

Abstract
The effects of radiation and cytotoxic agents on telomerase activity in lymphoma cells were analyzed by a polymerase chain reaction-based telomeric repeat amplification protocol coupled with an enzyme-linked immunosorbent assay, reverse transcriptase-polymerase chain reaction for the expression of the catalytic subunit of telomerase (hTERT), and by Western blot analysis in three lymphoma cell lines (Jurkat, Raji, CEM-6). Telomeric repeat amplification protocol-enzyme-linked immunosorbent assay demonstrated high basal levels of telomerase activity in all cell lines compared to normal and activated peripheral blood lymphocytes. A significant decrease in telomerase activity was observed in all cell lines after exposure to vincristine for 24 hours. The decrease in telomerase activity paralleled the decrease in cell viability in Jurkat and CEM-6 cells but not in Raji cells. Radiation exposure inhibited the telomerase activity of Jurkat and CEM-6 cells whereas Raji cells were unaffected. Cell cycle analysis demonstrated a significant G2/M arrest by cisplatin, VP-16, and vincristine. In contrast to the decline in telomerase activity, the level of hTERT RNA and protein increased. Furthermore, the induction of hTERT was preceded by increased expression of the cyclin-dependent kinase inhibitor, p27/Kip1 protein, and p53. These results indicate that telomerase activity is down-regulated by anti-neoplastic agents in lymphoma cells, however expression of hTERT may not be correlated with telomerase activity. We also show that p27/Kip1 may be involved in the G2/M growth arrest induced by anti-neoplastic agents.
The activation of telomerase, a ribonucleoprotein with reverse transcriptase activity results in the synthesis of telomeric DNA onto chromosomal ends. This leads to unlimited cell proliferation and is believed to play a critical role in the neoplastic process. Telomerase uses an internal RNA template to form the specially structured ends of chromosomal DNA (telomeres) by directing the synthesis of new hexameric nucleotide repeats (TTAGGG) onto the 3′ end of chromosomes. 1,2 Cells without telomerase activity display progressive shortening of telomeric repeats with each cell division because of the end-replication problem, leading to the stage when the cell is likely to exit from the cell cycle and become senescent. 3 Currently, it is thought that activation or up-regulation of telomerase allows tumor cells to escape cellular senescence and proliferate indefinitely. The absence of telomerase activity in most nonneoplastic tissues and somatic cells, 4 and its presence in almost all malignant tumors 4-11 has raised much interest in its potential diagnostic, prognostic, and therapeutic implications in the management of human cancer.
The telomerase holoenzyme is composed of the single-strand telomere-binding protein telomerase reverse transcriptase (hTERT), 12 telomerase-associated protein (TEP1), 13 and hnRNP A1. 14 The de novo transcription of the hTERT gene is considered the dominant, rate-limiting step in telomerase activation whereas the expression of hTEP1 and hTR is constitutive. 13,15 HTERT is only expressed in cells and tissues positive for telomerase activity, and is not detected in normal somatic cells that lack telomerase activity. 12,16
Regulation of telomerase activity is complex and involves control at the level of gene transcription, posttranslational protein-protein interactions, and protein phosphorylation. Proto-oncogenes and tumor suppressor genes have been implicated in the regulation of telomerase activity, both directly and indirectly including C-MYC, 17 BCL-2, 18 p21/Waf1, 19 pRB, 20 p53, 21 protein kinase C, 22 Akt/PKB, 23 and protein phosphatase 2A. 24 Evidence indicates that telomerase is reversibly regulated. Sun exposure to normal human skin results in telomerase activation whereas the process of terminal differentiation of various cells is associated with its down-regulation. 25-27 In vitro studies on human breast cancer cell lines showed that anti-neoplastic agents such as doxorubicin, cisplatinum, and tomozolomide decreased telomerase activity in a dose- and time-dependent manner. 28 HeLa cervical cancer cells and colorectal cancer cells also show down-regulated telomerase activity after exposure to ionizing radiation. 29 Cells resistant to these agents showed no decline in telomerase activity and cell growth. 29 These data suggest that telomerase activity may be used as a potential marker for assessing the efficacy of anti-neoplastic agents and test their potential for modulating telomerase activity.
Within the hematopoietic system peripheral blood lymphocytes have low to undetectable levels of telomerase activity whereas early progenitor stems cells in the bone marrow and thymocytes exhibit high activity. 30,31 On activation through T- or B-cell antigen receptors or phorbol myristate acetate (PMA) stimulation, a non-cell cycle-restricted induction of telomerase activity has been observed 32-35 High telomerase activity was also noted in non-Hodgkin’s lymphomas and cell lines compared with reactive lymph nodes. 36
In the current study, we investigated the effect of anti-neoplastic agents (γ-irradiation, cisplatin, VP-16, and vincristine) on telomerase activity using a sensitive biochemical method termed telomeric repeat amplification protocol (TRAP) modified for enzyme-linked immunosorbent assay (ELISA). The results were correlated with cell viability, cell cycle parameters, and levels of hTERT mRNA and protein. We show that various chemotherapeutic agents and radiation result in significant inhibition of telomerase activity in cell lines derived from T lymphoblastic lymphoma and Burkitt (B-lineage) lymphoma. Furthermore, an induction of hTERT transcript and protein was seen in response to these agents. In addition, we show that p27/Kip1 may be involved in the G2/M growth arrest induced by the anti-neoplastic agents.
Materials and Methods
Cell Cultures and Treatment
The cell lines used were the human T-cell leukemia Jurkat, human T-cell lymphoma CEM-6, and the human Burkitt lymphoma Raji. These cell lines were maintained in RPMI 1640 (Gibco Life Technologies, Inc.) supplemented with 10% heat-inactivated fetal calf serum, 2 mmol/L l-glutamine, and 100 U/ml of penicillin-streptomycin mixture (Gibco Life Technologies, Inc.).
For drugs or irradiation experiments, cells at late exponential growth phase were collected by centrifuging at 1200 rpm and resuspended in fresh medium with 5 × 10 5 cells per ml. Cells were incubated at 37°C in 5% CO2 with anti-tumor drugs or γ-ray irradiated. Cells were then collected at different time points for viability assay, proliferation assay, protein analysis (including Western and telomerase activity assays), and RNA analysis (RT-PCR). Control cells were not exposed to chemotherapeutic agents or radiation and otherwise maintained under the same conditions as the treated cells.
