

Abstract
α-Synuclein (α-SYN) is deposited in intraneuronal cytoplasmic inclusions (Lewy bodies, LBs) characteristic for Parkinson’s disease (PD) and LB dementias. α-SYN forms LB-like fibrils in vitro, in contrast to its homologue β-SYN. Here we have investigated the solubility of SYNs in human LB diseases and in transgenic mice expressing human wild-type and PD-associated mutant [A30P]α-SYN driven by the brain neuron-specific promoter, Thy1. Distinct α-SYN species were detected in the detergent-insoluble fractions from brains of patients with PD, dementia with LBs, and neurodegeneration with brain iron accumulation type 1 (formerly known as Hallervorden-Spatz disease). Using the same extraction method, detergent-insolubility of human α-SYN was observed in brains of transgenic mice. In contrast, neither endogenous mouse α-SYN nor β-SYN were detected in detergent-insoluble fractions from transgenic mouse brains. The nonamyloidogenic β-SYN was incapable of forming insoluble fibrils because amino acids 73 to 83 in the central region of α-SYN are absent in β-SYN. In conclusion, the specific accumulation of detergent-insoluble α-SYN in transgenic mice recapitulates a pivotal feature of human LB diseases.
α-Synuclein (α-SYN) has been identified as the precursor protein of a nonamyloid β-protein component (NAC) isolated from Alzheimer’s disease plaques. 1 α-SYN was detected immunohistochemically in Lewy bodies (LBs) and Lewy neurites that characterize Parkinson’s disease (PD), LB dementia (DLB), LB variant Alzheimer’s disease, 2-7 and neurodegeneration with brain iron accumulation type 1 (NBIA1). 8-11 Antibodies directed against both N-terminal and C-terminal epitopes recognized LB filaments, 12,13 and the presence of full-length α-SYN was biochemically proven on Western blots of isolated LBs. 6 Moreover, full-length α-SYN is the major fibrillar component of glial cytoplasmic inclusions in multiple system atrophy. 14
The formation of LB-like fibrils is an intrinsic property of α-SYN. Purified recombinant α-SYN, but not β-SYN, aggregated in vitro to amyloid fibrils resembling those extracted from LBs. 15-19 PD risk factors, namely α-SYN mutations 20-22 and oxidative stress, 23 accelerated α-SYN aggregation. The causal relationship between α-SYN fibrillization and PD are therefore subject to intense research. 24
Transgenic animals expressing human wild-type [wt]- as well as PD-associated mutant [A53T]α-SYN 25 and [A30P]α-SYN 26 were recently presented. Wild-type and mutant α-SYN assembled into LB-like fibrils in transgenic Drosophila, and a locomotor deficit became apparent with increasing age. 27 Somal and neuritic accumulations of wt and mutant α-SYN were observed in transgenic mouse brain. 28-30 Ubiquitination was occasionally detected, but the α-SYN accumulations did not meet ultrastructural criteria of LBs. 28,29 Masliah and colleagues 28 reported a modest reduction of locomotor performance and van der Putten and colleagues 29 found that age-dependent degeneration of neuromuscular junctions caused a severe locomotor deficit and premature death in their mice.
α-SYN and β-SYN have both been found in the synaptosomal fractions of rodent and human brain. 30-34 Synaptosomal α-SYN was released into the soluble fraction of human brain biopsies. 30 We now report that synaptosomal α-SYN was recovered from the particulate fraction in the case of frozen post mortem brain samples whereas β-SYN was released into the soluble synaptosomal fraction even from archived brain samples. To directly measure SYN solubility, differential detergent extractions were performed. Most of the α-SYN was highly soluble in aqueous buffer and the remainder easily extractable with sodium dodecyl sulfate (SDS). However, detergent-insoluble α-SYN monomers and aggregates were detected in urea extracts from LB disease patient brains, but not in controls. Likewise, some of the human α-SYN was detergent-insoluble in transgenic mouse brains, in sharp contrast to the endogenous mouse α-SYN and β-SYN. The nonamyloidogenic β-SYN failed to form aggregates in vitro because of the lack of amino acids 73 to 83 in the NAC domain. In conclusion, transgenic expression of human α-SYN in mouse brain neurons recapitulates an important aspect of human LB diseases, namely the accumulation of insoluble α-SYN.
Materials and Methods
Antibodies
Rat monoclonal anti-α-SYN 15G7 11,30 and mouse monoclonal anti-synaptophysin SY38 hybridoma supernatants were used as described previously. 30 Mouse monoclonal anti-α-SYN LB509 and Syn102 were described before. 6 The mouse monoclonal anti-α-SYN MC42 (working dilution, 1:1000) was purchased from Transduction Laboratories (Lexington, KY), and the rabbit polyclonal anti-α-SYN antiserum 3400 (working dilution, 1:20,000) from Affiniti (Mamhead, UK). Mouse-specific anti-α-SYN antiserum 7544 and anti-β-SYN antiserum 6485 have been described previously. 30 The rabbit polyclonal anti-NAC antiserum 7 was used at a working dilution of 1:1000. Mouse monoclonal anti-ubiquitin Ubi-1 (working dilution, 1 μg/ml) was purchased from Zymed (South San Francisco, CA). Goat anti-rat IgG peroxidase conjugate (working dilution, 1:1000) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG (working dilution, 1:5000) from Sigma (St. Louis, MO).
Brain Fractionation and Western Blotting
Subcellular fractionation of archived human cerebral cortex samples was performed as previously described for fresh tissue. 30 The detergent extraction method of Culvenor and colleagues 35 was applied to human and mouse brain with slight modifications. Approximately 0.5 g of brain tissue was homogenized in 10 volumes of TBS+ (Tris-buffered saline plus Complete protease inhibitor cocktail; Roche Diagnostics, Mannheim, Germany) and sonicated. After 5 minutes of centrifugation at 1000 × g, the supernatants were ultracentrifuged for 1 hour at 130,000 × g. The resulting supernatants represented the buffer-soluble fractions. The pellets were rinsed twice with TBS+ and extracted with 500 μl of 5% SDS in TBS+. All subsequent steps were performed at 24°C. After ultracentrifugation for 30 minutes at 130,000 × g the pellets were re-extracted twice with 5% SDS, and the detergent-soluble supernatants were collected. The bicucullinic acid (BCA) protein assay (Pierce, Rockford, IL) revealed concentrations >1 mg/ml in the first two SDS supernatants that were pooled. The extensively washed detergent-insoluble pellets were squashed in 100 μl of 8 mol/L urea/5% SDS in TBS+ and incubated for at least 10 minutes at room temperature. Then, 80 μl of the resulting suspension were mixed with 20 μl of trichloroacetic acid (TCA) (100%) and allowed to precipitate overnight at 4°C. Protein precipitates were collected by centrifugation, washed with acetone, and resuspended in protein gel-loading buffer containing 6 mol/L urea.
