observation suggests that the cell lines are composed of uniform cells, which have potential to differentiate to varying degrees in a more natural host environment. This differentiation takes the form of the beginnings of myogenic differentiation (as revealed by fusion into giant cells and expression of the intermediate filament desmin) and organization of cell-cell contacts to form clefts and intercellular spaces. It is interesting to speculate that the tumor cells that form immortalized cell lines in culture are therefore less differentiated and more primitive, possibly reflecting transformation of cells within developing or mature muscle at an early stage of myogenic differentiation.
Future studies will address the mechanism by which PAX3-FKHR contributes to the more malignant phenotype. Gene expression array profiling experiments in heterologous cells have identified a number of potential physiological targets for PAX3-FKHR. 28 It will be interesting to see how many of them are important in rhabdomyosaroma cells; and whether individual genes regulated by PAX3-FKHR are capable of mimicking the effect of ectopic PAX3-FKHR expression in ERMS cells. Ultimately, knowledge of the transciptional control mechanisms operated by PAX3-FKHR will reveal novel therapeutic targets.
Acknowledgments
We thank Professor Richard Carter for his contribution to the review of pathology of primary tumors. Research at the Institute of Child Health and Great Ormond Street Hospital for Children National Health Service trust benefits from Research and Development funding received from the NHS executive. The views expressed in this publication are those of the authors and are not necessarily those of the NHS executive. JA and KPJ are supported by the Cancer Research Campaign.
Footnotes
Address reprint requests to John Anderson, Clinical Lecturer in Pediatric Oncology, Institute of Child Health and Great Ormond Street Hospital NHS Trust, 30 Guilford Street, London WC1N 1EH, UK. E-mail: j.anderson@ich.ucl.ac.uk.
References
- 1.Tsokos M, Webber BL, Parham DM, Wesley RA, Miser A, Miser JS, Etcubanas E, Kinsella T, Grayson J, Glatstein E, et al: Rhabdomyosarcoma: a new classification scheme related to prognosis. Arch Pathol Lab Med 1992, 116:847-855 [PubMed] [Google Scholar]
- 2.Newton WA, Jr, Gehan EA, Webber BL, Marsden HB, van Unnik AJ, Hamoudi AB, Tsokos MG, Shimada H, Harms D, Schmidt D, et al: Classification of rhabdomyosarcomas and related sarcomas: pathologic aspects and proposal for a new classification–an Intergroup Rhabdomyosarcoma Study. Cancer 1995, 76:1073-1085 [DOI] [PubMed] [Google Scholar]
- 3.Asmar L, Gehan EA, Newton WA, Webber BL, Marsden HB, van Unnik AJ, Hamoudi AB, Shimada H, Tsokos M, Harms D, et al: Agreement among and within groups of pathologists in the classification of rhabdomyosarcoma and related childhood sarcomas: report of an international study of four pathology classifications. Cancer 1994, 74:2579-2588 [DOI] [PubMed] [Google Scholar]
- 4.Turc-Carel C, Lizard-Nacol S, Justrabo E, Favrot M, Philip T, Tabone E: Consistent chromosomal translocation in alveolar rhabdomyosarcoma. Cancer Genet Cytogenet 1986, 19:361-362 [DOI] [PubMed] [Google Scholar]
- 5.Douglass EC, Rowe ST, Valentine M, Parham DM, Berkow R, Bowman WP, Maurer HM: Variant translocations of chromosome 13 in alveolar rhabdomyosarcoma. Genes Chromosomes Cancer 1991, 3:480-482 [DOI] [PubMed] [Google Scholar]
- 6.Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ, 3rd, Emanuel BS, Rovera G, Barr FG: Fusion of a fork head domain gene to PAX3 in the solid tumor alveolar rhabdomyosarcoma [published erratum appears in Nat Genet 1994 Feb;6(2): 214]. Nat Genet 1993, 5:230-235 [DOI] [PubMed] [Google Scholar]
- 7.Shapiro DN, Sublett JE, Li B, Downing JR, Naeve CW: Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res 1993, 53:5108-5112 [PubMed] [Google Scholar]
- 8.Davis RJ, D’Cruz CM, Lovell MA, Biegel JA, Barr FG: Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res 1994, 54:2869-2872 [PubMed] [Google Scholar]
- 9.Weber-Hall S, Anderson J, McManus A, Abe S, Nojima T, Pinkerton R, Pritchard-Jones K, Shipley J: Gains, losses, and amplification of genomic material in rhabdomyosarcoma analyzed by comparative genomic hybridization. Cancer Res 1996, 56:3220-3224 [PubMed] [Google Scholar]