Cell Viability and Proliferation
Viable cell counts were performed using trypan blue solution (Sigma) as described by the manufacturer. Cells (Sigma Chemicals, St. Louis, MO) were counted by using the hemocytometer. Cell proliferation was evaluated by [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide assay] MTT assay as described previously. 28 Briefly, 0.1 ml cell cultures (0.5 × 10 5 cells) per well were seeded in the 96-well plate. MTT (10 mg/ml, 10 μl/well; Sigma) was added to each well and incubated at 37°C for 3 hours. Cells were then lysed by adding 0.1 ml per well of lysis solution (20% sodium dodecyl sulfate, 50% N,N-dimethyl formamide, pH 4.7). Absorbance was measured at 540 nm using an ELISA reader after overnight incubation of the mixture.
Telomerase Activity Assay
Telomerase activity was determined using the TRAP ELISA kit (Boehringer Mannheim, Mannheim, Germany). Briefly, cells were lysed in lysis buffer by incubating the cell suspensions on ice for 30 minutes. Samples were microcentrifuged at 15,000 rpm for 15 minutes at 4°C. Protein concentrations of cell extracts were measured using the bicinchoninic acid protein assay kit (Pierce, Rockford, IL). Unless specified otherwise, 0.03 μg of protein taken from the cell extracts was incubated at 25°C in the presence of biotin-labeled primers. The telomeric repeats were added onto the ends of the synthetic primers by the active telomerase present in the cell extracts. The synthetic products were further amplified by PCR. The denatured products were allowed to bind to a streptavidin-coated 96-well plate and hybridized to a digoxigenin-labeled, telomeric repeat-specific probe. The biotin-labeled PCR products were detected using peroxidase-conjugated antibodies against digoxigenin and subsequently visualized by adding Tris borate-ethylenediaminetetraacetic acid as the substrates of the peroxidase. The amount of the colored products representing the telomerase activity was measured at 450 nm using an ELISA reader. Telomerase activity was determined in triplicate. Negative controls were prepared by inactivating the telomerase activity present in the cell extracts with high temperature (95°C for 10 minutes) before the incubation step at 25°C.
Cell Cycle and Apoptosis Analysis
Cell cycle distribution was determined by staining with propidium iodide (Sigma) as previously described. 37 Briefly, 1 × 10 6 control or treated cells were pelleted and resuspended in 1.2 ml of propidium iodide solution (50 μg/ml in 0.1% sodium citrate plus 0.1% Triton X-100 (Sigma). All profiles were generated using Becton Dickinson FACScan flow cytometer (Becton Dickinson, San Diego, CA) using Cell Quest software.
Immunoblot Analysis
Cell extracts were prepared as described in the Telomerase Assay section. To each lane, 15 μg proteins were resolved in a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel using a BioRad minigel system. Separated proteins were subsequently transferred onto a nitrocellulose film using a semidry transfer apparatus (BioRad). The following antibodies were used for immunoblot analysis: rabbit polyclonal antibody against hTERT (Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal antibody against p27/Kip1 (Transduction Laboratories, Lexington, KY), mouse monoclonal antibody against p53 or p21/Waf1 (Calbiochem, La Jolla, CA) and rabbit polyclonal antibody against BCL-2 (Santa Cruz Biotechnology). Protein bands were visualized using the ECL kit (Amersham, Arlington Heights, IL).
RT-PCR
Total RNA was extracted using Trizol Reagent (Gibco Life Technologies, Inc.) following the manufacturer’s instructions. First-strand cDNA was synthesized from 2 μg of RNA in 20 μl of reaction solution using a random primer and Superscript II reverse transcriptase reagent (Gibco Life Technologies, Inc.) according to the manufacturer’s instructions.
The hTERT (human telomerase reverse transcriptase) cDNA was amplified using oligonucleotide primers LT5 and LT6. 12 The p27 cDNA was amplified using the hp27-7F (AACGTGCGAGTGTCTAACG) and HP27-234R (CTCTTGCCACTCGTACTTG) primers. The GAPDH cDNA was amplified using a forward primer (CGGAGTCAACGGATTTGGTCG) and a reverse primer (AGCCTTCTCCATGGTGGTGAAGAC) and used as a control. The thermal cycling condition for all PCR was: 94°C for 5 minutes, followed by 30 cycles of 94°C for 50 seconds, 59°C for 50 seconds, and 72°C for 90 seconds. The PCR products were resolved on a 2% agarose gel, stained with ethidium bromide (Sigma). Densitometry was performed using the Molecular Dynamics Imaging system and ImageQuant 3.3 software to quantitate relative amounts of protein detected on Western blots and ethidium bromide-stained gels.
Statistical Analyses
Descriptive statistics (fold increases and percentage) were used to present the distribution of the measured parameters. The SDs of mean values were used to measure the variability of the parameters. The significance of changes in parameters between untreated and treated samples were determined by the Wilcoxon matched-pairs test. A two-tailed α level of 0.05 was used as the criterion for statistical significance.
Results
Determination of Telomerase Activity in Lymphoma Cells Using TRAP-PCR ELISA
To determine conditions under which the TRAP-PCR ELISA assay would yield quantitative data, we used total protein lysates of peripheral blood lymphocytes and three malignant lymphoma cell lines and determined the range over which the telomerase assay products were proportional to the protein concentration. Jurkat and CEM-6 are T-lineage lymphoblastic lymphomas, whereas Raji is a B-lineage malignancy derived from a Burkitt lymphoma. For all cell lines, cell extracts at 0 to 0.05 μg protein per assay yielded a linear increase in telomerase activity. Telomerase activity plateaued at protein levels exceeding 0.05 μg per reaction (Figure 1) ▶ . All subsequent assays therefore used 0.03 μg of protein to ensure quantitative measurement of telomerase activity. Jurkat cells exhibited the highest level of telomerase activity at 0.03 μg [absolute value of 1.22 ± 0.024 optical density (OD) 450 nm] that was the protein concentration that yielded 75% of maximal telomerase activity. Raji cells expressed the lowest telomerase activity with absolute OD 450 nm reading of 0.43 ± 0.013 whereas CEM-6 cells demonstrated an intermediate level of telomerase activity (absolute OD 450 of 0.75 ± 0.015). Control peripheral blood lymphocytes demonstrated a negligible level of telomerase activity (absolute OD 450 of <0.1).
Figure 1.

Telomerase activity at different protein concentration of cell extracts. Values represent the mean ± SEM of triplicate. Cell extracts were from untreated cells in the mid-exponential growth phase. Telomerase activity of lymphoma cells plateaued at and above 0.05 μg of protein per assay. For subsequent assays, 0.03 μg of protein was used per assay.