Denaturing polyacrylamide gel electrophoresis (PAGE), Western blotting, and probing were done as described previously. 30 Equal loading was verified by Coomassie blue staining of the gels after transfer. Enhanced chemiluminescence was generated with SuperSignal (Pierce) or ECLplus (Amersham Pharmacia, Little Chalfont, UK). Human-specific 15G7 band intensities in 25-μg mouse brain cytosol samples were determined relative to 5 ng, 10 ng, 20 ng, and 40 ng recombinant human α-SYN (see below) on the same blot. Mouse-specific 7544 band intensities in 100-μg mouse brain cytosol samples were determined relative to four standards of 75- to 500-ng recombinant mouse α-SYN (see below) on the same blot. Band intensities from densitometric scans were quantified using NIH Image v1.62 freeware (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image). Linear regression of the α-SYN standard band intensities revealed correlation coefficients between 0.9 and 1.0.
Generation and Characterization of Transgenic Mice
The amplified human [wt]α-SYN-coding sequence was subcloned into the XhoI site of the Thy1 cassette of pTSC21k, and the NotI-linearized DNA was used to generate transgenic C57BL/6 mice as described for [A30P] α-SYN. 30 Five founders stably transmitted the transgene, as determined by tail biopsy polymerase chain reaction.
Transgene copy number was determined by Southern blotting using as references known amounts of transgene fragment mixed to genomic DNA isolated from nontransgenic littermates. Ten μg of genomic XbaI-KpnI restriction fragments were fractionated by gel electrophoresis and blotted onto Nylon membranes (Roche Molecular Biosciences). A 1.6-kb DNA probe (HindIII-EcoRV fragment of the transgene) was labeled with [33P]dCTP by the random primer method using the Ready-to-Go DNA labeling kit (Pharmacia Biotech). Hybridization was performed overnight at 65°C in 6× standard saline citrate, 10% dextran sulfate, 0.5% SDS. Blots were washed in 2× standard saline citrate (+0.1% SDS) at 65°C for 20 minutes followed by a second wash for 20 minutes at 65°C in 0.2× standard saline citrate (+0.1% SDS). The intensity of the bands was quantified using a phosphorimager scanner. Northern blotting using oligonucleotide probes specific for mRNA of the human α-SYN transgene and mouse β-actin was performed as described previously. 30
Fresh mouse brains were fixed in phosphate-buffered 4% paraformaldehyde and embedded in paraffin. Immunocytochemical detection of SYNs was performed as described previously. 30
Expression and in Vitro Aggregation of Recombinant SYNs
The β-SYN expression vector has been described by Jakes and colleagues. 36 The mouse α-SYN 37 coding region was amplified from whole brain RNA (High Pure RNA Isolation Kit; Boehringer Mannheim, Mannheim, Germany) by reverse transcriptase-polymerase chain reaction using outer mouse primers (5′-GGAATTCCATATGGATGTGTTCATGAAAGG-3′ and 5′-GGAAT-TCCATATGTTAGGCTTCAGGCTCAT-3′). The coding region of human α-SYN 38 was amplified by polymerase chain reaction with outer human primers (5′-TTCATTACATATGGATGTATTCATGAAAGG-3′ and 5′-GGAATTCCATATGTTAGGCTTCAGGTTCGTAG-3′). Codons 73 to 83 of α-SYN were deleted by 4-primer polymerase chain reaction using outer human primers and inner mutagenesis primers 5′-GGAGGAGCAGTGGTGACGGGAGCAGGGAGC-3′ and 5′-GCTCCCTGCTCCCGTCACCACTGCTCCTCC-3′. Amplimers were subcloned into the NdeI site of pET-5a (Promega, Madison, WI), and constructs used to transform Escherichia coli BL21(DE3) pLys. All constructs were sequenced (Medigenomix, Martinsried, Germany).
Bacterial cultures were induced with isopropyl-β-d-thiogalactoside for 4 hours, and lysed by freeze/thaw and sonication. After 15 minutes of boiling, the heat-stable 17,000 × g supernatant was loaded onto Q-Sepharose (Pharmacia, Uppsala, Sweden) and eluted with a 25-mmol/L to 500-mmol/L salt gradient. The pooled SYN peak fractions were desalted by Sephacryl S-200 (Pharmacia) gel filtration.
Characteristic electron-dense fibrils (data not shown) were formed after 7 days of incubation of 2 mg/ml of purified recombinant SYN proteins in 50 mmol/L HEPES or phosphate (pH 6.9) at 37°C under constant agitation. Aggregates were collected by 100,000 × g centrifugation, and subjected to the detergent extraction protocol described above.
Results
Recovery of α-SYN from the Particulate Synaptosomal Lysate Fraction Post Mortem
Previous subcellular fractionation experiments with human brain have demonstrated the presence of α-SYN in synaptosomes. 30,34 In accord with results from rapidly processed rodent brain, 30,33 α-SYN was released into the soluble fraction on hypotonic lysis of the synaptosomes prepared from human biopsy brain. 30 Using archived cortical tissue, Irizarry and colleagues 34 have found a significant portion of α-SYN in the particulate fraction of lysed synaptosomes. Indeed, when subcellular fractionations (Figure 1) ▶ were performed with frozen tissue, recovery of α-SYN from the soluble synaptosomal fraction (LS2) was decreased and instead a significant portion of α-SYN was detected in the particulate synaptosomal fraction (LP2). Interestingly, the subcellular fractionation profiles of β-SYN as well as of synaptophysin were the same as previously reported for rapidly processed human biopsy samples. 30 Thus, there seems to be a shift of α-SYN but not β-SYN into the pelletable synaptosomal fraction post mortem. This effect could be either because of altered membrane affinity and/or decreased solubility of α-SYN.
Figure 1.