- 10.Wurl P, Taubert H, Bache M, Kroll J, Meye A, Berger D, Siermann A, Holzhausen HJ, Hinze R, Schmidt H, Rath FW: Frequent occurrence of p53 mutations in rhabdomyosarcoma and leiomyosarcoma, but not in fibrosarcoma and malignant neural tumors. Int J Cancer 1996, 69:317-323 [DOI] [PubMed] [Google Scholar]
- 11.Keleti J, Quezado MM, Abaza MM, Raffeld M, Tsokos M: The MDM2 oncoprotein is overexpressed in rhabdomyosarcoma cell lines and stabilizes wild-type p53 protein. Am J Pathol 1996, 149:143-151 [PMC free article] [PubMed] [Google Scholar]
- 12.Zhan S, Shapiro DN, Helman LJ: Activation of an imprinted allele of the insulin-like growth factor II gene implicated in rhabdomyosarcoma. J Clin Invest 1994, 94:445-448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson J, Gordon A, McManus A, Shipley J, Pritchard-Jones K: Disruption of imprinted genes at chromosome region 11p15.5 in pediatric rhabdomyosarcoma. Neoplasia 1999, 1:340-348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qualman SJ, Coffin CM, Newton WA, Hojo H, Triche TJ, Parham DM, Crist WM: Intergroup Rhabdomyosarcoma Study: update for pathologists. Pediatr Dev Pathol 1998, 1:550-561 [DOI] [PubMed] [Google Scholar]
- 15.Gerdes J, Li L, Schlueter C, Duchrow M, Wohlenberg C, Gerlach C, Stahmer I, Kloth S, Brandt E, Flad HD: Immunobiochemical and molecular biologic characterization of the cell proliferation-associated nuclear antigen that is defined by monoclonal antibody Ki-67. Am J Pathol 1991, 138:867-873 [PMC free article] [PubMed] [Google Scholar]
- 16.Van Muijen GN, Ruiter DJ, Warnaar SO: Coexpression of intermediate filament polypeptides in human fetal and adult tissues. Lab Invest 1987, 57:359-369 [PubMed] [Google Scholar]
- 17.Goldhamer DJ, Brunk BP, Faerman A, King A, Shani M, Emerson CP, Jr: Embryonic activation of the myoD gene is regulated by a highly conserved distal control element. Development 1995, 121:637-649 [DOI] [PubMed] [Google Scholar]
- 18.Kelland LR, Bingle L, Edwards S, Steel GG: High intrinsic radiosensitivity of a newly established and characterized human embryonal rhabdomyosarcoma cell line. Br J Cancer 1989, 59:160-164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly KM, Womer RB, Sorensen PH, Xiong QB, Barr FG: Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol 1997, 15:1831-1836 [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Kumar P, Epstein J, Helman L, Moore JV, Kumar S: Insulin-like growth factor II and PAX3-FKHR cooperate in the oncogenesis of rhabdomyosarcoma. Cancer Res 1998, 58:4426-4433 [PubMed] [Google Scholar]
- 21.Bennicelli JL, Advani S, Schafer BW, Barr FG: PAX3 and PAX7 exhibit conserved cis-acting transcription repression domains and utilize a common gain of function mechanism in alveolar rhabdomyosarcoma. Oncogene 1999, 18:4348-4356 [DOI] [PubMed] [Google Scholar]
- 22.Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG, Rauscher FJ, III: The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol 1995, 15:1522-1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Epstein JA, Song B, Lakkis M, Wang C: Tumor-specific PAX3-FKHR transcription factor, but not PAX3, activates the platelet-derived growth factor alpha receptor. Mol Cell Biol 1998, 18:4118-4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Margue CM, Bernasconi M, Barr FG, Schafer BW: Transcriptional modulation of the anti-apoptotic protein BCL-XL by the paired box transcription factors PAX3 and PAX3/FKHR. Oncogene 2000, 19:2921-2929 [DOI] [PubMed] [Google Scholar]
- 25.Ayyanathan K, Fredericks WJ, Berking C, Herlyn M, Balakrishnan C, Gunther E, Rauscher FJ: Hormone-dependent tumor regression in vivo by an inducible transcriptional repressor directed at the PAX3-FKHR oncogene. Cancer Res 2000, 60:5803-5814 [PubMed] [Google Scholar]
- 26.Bernasconi M, Remppis A, Fredericks WJ, Rauscher FJ, III, Schafer BW: Induction of apoptosis in rhabdomyosarcoma cells through down-regulation of PAX proteins. Proc Natl Acad Sci USA 1996, 93:13164-13169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borycki AG, Li J, Jin F, Emerson CP, Epstein JA: Pax3 functions in cell survival and in pax7 regulation. Development 1999, 126:1665-1674 [DOI] [PubMed] [Google Scholar]
- 28.Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, Pavan WJ, Trent JM, Meltzer PS: cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci USA 1999, 96:13264-13269 [DOI] [PMC free article] [PubMed] [Google Scholar]