Anti-Neoplastic Agents and Radiation Treatment Results in Inhibition of Telomerase Activity
To determine whether the level of telomerase activity would be modulated by radiation and chemotherapy and potentially useful in monitoring residual cancer cell survival, we studied the effect of 137Cesium γ-rays and a variety of hematopoietic anti-neoplastic agents on telomerase activity. Preliminary experiments were performed to determine dosage and concentration of anti-neoplastic agents that were effective cytotoxic agents based on previously published studies. 38 Cells growing at late-exponential phase were exposed to 30 Gy of ionizing radiation and incubated at 37°C for up to 72 hours. Cells collected at different time points after irradiation were examined for cell viability and telomerase activity.
Analysis of telomerase activity by TRAP-PCR ELISA after exposure to anti-neoplastic agents showed variable responses in different cell lines. Raji cells demonstrated significant reduction of telomerase activity after cisplatin, VP-16, and vincristine treatment. As early as 24 hours after treatment there was a 51% reduction in telomerase activity after exposure to cisplatin compared to untreated cells (P < 0.0001), a 44% reduction by VP-16 (P < 0.0001), and a 32% reduction by vincristine (<0.001) (Figure 2) ▶ . There was sustained reduction of telomerase activity after vincristine treatment in the Raji cells with maximal inhibition of 80% at 48 hours (<0.0001) whereas other treatments showed transient inhibition with subsequent increases seen at 48 hours. Jurkat and CEM-6 cells showed significant and sustained reduction of telomerase activity after vincristine (82%, P < 0.0001; 93%, P < 0.0001, respectively) and γ-irradiation (82%, P < 0.0001; 93%, P < 0.0001, respectively) at 48 hours. Telomerase activity decreased only minimally by cisplatin and VP-16 in the T lymphoblastic cells. No treatments resulted in an increase in telomerase activity.
Figure 2.
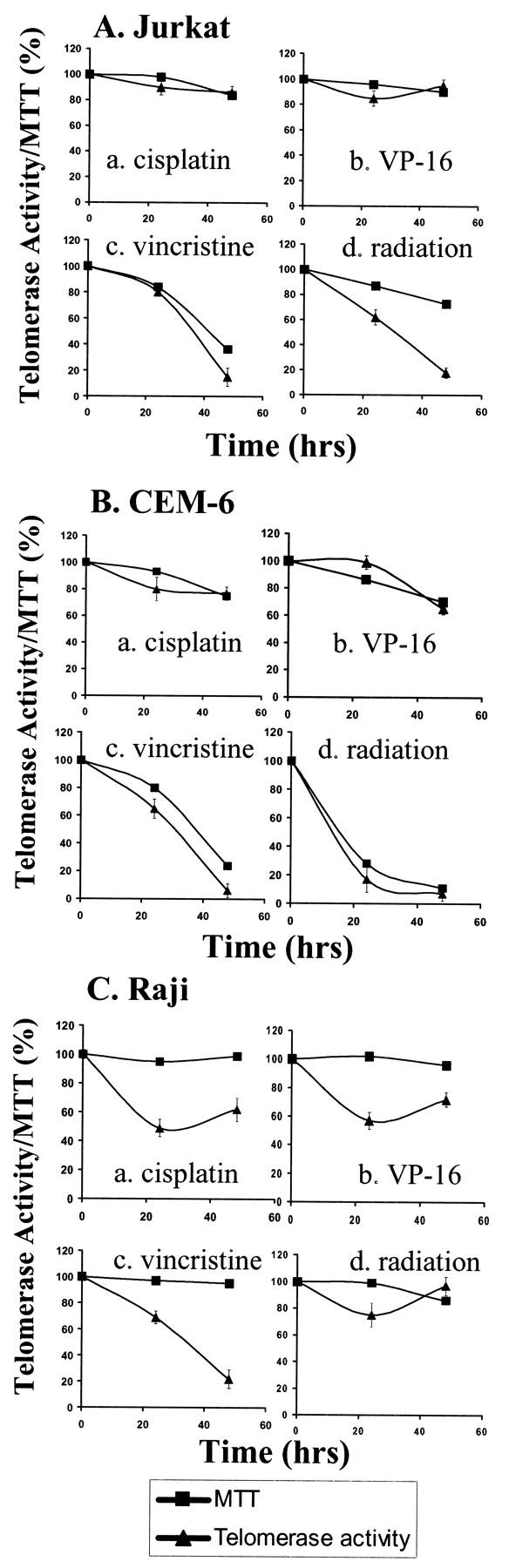
Telomerase activity and cell viability after anti-neoplastic drug treatments. Jurkat (A), CEM-6 (B), and Raji cell viability (MTT) and telomerase activity (TRAP ELISA) (C) after anti-neoplastic drug treatments [0.8 μg/ml cisplatin (a); 0.2 μg/ml VP-16 (b), and 0.1 μg/ml vincristine (c)] and γ-ray irradiation [30 GY (d)]. Each telomerase activity value represents the mean ± SEM of triplicate determinations.
Chemotherapeutic agents and irradiation also had variable effects on cell viability (Figure 2) ▶ . The relationship between viability and decrease in telomerase activity was also variable between cells and specific anti-neoplastic agents. Jurkat and CEM-6 cells that are high-grade T-precursor lymphoblastic lymphoma cells were most sensitive to the effects of vincristine and γ-irradiation. Growth impairments induced by these agents paralleled their effect on suppression of telomerase activity except for CEM-6 cells treated with radiation. Cisplatin and VP-16 had minimal effects on the viability of both Jurkat and CEM-6 cells and had parallel effects on telomerase activity. In contrast, Raji, a B-lineage Burkitt lymphoma cell line was resistant to all agents with no impairment of cell growth at 48 hours (Figure 2C; a, b, c, and d ▶ ) and had no effects even at 96 hours after treatment (data not shown). In contrast to the responses of the Jurkat and CEM-6 cells, there was a dramatic decrease in the telomerase activity by all treatments that was not paralleled with growth impairment.