Subcellular fractionation of frozen human brain tissue. A: Schematic representation of the subcellular fractionation steps, annotations in B correspond to the fractions outlined in A. B: Parietal cortex (0.4 g) from a human control individual was homogenized and subjected to subcellular fractionation. Twenty μg of each fraction was TCA precipitated (except postnuclear fraction S1 that was loaded directly) and subjected to denaturing 12.5% PAGE. Sequential Western probing was done with 15G7 anti-α-SYN, SY38 anti-synaptophysin (SPH), and 6584 anti-β-SYN, as indicated. Data are representative for three different control cortex fractionations.
Detergent-Insoluble α-SYN in LB Disease Brain
Sequential detergent extraction methods have been successfully used to detect α-SYN in brains of patients with α-synucleinopathies. 6,10,35,39 We have adapted the method of Culvenor and colleagues 35 (Figure 2A) ▶ to detect insoluble α-SYN molecules in human LB disease brain and in transgenic mice expressing human α-SYN.
Figure 2.

SDS-insoluble α-SYN species in LB diseases. A: Schematic representation of the differential extraction steps. See Materials and Methods for details. B: Structure of α-SYN and epitopes recognized by anti-α-SYN antibodies. The imperfect KTKEGV repeats are numbered, the sixth repeat missing in β-SYN (see below) is stippled. C: Extracts from temporal cortex of control and DLB brain were prepared, and 10 μg of TBS-soluble material, 10 μg of SDS-soluble material, and 10 μl of urea extracts or TCA precipitates from 50-μl urea extracts (as indicated at the bottom) were subjected to denaturing 12.5% PAGE. Western blots were sequentially probed with three different antibodies against α-SYN (15G7, 3400, MC42) and anti-ubiquitin (Ubi-1), as indicated on the top. Immunoreactivity was visualized with SuperSignal (for 15G7) or ECLplus. D: TCA precipitates from 50-μl urea extracts from temporal cortex of control and DLB brain (left) and from parietal cortex of control and PD brain (right) were loaded on 10 to 20% Tris-tricine gels. The corresponding Western blots were probed with anti-NAC and developed with ECLplus. E: Parietal cortex samples from two controls and two NBIA1 patients were extracted in parallel. TBS-soluble material (10 μg, left), SDS-soluble material (25 μg, middle), and urea extracts (80 μl, right) were separated by SDS-PAGE (TBS-soluble, 15%; SDS and urea extracts, 4 to 20% gradient). MC42 and ECLplus were used for Western detection of α-SYN. Note that sample NBIA1 no. 2 with higher LB density than NBIA1 no. 1 had also much stronger α-SYN immunoreactivity in the urea extract. Nevertheless, the α-SYN immunoreactive band pattern of NBIA1 no. 1 was qualitatively the same as of NBIA1 no. 2, as evidenced by a longer exposure of the blot to the far right. Each experiment is representative for two to three independent extractions. Positions of prestained molecular weight standards are indicated to the left. See text for description of α-SYN species denoted to the right.
Buffer- and detergent-soluble monomeric α-SYN was detected in brains from human controls as well as from LB disease patients (Figure 2) ▶ . In contrast to control brain urea extracts that were virtually devoid of α-SYN, strong immunoreactivity was found in urea extracts from PD and DLB patients (Figure 2, C and D) ▶ . The detergent-insoluble α-SYN was characterized using four different antibodies raised against distinct epitopes (Figure 2B) ▶ . Monomeric α-SYN migrated as a 16- to 19-kd band [(α-SYN)1]. Anti-NAC, but not C-terminal antibodies detected a previously unrecognized α-SYN species with slightly retarded electrophoretic motility (α-SYN)p17) (Figure 2D) ▶ . This band was unlikely to be cross-reactive β-SYN, because specific anti-β-SYN did not reveal any signal in urea extracts (data not shown). In addition, all four anti-α-SYN antibodies recognized ∼40- to 45-kd double bands [(α-SYN)2; consistent with α-SYN dimers and/or a recently described membrane-bound form of α-SYN 40 ], multiple bands in the 60- to 80-kd range [(α-SYN)n; putative α-SYN oligomers), and higher molecular weight aggregates (Figure 2) ▶ . Interestingly, an ∼25-kd band [(α-SYN)p25] was consistently observed in urea extracts from LB disease patients (Figure 2) ▶ . This band was reminiscent of an O-glycosylated form of α-SYN recently described by Shimura and colleagues. 41 Consistent with this notion was the absence of ubiquitin immunoreactivity of (α-SYN)p25 (Figure 2C) ▶ , indicating that this species did not correspond to mono-ubiquitinated α-SYN.
A similar differential extraction pattern for α-SYN was found for another LB disease. NBIA1 is a neurodegenerative synucleinopathy formerly known as Hallervorden-Spatz disease that is characterized by α-SYN inclusions similar to LBs as well as by axonal spheroids that contain immunoreactive α-, β-, and γ-SYN. 8-11 We have analyzed brain samples from two NBIA1 patients described elsewhere (case no. 1, 42 case no. 2 10 ). Monomeric α-SYN was detected in the buffer-soluble and detergent-soluble fractions. The decrease in the relative amount of buffer-soluble α-SYN in both NBIA1 samples (Figure 2E) ▶ may be indicative of defective synaptic integrity in these patients. 10 Alternatively, the soluble pool of α-SYN might be depleted as α-SYN monomers and aggregates accumulated in the detergent-insoluble fraction (Figure 2E) ▶ . Limited N-terminal degradation to a 14-kd band ([ΔN]α-SYN) was occasionally noted, but the bulk of insoluble α-SYN was full-length protein. Higher molecular weight α-SYN bands were observed even in the SDS fractions from brain of NBIA1 case no. 2, who also had much more intense α-SYN immunoreactivity in the urea extracts than NBIA1 case no. 1 (Figure 2E) ▶ . In fact, there was a positive correlation between the α-SYN immunoreactivity in urea extracts and LB density in the two NBIA1 patients and the three DLB patients (Table 1) ▶ .
Table 1.