Effect of Chemotherapeutic Agents and Radiation Treatment on Cell-Cycle Distribution and Apoptosis
The decrease in telomerase activity paralleled cell growth impairment evaluated by trypan blue dye exclusion or by measurement of cell ability to convert tetrazolium salt to colored formazan [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide assay] (MTT) in the T-lymphoblastic lymphoma cells Jurkat and CEM-6. To determine the relationship between the reduction in telomerase activity and cell cycle distribution we performed cell cycle analysis on CEM-6 cells that had been exposed to 30 Gy of irradiation and to cisplatin, vincristine, and VP-16. Figure 3 ▶ and Figure 4 ▶ show the cell cycle distribution at 0 hours, 24 hours, and 72 hours after exposure to anti-neoplastic agents. Cells undergoing apoptosis are identified by the subdiploid peak on staining with propidium iodide. This is attributed to the loss of oligonucleotide size DNA fragments by DNase digestion in the early stages of programmed cell death. Flow cytometric analysis of propidium iodide-stained cells is a well-accepted method for quantitative assessment of apoptosis. 39 As shown in Figure 3A ▶ untreated control cells were composed of 50.6% cells in G0/G1, 16.5% cells in S, and 28% cells in G2/M, whereas only 2.5% of cells are in the subdiploid apoptotic population. Treatment of CEM-6 cells with cisplatin, VP-16, and vincristine resulted in a G2/M arrest followed by apoptosis (Figure 3; B, C, and D ▶ ). Irradiation however resulted in rapid increase of apoptotic cells with >50% cells in the subdiploid peak at 24 hours (Figure 3E) ▶ . CEM-6 cells were most susceptible to vincristine and radiation-mediated apoptosis (Figure 3, D and E) ▶ whereas VP-16 and cisplatin did not significantly increase the number of apoptotic cells even at 72 hours (1.5- and 3.1-fold, respectively).
Figure 3.
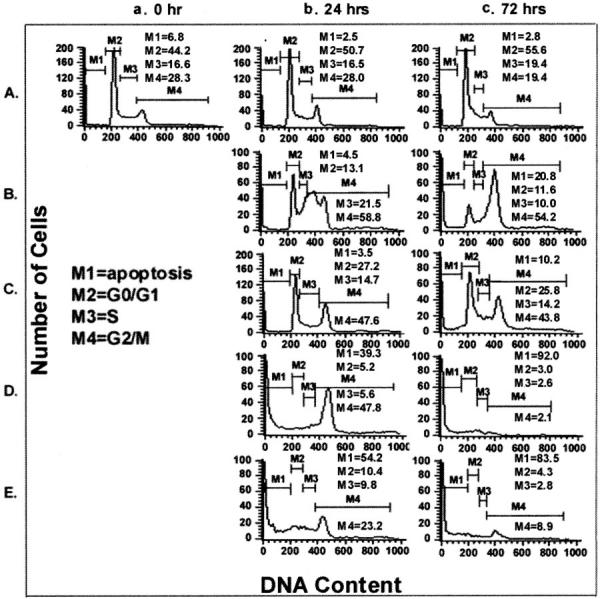
Cell-cycle analysis of CEM-6 cells after various treatments. CEM-6 cells were exposed to various agents as shown in Figure 2 ▶ [untreated controls (A), cisplatin (B), VP-16 (C), vincristine (D), and irradiation (E)]. Cells were collected for analysis at 0 (a), 24 (b), and 72 (c) hours after treatment.
Figure 4.

Time course of cell-cycle distribution changes induced by anti-neoplastic agents and γ-irradiation of CEM-6 cells. CEM-6 cells were exposed to various agents as shown in Figure 2 ▶ [untreated controls (A), cisplatin (B), VP-16 (C), vincristine (D), and irradiation (E)]. Cells were collected for analysis at 0 (a), 24 (b), and 72 (c) hours after treatment.
Effect of Radiation and Chemotherapy on Expression of Telomerase hTERT mRNA and Protein
Reverse transcriptase (RT)-PCR and Western blot analyses were used to determine whether the decrease in telomerase activity correlated with the expression of the hTERT mRNA and its protein product. Surprisingly, all anti-neoplastic agents consistently induced the expression of hTERT protein in all cell lines despite inhibition of telomerase activity. Figure 5 ▶ shows that the level of hTERT protein induction is both cell-type-dependent and time-dependent. All treatments resulted in significant induction of hTERT protein expression in Jurkat cells. The relative density of hTERT protein compared to control untreated cells was induced 4.3-fold by cisplatin, 4.1-fold by VP-16, 5.6-fold by vincristine, and 5.8-fold by radiation at 72 hours. Significant induction of hTERT protein expression was seen in CEM-6 cells by vincristine (5.1-fold) and radiation (3.6-fold) at 72 hours whereas cisplatin and VP-16 failed to induce its expression (1.6-fold and 1.2-fold, respectively). Vincristine and radiation also induced hTERT protein expression by 4.8- and 3.7-fold in Raji cells at 72 hours whereas cisplatin and VP-16 had minimal effects (1.7- and 2.3-fold respectively). Furthermore, RT-PCR analysis of hTERT transcript demonstrated that the induction of hTERT protein observed for the CEM-6 cells correlated with the induction of hTERT mRNA expression by threefold to fourfold by cisplatin and VP16 (Figure 6, A and B) ▶ . By contrast, RT-PCR analysis of p27/Kip1 mRNA expression showed absence of induction in response to these agents. Thus, the inhibition of telomerase activity was seen concurrent with the induction of hTERT mRNA and protein expression.
Figure 5.

Western blot analysis of hTERT, p53, p27/Kip1, and p21/Waf1 in Jurkat, CEM-6, and Raji lymphoma cells after anti-neoplastic drug treatments and γ-irradiation. Jurkat, CEM-6, and Raji lymphoma cells were exposed to the following anti-neoplastic drug treatments (0.8 μg/ml cisplatin, 0.2 μg/ml VP-16, and 0.1 μg/ml vincristine) and γ-irradiation (30 GY) for 0, 24, 48, and 72 hours. Western blot analysis was performed using equal amount of protein obtained from total cell lysates. Cell lysates were the same as those used in the telomerase activity assays.
Figure 6.
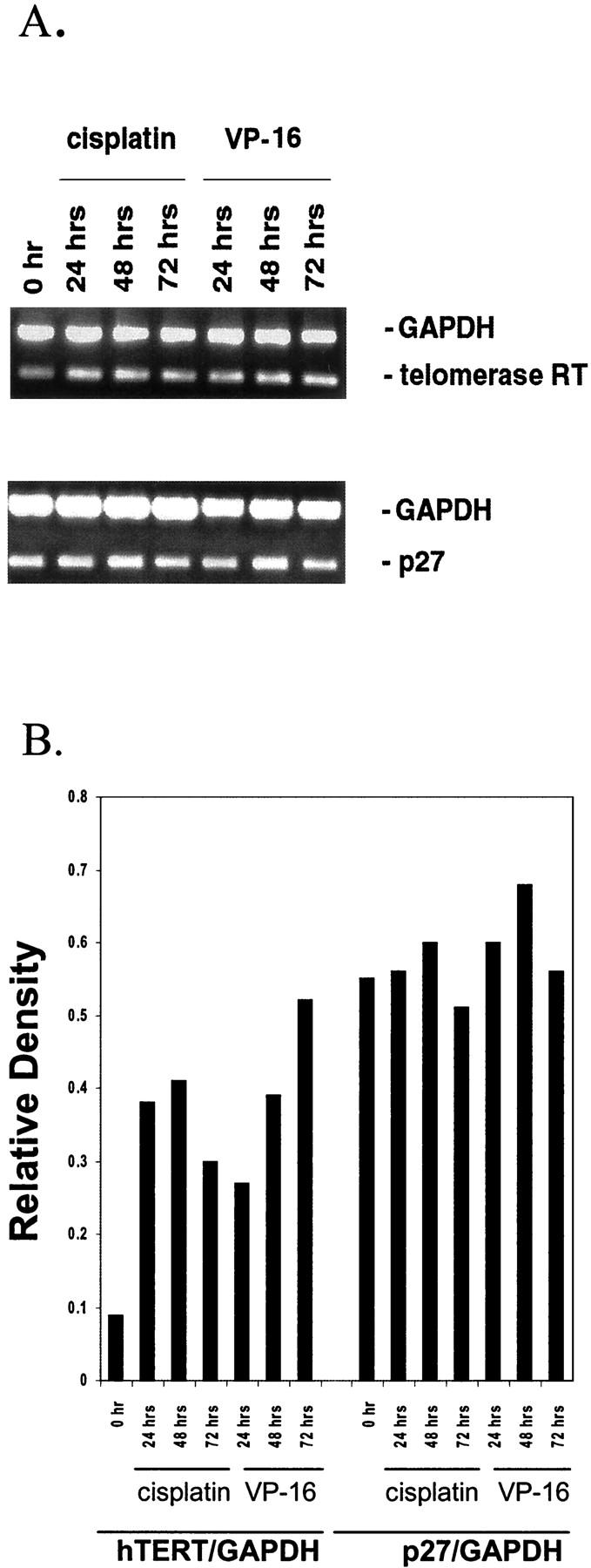
A: Duplex RT-PCR analyses of hTERT and p27/Kip1 mRNA expression in CEM-6 cells after cisplatin and VP-16. Expression of hTERT and p27/Kip1 mRNA were compared to GAPDH in CEM-6 cells treated with cisplatin (0.8 μg/ml) and VP-16 (0.2 μg/ml) as determined by duplex RT-PCR. B: Relative expression of hTERT/GAPDH and p27/Kip1/GAPDH by densitometric analysis of RT-PCR products.
Expression of p53, p27/Kip1, and p21/Waf1 in Lymphoma Cells Treated with Chemotherapy and Radiation
Expression of cell cycle-associated proteins was performed by Western blot analysis of cell lysates after treatment with radiation and chemotherapy. Significant induction of p27/Kip1 protein was seen in cells treated with chemotherapeutic and radiation (Figure 5) ▶ that was detectable as early as 24 hours. Even in Jurkat cells with high basal levels of p27/Kip1 protein there was a 2.8-fold to 4-fold induction by vincristine and cisplatin (Figure 5) ▶ . In contrast to hTERT, RT-PCR analysis of the p27/Kip1 transcript revealed no significant differences in expression in treated cell lines (Figure 6, A and B) ▶ . This is consistent with the notion that p27/Kip1 is regulated at the posttranslational levels by phosphorylation and ubiquitination. 40-42 Induction of p21/Waf 1 was inconsistent except for Raji cells exposed to vincristine (3.9-fold at 24 hours). In all cell lines p53 protein levels were induced most significantly by radiation treatment. As the expression of p53 is regulated predominantly at the posttranslational level, 43,44 levels of mRNA were not determined. Western blotting for BCL-2 demonstrated up-regulation by cisplatin in Jurkat cells and by vincristine in CEM-6 cells (data not shown).
Discussion
Regulation of telomerase activity is a highly controlled process as evidenced by its selective expression in malignant cells and in limited types of somatic cells. Elucidating the mechanism of telomerase activation or repression in vivo is an important step in developing diagnostic and therapeutic applications.
To test whether DNA damaging agents that block cells at specific checkpoints of the cell cycle would have an effect on modulating telomerase activity we studied the effect of these agents and γ-irradiation in three aggressive lymphoma cell lines of both B-cell (Raji) and T-cell (Jurkat and CEM-6) lineage. We found that there was significant reduction of telomerase activity in lymphoma cells exposed to chemotherapeutic agents and radiation. The reduction was demonstrable as early as 24 hours after treatment and was sustained to beyond 72 hours. The down-regulation of telomerase activity in T-lymphoblastic lymphoma cells was specific to vincristine and radiation with no effect with cisplatin and VP-16. In addition, the decrease in telomerase activity was not attributed to decreased numbers of cells as the percentage of viable cells did not decrease with time for most treatment except for CEM-6 cells treated with vincristine and radiation in which the decrease in telomerase activity was directly proportional to effect on cell viability. The cell-type-specific reduction suggests that regulation of telomerase activity in T-lymphoblastic lymphomas may be different from that of Burkitt lymphomas. This may be attributed to additional genetic alterations such as c-myc overexpression secondary to the t(8;14) translocation and/or the oncogenic properties of Epstein-Barr virus that may render these cells more resistant to cytotoxic agents. These results are similar to those reported in HeLa cells, colorectal carcinoma cells, and ataxia-telangiectasia cells 29 where a dose-dependent decrease in telomerase activity was observed in proportion to cell kill and tumor regression after ionizing radiation treatment. Specific cellular context may be important in regulating the telomerase activity in response to irradiation as contradictory data were reported in which low doses of radiation (2 to 10 Gy) resulted in an increase in telomerase activity in certain lymphoma and myeloma cell lines. 45 This therefore may highlight the lymphoma-specific regulation of telomerase activity. Alternatively, lower doses of radiation may induce a different cellular response.
Telomerase activity may also be an indicator of responsiveness to cytotoxic agents. Inhibition of telomerase activity with an anti-sense telomerase expression vector has been shown to not only decrease telomerase activity but also increase susceptibility to cisplatin-induced apoptotic cell death in cisplatin-sensitive malignant glioblastoma cells. Others have shown that cells with higher telomerase activity tended to be more sensitive to 5-FU and cisplatin in human esophageal cancer cells. 46 Our data supports this notion because Jurkat and CEM-6 exhibited a higher basal level of telomerase activity compared to Raji cells. These two cells were more sensitive to vincristine and radiation than Raji cells. These findings suggest a correlation between basal level of telomerase activity and response to anti-neoplastic agents in lymphoma cells. A close link between telomerase activity and factors that regulate cell apoptosis and survival may yet exist.