The Amount of Insoluble α-SYN Correlates with Severity of LB Diseases
| Brain sample | Insoluble α-SYN | LB count |
|---|---|---|
| Temporal cortex | ||
| CON no. 1 | − | − |
| CON no. 2 | − | − |
| CON no. 3 | − | − |
| DLB no. 1 | ++ | 22.8/mm2 |
| DLB no. 2 | + | 6.0/mm2 |
| DLB no. 3 | + | 6.6/mm2 |
| Parietal cortex | ||
| CON no. 4 | − | − |
| CON no. 5 | − | − |
| NBIA1 no. 1 | + | 5.3/mm2 |
| NBIA1 no. 2 | ++ | 13.5/mm2 |
The amount of insoluble α-SYN was estimated from the band strength on urea extract blots (see Figure 2 ▶ ). Band intensity: ++, strong; +, weak; −, absent. To determine LB density, vibratome sections of 50 μ thickness from the NBIA1 brains were immunostained with Syn102, and 5-μ-thick sections of DLB brains with 15G7. Five different visual fields (each 0.98 mm2) were examined.
Expression Pattern of α-SYN in Transgenic Mice
To generate a rodent model of α-synucleinopathy, we have generated transgenic mice expressing human wt and PD-associated mutant [A30P]α-SYN under the control of a brain-specific pan-neuronal promoter, Thy1. The (Thy1)-[A30P]α-SYN mice described previously 30 and newly generated strains of (Thy1)-[wt]α-SYN mice were of identical genetic background.
As expected for the Thy1 cassette, 43 expression of transgenic α-SYN was undetectable in the first postnatal week, then increased sharply to reach a plateau around 1 month (Figure 3) ▶ . The developmental onset of transgene expression paralleled that of endogenous α-SYN, except for the low but significant early postnatal expression of endogenous α-SYN (Figure 3) ▶ . The putative embryonic/perinatal function of α-SYN in mouse brain 37,44 remains to be elucidated.
Figure 3.

Developmental expression of α-SYN. Mouse brains were collected from mice at the indicated age, and Western blots prepared from 25-μg cytosolic extracts. Wild-type mouse blots were probed with MC42 (top), representative [(Thy1)-h[A30P]α-SYN line 18] transgenic mouse blots probed with 3400 (bottom), and developed with ECLplus.
In adult animals, expression levels of transgenic mRNA and protein generally correlated with each other (except in the [A30P]α-SYN-expressing mouse line 8, which had very high mRNA levels but intermediate protein amounts) (Table 2) ▶ . In contrast, the transgene copy number was no predictor of expression levels (Table 2) ▶ . Approximately 0.1% (w/w) of total adult transgenic mouse brain cytosolic protein was α-SYN, as determined by quantitative Western blot analysis (Table 2) ▶ . Individual quantification of mouse and human α-SYN revealed up to threefold overexpression levels of the transgenic protein.
Table 2.
α-SYN Expression Levels in Transgenic Mice
| Transgenic construct | Line | α-SYN transgene copy number | Transgenic α-SYN mRNA [% actin] | Transgenic α-SYN protein [ng/μg] | Endogenous α-SYN protein [ng/μg] |
|---|---|---|---|---|---|
| (Thy1)-h[wt]α-SYN | 6 | 7 | 175 | 1.93 ± 0.31 | 1.35 ± 0.10 |
| (Thy1)-h[wt]α-SYN | 10 | 85 | 65 | 0.80 ± 0.31 | 1.61 ± 0.14 |
| (Thy1)-h[wt]α-SYN | 14 | 13 | 190 | 2.02 ± 0.11 | 0.65 ± 0.06 |
| (Thy1)-h[wt]α-SYN | 23 | 2 | 130 | 1.32 ± 0.17 | 0.43 ± 0.21 |
| (Thy1)-h[wt]α-SYN | 38 | 5 | 130 | 1.58 ± 0.60 | 0.81 ± 0.28 |
| Nontransgenic | N/A† | N/A | N/A | 1.77 ± 0.55 | |
| (Thy1)-h[A30P]α-SYN | 8 | 15–25 | 370 | 1.20 ± 0.60 | 1.42 ± 0.99 |
| (Thy1)-h[A30P]α-SYN | 9 | 5 | 30 | 0.39 ± 0.05 | 1.64 ± 0.05 |
| (Thy1)-h[A30P]α-SYN | 14 | 10 | 105 | 1.2* | n.d. |
| (Thy1)-h[A30P]α-SYN | 18 | 10–15 | 170 | 1.43 ± 0.18 | 1.59 ± 0.00 |
| (Thy1)-h[A30P]α-SYN | 31 | 15–20 | 155 | 1.69 ± 0.31 | 1.26 ± 0.01 |
Five lines each of [wt]α-SYN and [A30P]α-SYN-expressing transgenic mice stably transmitted the transgene. The integrated transgene copy number was determined by quantitative Southern blotting, transgenic α-SYN mRNA expression levels relative to β-actin by quantitative Northern blotting, and the amount of transgenic and endogenous α-SYN protein by quantitative Western blotting. α-SYN protein was measured in three different mouse whole-brain cytosol samples on the same blot. Results are expressed as mean ± SD.
*Determined indirectly; expression levels relative to the other lines.
†N/A: not applicable.
At these rather moderate overexpression levels, aberrant subcellular localization of human α-SYN was apparent in transgenic mouse brain sections. The human (transgene)-specific antibody showed somal and neuritic accumulations in both [wt]α-SYN and [A30P]α-SYN mice (Figure 4, A and D) ▶ , in addition to neuropil (presumably synaptic) staining. Compact LBs were not observed in transgenic mice. Nevertheless, swollen α-SYN-positive neurites as observed in [wt]α-SYN and [A30P]α-SYN transgenic mice (Figure 4, A and D ▶ , inserts) were a prominent feature in brain sections from LB disease patients. 2,9,30 In sharp contrast, endogenous mouse α-SYN showed only the normal neuropil staining pattern (Figure 4, C and F) ▶ . Likewise, our β-SYN antibody detected only a normal synaptic staining pattern in sections from transgenic mouse brain (Figure 4, B and E) ▶ .
Figure 4.
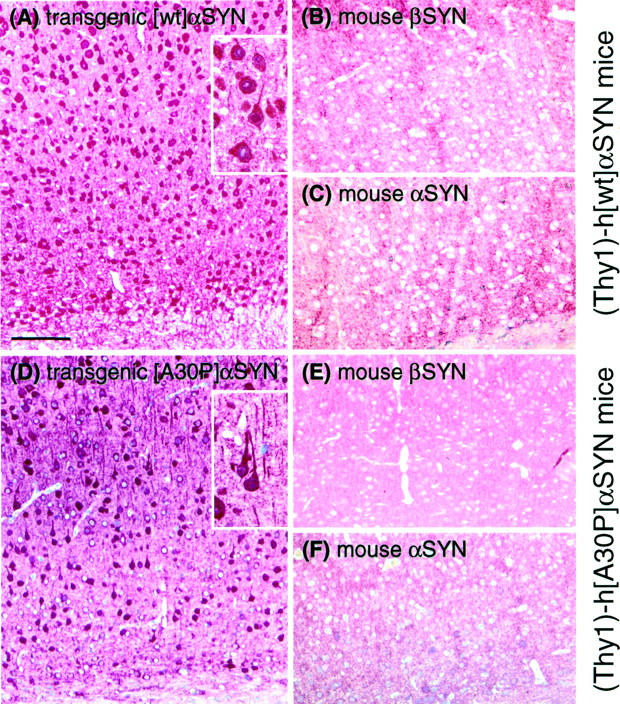
Immunostainings of brain slices (motor cortex) show specific accumulation of α-SYN but not of β-SYN in transgenic mice. Animals expressing either [wt]α-SYN (A–C) or [A30P]α-SYN (D–F) showed a strong cytosolic labeling of neuronal cells with the human-specific α-SYN antibody 15G7 (A and D). A section from a 1-month-old [wt]α-SYN-expressing mouse is shown in A to demonstrate the early onset of accumulation of transgenic protein. In contrast, immunostainings with the β-SYN-specific antiserum (6485) and the murine-specific α-SYN antiserum (7544) only revealed a synaptic staining pattern. Neither accumulation of endogenous murine α-SYN (C and F) nor β-SYN (B and E) was detectable in neuronal cell bodies. Scale bar in A, 100 μm.
Transgenic Human α-SYN But Not Endogenous Mouse SYNs in Detergent-Insoluble Fractions
As the presence of SDS-insoluble α-SYN seemed to be a diagnostic criterion for LB diseases in human brain, we applied the above method to transgenic mouse brains expressing human α-SYN in brain neurons. Although the overexpression levels of transgenic α-SYN were rather moderate, a portion of human α-SYN was specifically detected in urea extracts of detergent-insoluble fractions from transgenic mouse brains (Figure 5) ▶ . Both transgenic [A30P] α-SYN and [wt] α-SYN were found in SDS-insoluble fractions (Figure 5A) ▶ . Insoluble transgenic α-SYN became detectable parallel to the onset of transgene expression (Figure 3) ▶ and persisted for at least 1 year (Figure 5B) ▶ . The onset of expression of insoluble transgenic α-SYN was concomitant with the appearance of cytosolic accumulations (Figure 4A) ▶ .
Figure 5.

Detergent-insoluble α-SYN in transgenic mouse brains. Whole brains of transgenic mice (A: 3- to 4-month-old [wt]α-SYN lines 14 and 23, and [A30P]α-SYN lines 18 and 31; B: 1-month-old and 1-year-old [A30P]α-SYN lines 18 and 31; as indicated at the bottom) and age-matched nontransgenic littermates (lm) were differentially extracted. Buffer- and detergent-soluble proteins (10 μg for transgenic human α-SYN, 50 μg for endogenous mouse SYNs), and TCA precipitate of urea extracts were subjected to denaturing 15% PAGE. Western blots were probed with human (transgene)-specific anti-hα-SYN 15G7, endogenous mouse-specific anti-mα-SYN 7544, and anti-β-SYN 6485, as indicated to the left. 15G7-immunoreactive bands were developed with SuperSignal, polyclonal antibody immunoreactivity with ECLplus. A control lane on the urea extract blots contained 10 μg of cytosol from a transgenic [A30P]α-SYN mouse. The positions of prestained molecular weight markers are indicated to the right. Individual variance of transgenic α-SYN expression levels may account for the apparently higher amount of urea extractable mutant h[A30P]α-SYN compared to h[wt]α-SYN in one experiment (A, exp. 2), but not in two additional experiments (one of them shown as A, exp. 1), and for the apparent increase with age of SDS-soluble α-SYN (B) that was not seen in an additional experiment.
In sharp contrast, endogenous mouse α-SYN as well as β-SYN were entirely soluble in buffer and detergent. In a semiquantitative manner, Western blots were sequentially probed with human (transgene)-specific anti-α-SYN, mouse (endogenous)-specific anti-α-SYN, and anti-β-SYN, and the signal of a control lane on the same blot (10 μg transgenic mouse brain cytosol) was used for normalization. Exposure times yielding comparable signals from the control lane demonstrated that SDS-insoluble endogenous α-SYN or β-SYN in transgenic α-SYN-positive urea extracts were undetectable (Figure 5) ▶ .
Taken together, cytoplasmic accumulation and detergent-insolubility of transgenic human α-SYN represent specific pathological alterations in transgenic mouse brain that are reminiscent of human LB diseases.
Lack of an Aggregation-Promoting Stretch of Amino Acids in the NAC Domain of β-SYN
To determine whether the detergent insolubility of α-SYN in LB disease brain and transgenic mice was a consequence of aggregation, in vitro formed aggregates of α-SYN were subjected to a similar differential extraction procedure used for brain tissue (see above). α-SYN aggregated at a concentration of 2 mg/ml was partially solubilized by resuspension in 15 volumes of TBS+. However, 5% SDS was required for complete solubilization (Figure 6A) ▶ . In contrast, β-SYN failed to form insoluble aggregates in vitro (Figure 6B) ▶ .
Figure 6.