Telomerase holoenzyme is composed of hTERT, the catalytic subunit of telomerase that is concomitantly expressed with the activation of telomerase during cellular immortalization and tumor progression, 47 and is the rate-limiting enzyme in telomerase activity. In vitro reconstitution of telomerase also shows that hTERT and the telomerase RNA subunit constitute a minimum core structure of telomerase. 48 A positive correlation between telomerase activity and telomere length and a negative relationship between telomerase activity and hTERT expression has been reported. 49 Our data show that telomerase activity is negatively correlated with the hTERT mRNA and protein expression. This is consistent with observations made in human esophageal cancer cells 46 as well as human lymphocytes. 49 Possible explanations for this include presence of hTERT splice variants and inhibitors of telomerase activity that were excluded by Liu and colleagues. 49 We can further speculate that the transcriptional regulation of hTERT may not be sufficient to account for telomerase activity and may include other posttranscriptional factors in the control of the holoenzyme. Time-course analysis of telomerase activity and hTERT protein expression after exposure to chemotherapeutic agents and radiation indicate that whereas significant inhibition of telomerase activity was evident by 24 hours, the induction in hTERT protein expression was not seen until 48 hours. This suggests the possible existence of a feedback mechanism whereby a decrease in telomerase activity results in the induction of telomerase protein. RT-PCR analysis shows that increased hTERT transcript is detectable at 24 hours consistent with the notion that there is transcriptional up-regulation of hTERT in response to chemotherapeutics and radiation treatment. It is accepted that de novo activation of hTERT gene expression is a first rate-limiting step in telomerase activation in cancer. The induction of hTERT mRNA and protein expression seen in our data also suggests the possibility of residual viable cells that are resistant to the cytotoxic agents. Moreover these results suggest that up-regulation of hTERT expression may be a marker of resistance.
We hypothesized that the down-regulation of telomerase activity by DNA-damaging agents may be mediated by cyclin-dependent kinase inhibitors (CDKI) that are induced during cell cycle arrest in G2/M. Our current study shows that the G2/M arrest observed in CEM-6 cells in response to cisplatin, VP-16, and vincristine correlated with the induction of p27/Kip1 protein expression but not p21/Waf1. These data suggest that p21/Waf1 and p27/Kip1 CDK inhibitors are discoordinately regulated in these cells. Although p21/Waf1 and p27/Kip1 share a similar domain involved in cyclin binding and mediate growth arrest when overexpressed, 50 others have shown that there is lack of coordination in the expression and function of CDKIs. There was differential induction of p27/Kip1 protein in a cell type-dependent and in a time-dependent manner. The constitutive expression of p27/Kip1 mRNA as determined by RT-PCR is consistent with the previously established posttranscriptional mode of regulation of this gene. 51 These results suggest that heterogeneity in chemotherapy and radiation-mediated growth effects may be attributed to differential induction of p27/Kip1 expression. Although p21/Waf1 has been implicated in regulation of telomerase activity 19 a similar role for p27/Kip1 has not been reported. Although the up-regulation of p27/Kip1 may reflect an indirect effect of radiation and chemotherapeutic agents on telomerase activity, the role of p27/Kip1 as a modulator of telomerase expression remains to be determined.
p27/Kip1 has been shown to be induced by a number of agents that induce G1 growth arrest including lovastatin, 52 transforming growth factor-β1, 53 after cell-to-cell contact inhibition, rapamycin, and by agents that induce cAMP-mediated growth arrest. 54,55 This is the first report of p27/Kip1 up-regulation in response to γ-irradiation and chemotherapeutic agents. These findings provide support for the central role of p27/Kip1 in mediating growth arrest and apoptosis in lymphoma cells.
Some investigators have reported an inverse relationship between p53 expression and telomerase activity. Induced expression of wild-type p53 in immortalized fibroblasts resulted in the down-regulation of telomerase activity. 56 Adenovirus-mediated p53 gene transduction has also been shown to result in down-regulation of telomerase activity in human pancreatic cancer cells 57 with concomitant G1 cell cycle arrest and apoptosis. In vitro studies have provided evidence for direct molecular interactions between telomerase associated protein 1 (TEP1) and p53. 21 All of the three cell lines in our study express mutant p53 protein. 58-61 Although the induction of p53 protein in response to chemotherapeutic and radiation exposure is correlated with down-regulation of telomerase activity consistent with previously reported observations, 62 there is no conclusive evidence that mutant p53 can regulate telomerase activity.
These data suggest that telomerase activity not only varies among different telomerase-positive cell lines, but also responses to chemotherapeutic agents and radiation exposure vary. The reduction in telomerase activity observed in our in vitro experiments paralleled cell kill by some agents whereas in others it was independent of cytotoxic effect. Determination of telomerase activity during or after treatment with anti-neoplastic agents might be a useful indicator of tumor regression. Our data also suggests the potential relationship between p27/Kip1 and telomerase activity. Whether telomerase activity is regulated by p27/Kip1 as has been suggested for p21/Waf1 19 remains to be determined.
Footnotes
Address reprint requests to Megan S. Lim, MD, Ph.D., FRCP(C), Department of Pathology, University of Utah, Rm A565, 50 North Medical Dr., Salt Lake City, Utah 84132. E-mail: megan.lim@path.utah.edu.
Supported by a grant from the Sunnybrook Trust for Research.
Current address of Zhaosheng Lin: ARUP Laboratories, 500 Chipeta Way, Salt Lake City, UT 84108.
References
- 1.Greider CW, Blackburn EH: Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43:405-413 [DOI] [PubMed] [Google Scholar]
- 2.Blackburn EH: Telomerases. Annu Rev Biochem 1992, 61:113-129 [DOI] [PubMed] [Google Scholar]
- 3.Allsopp RC, Vaziri H, Patterson C, Goldstein S, Youglai EV, Futcher AB, Greider CW, Harley CB: Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA 1992, 89:10114-10118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviellow GM, Wright WE, Weinrich SL, Shay JW: Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266:2011-2015 [DOI] [PubMed] [Google Scholar]
- 5.Broccoli D, Young JW, de Lange T: Telomerase activity in normal and malignant hematopoietic cells. Proc Natl Acad Sci USA 1995, 92:9082-9086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Counter CM, Gupta J, Harley CB, Leber B, Bacchetti S: Telomerase activity in normal leukocytes and in hematological malignancies. Blood 1995, 85:2315-2320 [PubMed] [Google Scholar]
- 7.Langford LA, Piatyszek MA, Xu R, Schold SC, Jr, Shay JW: Telomerase activity in human brain tumours. Lancet 1995, 346:1267-1268 [DOI] [PubMed] [Google Scholar]
- 8.Hiyama E, Yokoyama T, Tatsumoto N, Hiyama K, Imamura Y, Murakami Y, Kodama T, Piatyszek M, Shay JW, Matsurra Y: Telomerase activity in gastric cancer. Cancer Res 1995, 55:3258-3262 [PubMed] [Google Scholar]
- 9.Hiyama E, Hiyama K, Tatsumoto N, Kodama T, Shay JW, Yokoyama T: Telomerase activity in human intestine. Int J Oncol 1996, 9:453-458 [DOI] [PubMed] [Google Scholar]
- 10.Hiyama E, Gollahon L, Kataoka T, Kuroi K, Yokoyama T, Gazdar AF, Hiyama K, Piatyszek MA, Shay JW: Telomerase activity in human breast tumors. J Natl Cancer Inst 1996, 88:116-122 [DOI] [PubMed] [Google Scholar]
- 11.Lin Y, Miyamoto N, Fujinami K, Uemura H, Hosaka M, Iwasaki Y, Kubota Y: Telomerase activity in human bladder cancer. Clin Cancer Res 1996, 2:929-932 [PubMed] [Google Scholar]
- 12.Nakamura TM, Morin GB, Chapman B, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR: Telomerase catalytic subunit homologs from fission yeast and humans. Science 1997, 277:955-959 [DOI] [PubMed] [Google Scholar]
- 13.Harrington L, McPhail T, Mar V, Zhou W, Oulton R, Bass MB, Arruda I, Robinson MO: A mammalian telomerase-associated protein. Science 1997, 275:973-977 [DOI] [PubMed] [Google Scholar]
- 14.LaBranche H, Dupuis S, Ben-David Y, Bani MR, Wellinger RJ, Chabot B: Telomere elongation by hnRNP A1 and a derivative that interacts with telomeric repeats and telomerase. Nat Genet 1998, 19:199-202 [DOI] [PubMed] [Google Scholar]
- 15.Blasco MA, Rizen M, Greider CW, Hanahan D: Differential regulation of telomerase activity and telomerase RNA during multi-stage tumorigenesis. Nat Genet 1996, 12:200-204 [DOI] [PubMed] [Google Scholar]
- 16.Kilian A, Bowtell DDL, Abud HE, Hime GR, Venter DJ, Keese PK, Duncan EL, Reddel RR, Jefferson RA: Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet 1997, 6:2011-2019 [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Xie LY, Allan S, Beach D, Hannon GJ: Myc activates telomerase. Genes Dev 1998, 12:1769-1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandal M, Kumar R: Bcl-2 modulates telomerase activity. J Biol Chem 1997, 272:14183-14187 [DOI] [PubMed] [Google Scholar]
- 19.Terasaki M, Kato S, Sugita Y, Shigemori M, Morimatsu M: Adenovirus-mediated over-expression of p21Waf-1 differentially affects the proliferation, telomerase activity, and expression of glial fibrillary acidic protein in human glioma cells. Int J Oncol 1999, 14:63-69 [DOI] [PubMed] [Google Scholar]
- 20.Xu H-J, Shou Y, Ji W, Pereng G-S, Kruzelock R, Kong C-T, Bast RC, Mills GB, Li J, Hu S-X: Reexpression of the retinoblastoma protein in tumor cells induces senescence and telomerase inhibition. Oncogene 1997, 15:2589-2596 [DOI] [PubMed] [Google Scholar]
- 21.Li H, Cao Y, Berndt MC, Funder JW, Liu J-P: Molecular interactions between telomerase and the tumor suppressor protein p53 in vitro. Oncogene 1999, 18:6785-6794 [DOI] [PubMed] [Google Scholar]
- 22.Ku WC, Cheng AJ, Wang TC: Inhibition of telomerase activity by PKC inhibitors in human nasopharyngeal cancer cells in culture. Biochem Biophys Res Commun 1997, 241:730-736 [DOI] [PubMed] [Google Scholar]
- 23.Kang SS, Kwon T, Kwon DY, Do SI: Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem 1999, 274:13085-13090 [DOI] [PubMed] [Google Scholar]
- 24.Li H, Zhao LL, Funder JW, Liu JP: Protein phosphatase 2A inhibits nuclear telomerase activity in human breast cancer cells. J Biol Chem 1997, 272:16729-16732 [DOI] [PubMed] [Google Scholar]
- 25.Sharma HW, Sokoloski JA, Perez JR, Maltese JY, Sartorelli AC, Stein CA, Nichols G, Khaled Z, Telang NT, Narayanan R: Differentiation of immortal cells inhibits telomerase activity. Proc Natl Acad Sci USA 1995, 92:12343-12346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albanell J, Han W, Mellado B, Gunawardane R, Scher HI, Dmitrovsky E, Moore MAS: Telomerase activity is repressed during differentiation of maturation-sensitive but not resistant human tumor cell lines. Cancer Res 1996, 56:1503-1508 [PubMed] [Google Scholar]
- 27.Reichman TW, Albanell J, Wang X, Moore MAS, Studzinski GP: Downregulation of telomerase activity in HL60 cells by differentiating agents is accompanied by increased expression of telomerase-associated protein. J Cell Biochem 1997, 67:13-23 [PubMed] [Google Scholar]
- 28.Faraoni I, Turriziani M, Masci G, De Vecchis L, Shay JW, Bonmassar E, Graziani G: Decline in telomerase activity as a measure of tumor cell killing by antineoplastic agents in vitro. Clin Cancer Res 1997, 3:579-585 [PubMed] [Google Scholar]
- 29.Sawant SG, Gregoire V, Dhar S, Umbricht CB, Cvilic S, Sukumar S, Pandita TK: Telomerase activity as a measure for monitoring radiocurability of tumor cells. FASEB J 1999, 13:1047-1054 [DOI] [PubMed] [Google Scholar]
- 30.Norrback K-F, Dahlenborg K, Carlssson R, Roos G: Telomerase activation in normal B lymphocytes and non-Hodgkin’s lymphomas. Blood 1996, 88:222-229 [PubMed] [Google Scholar]
- 31.Leber BBS: Telomeres and telomerase in normal and malignant haematologic cells. Leuk Lymphoma 1996, 24:1-9 [DOI] [PubMed] [Google Scholar]
- 32.Bodnar AG, Kim NW, Effros RB, Chiu C-P: Mechanism of telomerase induction during T cell activation. Exp Cell Res 1996, 228:58-64 [DOI] [PubMed] [Google Scholar]