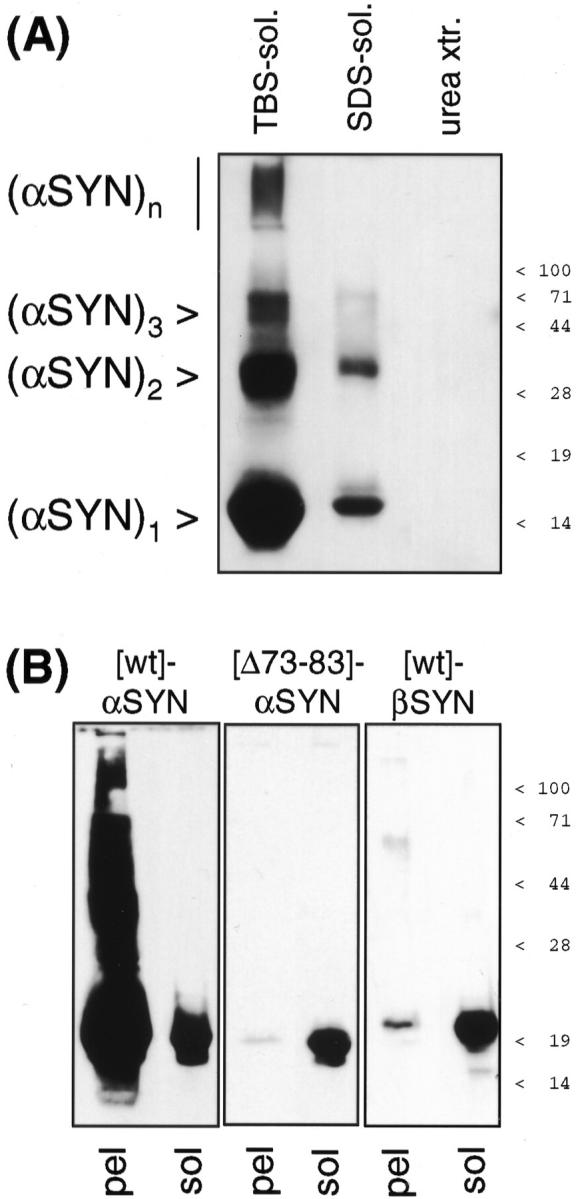
α-SYN, but not β-SYN aggregates in vitro because of a critical determinant in the NAC domain. A: Recombinant human α-SYN aggregates were collected by 100,000 × g centrifugation and redissolved in 15 volumes of TBS+. The buffer-insoluble material was extracted like the brain samples above. All fractions were TCA precipitated and separated by denaturing 15% PAGE. Western blots were probed with 3400 anti-α-SYN and developed with SuperSignal. This experiment was repeated three times with the same result. B: Solutions (2 mg/ml) of [wt]α-SYN (left), [Δ73–83]α-SYN (middle), and [wt]β-SYN (right) were aggregated for 7 days. After ultracentrifugation, the 100,000 × g pellets (pel) were subjected to denaturing 12.5% PAGE. α-SYN immunoblots were probed with MC42 and β-SYN immunoblots with 6485, and developed with SuperSignal until the band intensities of 1-μg freshly dissolved, nonaggregated protein (sol) were comparable. Note the typical retarded electrophoretic motility of recombinant β-SYN. 36,38 Positions of prestained molecular weight markers are indicated to the right.
β-SYN lacks amino acids homologous to residues 73 to 83 of α-SYN. Codons 73 to 83 were specifically deleted by site-directed mutagenesis yielding [Δ73–83]α-SYN. In parallel in vitro aggregation assays, [Δ73–83]α-SYN behaved like β-SYN in that it practically lost its capability to form 100,000 × g pellets after 1 week incubation at 37°C (Figure 6B) ▶ . Thus, amino acids 73 to 83 in the N-terminal half of NAC are critical for α-SYN aggregation, and their absence in β-SYN accounts for the loss of aggregation capacity of the nonamyloidogenic β-SYN.
Discussion
PD, DLB, and NBIA1 are characterized immunohistochemically by α-SYN-immunoreactive intraneuronal inclusions (LBs) and dystrophic neurites. Biochemically, detergent-insoluble α-SYN was found to be diagnostic for these diseases. We have performed differential detergent extractions to evaluate the potential development of α-synucleinopathy in transgenic mice expressing human α-SYN in brain neurons. Like in human LB diseases, detergent-insoluble human α-SYN was detected in transgenic mouse brain. In striking contrast, endogenous mouse α-SYN and β-SYN were not found in the urea extracts. These results demonstrate that a transgenic mouse model recapitulates some specific features of α-synucleinopathies.
The source of detergent-insoluble α-SYN may not only be solid LBs. It is noteworthy that the NBIA1 case with much urea-extractable α-SYN had abundant dystrophic neurites (not shown). Moreover, diffuse accumulation of α-SYN in neuronal cell bodies was occasionally reported for human α-synucleinopathies. 45,46 The accumulations of human α-SYN in transgenic mouse brain neurons did not meet ultrastructural criteria of LBs. Nevertheless, detergent-insoluble transgenic α-SYN was specifically detectable in these mice. Thus, a portion of transgenic α-SYN is converted to a less soluble form that might represent an early form of α-synucleinopathy. It remains to be shown if the decreased solubility of transgenic α-SYN is a mere consequence of overexpression, or if there is some secondary processing that is peculiar to human transgenic α-SYN in mouse brain.
Both PD-associated [A30P]α-SYN and human [wt]α-SYN were detected in detergent-insoluble fractions. This is of note because the overwhelming majority of PD patients have no mutation in the α-SYN gene. 47 The faster in vitro aggregation rate of concentrated solutions of mutant α-SYN 22 was apparently not reflected by greater pathology of human mutant α-SYN compared to [wt]α-SYN in transgenic mice. 29,30 The rather moderate expression levels of transgenic α-SYN (Table 2) ▶ make it unlikely that the critical concentration required for recombinant α-SYN aggregation in vitro (28 μmol/L) 48 was reached in neuronal cytosol. Perhaps the differences in aggregation kinetics between wt and mutant α-SYN are not evident at concentrations reached in transgenic mouse neurons. Because α-SYN expression is not elevated enough in PD patients to allow spontaneous aggregation, additional risk factors are likely to exist that favor the aggregation at subcritical α-SYN concentrations. Similar risk factors may act in rodents. For example, the mitochondrial complex I inhibitor, rotenone, was recently shown to elicit PD-like alterations in rat brain, including the formation of α-SYN inclusions and selective loss of striatonigral dopaminergic neurons. 49
Potentially aggregation-promoting posttranslational modifications of α-SYN include phosphorylation, 38 nitration, 50 and glycation. 51 Moreover, perturbation of proteosomal degradation should be considered. LBs contain ubiquitinated α-SYN. It was reported that in human brain, α-SYN needs to be converted to a slower migrating species (termed “αSp22”) to become a substrate for the ubiquitin ligase, parkin, 41 We have found an α-SYN species with similar retarded electrophoretic motility running at apparent 25-kd [(α-SYN)p25] in the detergent-insoluble fractions from LB disease brain (Figure 2) ▶ , but not in transgenic mice (Figure 5) ▶ . It is possible that posttranslational modifications characteristic for human LB diseases have to occur in transgenic mice to allow true LB formation in an animal model.