- 33.Igarashi H, Sakaguchi N: Telomerase activity is induced in human peripheral B lymphocytes by the stimulation to antigen receptor. Blood 1997, 89:1299-1307 [PubMed] [Google Scholar]
- 34.Weng N-P, Levine BL, June CH, Hodes RJ: Regulation of telomerase RNA template expression in human T lymphocyte development and activation. J Immunol 1997, 158:3215-3220 [PubMed] [Google Scholar]
- 35.Weng NP, Levine BL, June CH, Hodes RJ: Regulated expression of telomerase activity in human T lymphocyte development and activation. J Exp Med 1996, 183:2471-2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brousset P, al Saati T, Chaouche N, Zenou RC, Schlaifer D, Chittal S, G D: Telomerase activity in reactive and neoplastic lymphoid tissues: infrequent detection of activity in Hodgkin’s disease. Blood 1997, 89:26–31 [PubMed]
- 37.Kumar R, Atlas I: Interferon alpha induces the expression of retinoblastoma gene product in human Burkitt lymphoma Daudi cells: role in growth regulation. Proc Natl Acad Sci USA 1992, 89:6599-6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piovesan B, Pennell N, Berinstein NL: Human lymphoblastoid cell lines expressing mutant p53 exhibit decreased sensitivity to cisplatin-induced cytotoxicity. Oncogene 1998, 17:2339-2350 [DOI] [PubMed] [Google Scholar]
- 39.Zinzani PL, Tosi P, Visani G, Martinelli G, Farabegoli P, Buzzi MEO, Salvucci M, Bendandi M, Zaccaria A: Apoptosis induction with three nucleoside analogs on freshly isolated B-chronic lymphocytic leukemia cells. Am J Hematol 1994, 47:301-306 [DOI] [PubMed] [Google Scholar]
- 40.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M: Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995, 269:682-685 [DOI] [PubMed] [Google Scholar]
- 41.Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE: Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 1997, 1:1464-1478 [DOI] [PubMed] [Google Scholar]
- 42.Vlach J, Hennecke S, Amati B: Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J 1997, 16:5334-5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaulsky G, Goldfinger N, Tosky MS, Levine AJ, Rotter V: Nuclear localization is essential for the activity of p53 protein. Oncogene 1991, 6:2055-2065 [PubMed] [Google Scholar]
- 44.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW: Participation of p53 protein in the cellular response to DNA damage. Cancer Res 1991, 51:6304-6311 [PubMed] [Google Scholar]
- 45.Terashima M, Ogawa Y, Toda K, Nishioka A, Inomata T, Kubonishi I, Taguchi H, Yoshida S, Shizuta Y: Effects of irradiation on telomerase activity in human lymphoma and myeloma cell lines. Int J Mol Med 1998, 2:567-571 [DOI] [PubMed] [Google Scholar]
- 46.Asai A, Kiyozuka Y, Yoshida R, Fujii T, Hioki K, Tsubura A: Telomere length, telomerase activity and telomerase RNA expression in human esophageal cancer cells: correlation with cell proliferation, differentiation and chemosensitivity to anticancer drugs. Anticancer Res 1998, 18:1465-1472 [PubMed] [Google Scholar]
- 47.Harrington L, Zhou W, McPhail T, Oulton R, Yeung DSK, Mar V, Bass MB, Robinson MO: Human telomerase contains evolutionary conserved catalytic and structural subunits. Genes Dev 1997, 11:3109-3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beattie TL, Zhou W, Robinson MO, Harrington L: Reconstitution of human telomerase in vitro. Curr Biol 1998, 8:177-180 [DOI] [PubMed] [Google Scholar]
- 49.Liu K, Schoonmaker MM, Levine BL, June CH, Hodes RJ, Went N: Constitutive and regulated expression of telomerase reverse transcriptase (hTERT) in human lymphocytes. Proc Natl Acad Sci USA 1999, 96:5147-5152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yaroslavskiy B, Watskins S, Donnenberg AD, Patton TJ, Steinman RA: Subcellular and cell-cycle expression profiles of cdk-inhibitors in normal differentiating myeloid cells. Blood 1999, 93:2907-2917 [PubMed] [Google Scholar]
- 51.Hengst L, Reed SI: Translational control of p27Kip1 accumulation during the cell cycle. Science 1996, 271:1861-1864 [DOI] [PubMed] [Google Scholar]
- 52.Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI: A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci USA 1994, 91:5291-5295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A: p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Cell 1994, 78:59-66 [DOI] [PubMed] [Google Scholar]
- 54.Kato JY, Matsuoka M, Polyak K, Massague J, Sherr CJ: Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase 4 activation. Cell 1994, 79:487-496 [DOI] [PubMed] [Google Scholar]
- 55.Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH, Massague J, Crabtree GR, Roberts JM: Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature 1994, 372:570-573 [DOI] [PubMed] [Google Scholar]
- 56.Gollahon LS, Kraus E, Wu TA, Yim SO, Strong LC, Shay JW, Tainsky MA: Telomerase activity during spontaneous immortalization of Li-Fraumeni syndrome skin fibroblasts. Oncogene 1998, 17:709-717 [DOI] [PubMed] [Google Scholar]
- 57.Kusumoto M, Ogawa T, Mizumoto K, Ueno H, Niiyama H, Sato N, Nakamura M, Tanaka M: Adenovirus-mediated p53 gene transduction inhibits telomerase activity independent of its effects on cell cycle arrest and apoptosis in human pancreatic cancer cells. Clin Cancer Res 1999, 5:2140-2147 [PubMed] [Google Scholar]
- 58.Cherney BW, Bhatia KG, Sgadari C, Gutierrez MI, Mostowski H, Pike SE, Gupta G, Magrath IT, Tosato G: Role of the p53 tumor suppressor gene in the tumorigenicity of Burkitt’s lymphoma cells. Cancer Res 1997, 57:2508-2515 [PubMed] [Google Scholar]
- 59.Vigorito E, Plaza S, Mir L, Mongay L, Vinas O, Serra-Pages C, Vives J: Contributions of p53 and PMA to gamma-irradiation induced apoptosis in Jurkat cells. Hematol Cell Ther 1999, 41:153-161 [DOI] [PubMed] [Google Scholar]
- 60.Morgan SE, Kim R, Wang PC, Bhat UG, Kusumoto H, Lu T, Beck WT: Differences in mutant p53 protein stability and functional activity in teniposide-sensitive and -resistant human leukemic CEM cells. Oncogene 2000, 19:5010-5019 [DOI] [PubMed] [Google Scholar]
- 61.Naujokat C, Sezer O, Zinke H, Leclere A, Hauptmann S, Possinger K: Proteasome inhibitors induced caspase-dependent apoptosis and accumulation of p21WAF1/Cip1 in human immature leukemic cells. Eur J Haematol 2000, 65:221-236 [DOI] [PubMed] [Google Scholar]
- 62.Zhang N, Song Q, Lu H, Lavin MF: Induction of p53 and increased sensitivity to cisplatin in ataxia-telangiectasia cells. Oncogene 1996, 13:655-659 [PubMed] [Google Scholar]