In contrast to the amyloidogenic α-SYN, the close homologue β-SYN was absent from amyloid deposits. β-SYN has no intrinsic capacity to form amyloid fibrils. β-SYN lacks the sequence GVTAVAQKTVE corresponding to amino acids 73 to 83 of α-SYN in the NAC domain. Indeed, the deletion mutant protein [Δ73–83]α-SYN was aggregation-deficient. Similar findings were recently reported by Giasson and colleagues 52 for a slightly shifted deletion mutant, namely [Δ71–82]α-SYN. Interestingly, the region critical for α-SYN aggregation, which is missing in β-SYN, overlaps with one of the imperfectly conserved KTKEGV repeats. Such repeats are characteristic for the lipid-binding N-terminal half of α-SYN that has a predicted amphipathic helix structure comparable with apolipoprotein A. 53,54 It will be interesting to determine whether the aggregation-promoting region of α-SYN acquires an amphipathic β-strand conformation upon fibrillization.
In vitro aggregates of α-SYN clearly required harsher solubilization methods than β-SYN. Nevertheless, hardly any SDS-insoluble protein was found in urea extracts of in vitro aggregated recombinant α-SYN. Thus, the massive amounts of α-SYN in urea extracts from LB disease and transgenic mouse brain represent a pathological form of α-SYN that may not be fully reproduced by simple aggregation in vitro. Thus, α-SYN transgenic mice provide an in vivo model that exhibits an important aspect of human α-synucleinopathy, namely selective insolubility of α-SYN.
Acknowledgments
We thank R. Jakes and M. Goedert for the gift of β-SYN expression plasmid, K. Beyreuther and T. Hartmann for providing anti-NAC, W. Franke and R. Leube for the donation of anti-synaptophysin, and H. Schubert for animal care.
Footnotes
Address reprint requests to Philipp Kahle or Christian Haass, Laboratory for Alzheimer’s and Parkinson’s Disease Research, Department of Biochemistry, Ludwig Maximilians University of Munich, Schillerstrasse 44, D-80336 Munich, Germany. E-mail: pkahle@pbm.med.uni-muenchen.de or
Supported by grants from the Deutsche Forschungsgemeinschaft (HA 1737/4-1) and the Bavaria California Technology Center (to C. H.).
References
- 1.Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T: Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA 1993, 90:11282-11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M: α-Synuclein in Lewy bodies. Nature 1997, 388:839-840 [DOI] [PubMed] [Google Scholar]
- 3.Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H: NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci Lett 1997, 239:45-48 [DOI] [PubMed] [Google Scholar]
- 4.Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E: Abnormal accumulation of NACP/α-synuclein in neurodegenerative disorders. Am J Pathol 1998, 152:367-372 [PMC free article] [PubMed] [Google Scholar]
- 5.Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF, Hyman BT: Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain α-synuclein immunoreactivity. J Neuropathol Exp Neurol 1998, 57:334-337 [DOI] [PubMed] [Google Scholar]
- 6.Baba M, Nakajo S, Tu P-H, Tomita T, Nakaya K, Lee VM-Y, Trojanowski JQ, Iwatsubo T: Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 1998, 152:879-884 [PMC free article] [PubMed] [Google Scholar]
- 7.Bayer TA, Jäkälä P, Hartmann T, Havas L, McLean C, Culvenor JG, Li QX, Masters CL, Falkai P, Beyreuther K: α-Synuclein accumulates in Lewy bodies in Parkinson’s disease and dementia with Lewy bodies but not in Alzheimer’s disease β-amyloid plaque cores. Neurosci Lett 1999, 266:213-216 [DOI] [PubMed] [Google Scholar]
- 8.Arawaka S, Saito Y, Murayama S, Mori H: Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for α-synuclein. Neurology 1998, 51:887-889 [DOI] [PubMed] [Google Scholar]
- 9.Wakabayashi K, Yoshimoto M, Fukushima T, Koide R, Horikawa Y, Morita T, Takahashi H: Widespread occurrence of α-synuclein/NACP-immunoreactive neuronal inclusions in juvenile and adult-onset Hallervorden-Spatz disease with Lewy bodies. Neuropathol Appl Neurobiol 1999, 25:363-368 [DOI] [PubMed] [Google Scholar]
- 10.Galvin JE, Giasson B, Hurtig HI, Lee VM-Y, Trojanowski JQ: Neurodegeneration with brain iron accumulation, type 1 is characterized by α-, β-, and γ-synuclein neuropathology. Am J Pathol 2000, 157:361-368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neumann M, Adler S, Schlüter O, Kremmer E, Benecke R, Kretzschmar HA: α-Synuclein accumulation in a case of neurodegeneration with brain iron accumulation type 1 (NBIA-1, formerly Hallervorden-Spatz syndrome) with widespread cortical and brainstem-type Lewy bodies. Acta Neuropathol 2000, 100:568-574 [DOI] [PubMed] [Google Scholar]
- 12.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M: α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 1998, 95:6469-6473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arima K, Uéda K, Sunohara N, Hirai S, Izumiyama Y, Tonozuka-Uehara H, Kawai M: Immunoelectron-microscopic demonstration of NACP/α-synuclein-epitopes on the filamentous component of Lewy bodies in Parkinson’s disease and in dementia with Lewy bodies. Brain Res 1998, 808:93-100 [DOI] [PubMed] [Google Scholar]
- 14.Dickson DW, Lin W, Liu WK, Yen SH: Multiple system atrophy: a sporadic synucleinopathy. Brain Pathol 1999, 9:721-732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto M, Hsu LJ, Sisk A, Xia Y, Takeda A, Sundsmo M, Masliah E: Human recombinant NACP/α-synuclein is aggregated and fibrillated in vitro: relevance for Lewy body disease. Brain Res 1998, 799:301-306 [DOI] [PubMed] [Google Scholar]
- 16.Giasson BI, Uryu K, Trojanowski JQ, Lee VM-Y: Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem 1999, 274:7619-7622 [DOI] [PubMed] [Google Scholar]
- 17.Conway KA, Harper JD, Lansbury PT, Jr: Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000, 39:2552-2563 [DOI] [PubMed] [Google Scholar]
- 18.Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA: Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc Natl Acad Sci USA 2000, 97:4897-4902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biere AL, Wood SJ, Wypych J, Steavenson S, Jiang Y, Anafi D, Jacobsen FW, Jarosinski MA, Wu G-M, Louis J-C, Martin F, Narhi LO, Citron M: Parkinson’s disease-associated α-synuclein is more fibrillogenic than β- and γ-synuclein and cannot cross-seed its homologs. J Biol Chem 2000, 275:34574-34579 [DOI] [PubMed] [Google Scholar]
- 20.Conway KA, Harper JD, Lansbury PT: Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med 1998, 4:1318-1320 [DOI] [PubMed] [Google Scholar]
- 21.El-Agnaf OMA, Jakes R, Curran MD, Wallace A: Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of α-synuclein protein implicated in Parkinson’s disease. FEBS Lett 1998, 440:67-70 [DOI] [PubMed] [Google Scholar]
- 22.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis J-C, Wypych J, Biere AL, Citron M: Both familial Parkinson’s disease mutations accelerate α-synuclein aggregation. J Biol Chem 1999, 274:9843-9846 [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, Masliah E: Oxidative stress induces amyloid-like aggregate formation of NACP/α-synuclein in vitro. NeuroReport 1999, 10:717-721 [DOI] [PubMed] [Google Scholar]
- 24.Goldberg MS, Lansbury PT, Jr: Is there a cause-and-effect relationship between α-synuclein fibrillization and Parkinson’s disease? Nat Cell Biol 2000, 2:E115-E119 [DOI] [PubMed] [Google Scholar]
- 25.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL: Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276:2045-2047 [DOI] [PubMed] [Google Scholar]
- 26.Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O: Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet 1998, 18:106-108 [DOI] [PubMed] [Google Scholar]
- 27.Feany MB, Bender WW: A Drosophila model of Parkinson’s disease. Nature 2000, 404:394-398 [DOI] [PubMed] [Google Scholar]
- 28.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L: Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 2000, 287:1265-1269 [DOI] [PubMed] [Google Scholar]
- 29.van der Putten H, Wiederhold K-H, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WPJM, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G: Neuropathology in mice expressing human α-synuclein. J Neurosci 2000, 20:6021-6029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kahle PJ, Neumann M, Ozmen L, Müller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C: Subcellular localization of wild-type and Parkinson’s disease-associated mutant α-synuclein in human and transgenic mouse brain. J Neurosci 2000, 20:6365-6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maroteaux L, Scheller RH: The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Mol Brain Res 1991, 11:335-343 [DOI] [PubMed] [Google Scholar]
- 32.Shibayama-Imazu T, Okahashi I, Omata K, Nakajo S, Ochiai H, Nakai Y, Hama T, Nakamura Y, Nakaya K: Cell and tissue distribution and developmental change of neuron specific 14 kDa protein (phosphoneuroprotein 14). Brain Res 1993, 622:17-25 [DOI] [PubMed] [Google Scholar]
- 33.George JM, Jin H, Woods WS, Clayton DF: Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15:361-372 [DOI] [PubMed] [Google Scholar]
- 34.Irizarry MC, Kim T-W, McNamara M, Tanzi RE, George JM, Clayton DF, Hyman BT: Characterization of the precursor protein of the non-Aβ component of senile plaques (NACP) in the human central nervous system. J Neuropathol Exp Neurol 1996, 55:889-895 [DOI] [PubMed] [Google Scholar]
- 35.Culvenor JG, McLean CA, Cutt S, Campbell BCV, Maher F, Jäkälä P, Hartmann T, Beyreuther K, Masters CL, Li Q-X: Non-Aβ component of Alzheimer’s disease amyloid (NAC) revisited: NAC and α-synuclein are not associated with Aβ amyloid. Am J Pathol 1999, 155:1173-1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jakes R, Spillantini MG, Goedert M: Identification of two distinct synucleins from human brain. FEBS Lett 1994, 345:27-32 [DOI] [PubMed] [Google Scholar]
- 37.Hong L, Ko HW, Gwag BJ, Joe E, Lee S, Kim Y-T, Suh Y-H: The cDNA cloning and ontogeny of mouse α-synuclein. NeuroReport 1998, 9:1239-1243 [DOI] [PubMed] [Google Scholar]
- 38.Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C: Constitutive phosphorylation of the Parkinson’s disease associated α-synuclein. J Biol Chem 2000, 275:390-397 [DOI] [PubMed] [Google Scholar]
- 39.Dickson DW, Liu W-K, Hardy J, Farrer M, Mehta N, Uitti R, Mark M, Zimmerman T, Golbe L, Sage J, Sima A, D’Amato C, Albin R, Gilman S, Yen S-H: Widespread alterations of α-synuclein in multiple system atrophy. Am J Pathol 1999, 155:1241-1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leng Y, Chase TN, Bennett MC: Muscarinic receptor stimulation induces translocation of an α-synuclein oligomer from plasma membrane to a light vesicle fraction in cytoplasm. J Biol Chem 2001, 276:28212-28218 [DOI] [PubMed] [Google Scholar]
- 41.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ: Ubiquitination of a new form of α-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 2001, 293:263-269 [DOI] [PubMed] [Google Scholar]
- 42.Wakabayashi K, Fukushima T, Koide R, Horikawa Y, Hasegawa M, Watanabe Y, Noda T, Eguchi I, Morita T, Yoshimoto M, Iwatsubo T, Takahashi H: Juvenile-onset generalized neuroaxonal dystrophy (Hallervorden-Spatz disease) with diffuse neurofibrillary and Lewy body pathology. Acta Neuropathol 2000, 99:331-336 [DOI] [PubMed] [Google Scholar]
- 43.Kollias G, Spanopoulou E, Grosveld F, Ritter M, Beech J, Morris R: Differential regulation of a Thy-1 gene in transgenic mice. Proc Natl Acad Sci USA 1987, 84:1492-1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsu LJ, Mallory M, Xia Y, Veinbergs I, Hashimoto M, Yoshimoto M, Thal LJ, Saitoh T, Masliah E: Expression pattern of synucleins (non-Aβ component of Alzheimer’s disease amyloid precursor protein/α-synuclein) during murine brain development. J Neurochem 1998, 71:338-344 [DOI] [PubMed] [Google Scholar]
- 45.Gómez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT: α-Synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol 2000, 99:352-357 [DOI] [PubMed] [Google Scholar]
- 46.Gai WP, Yuan HX, Li XQ, Power JTH, Blumbergs PC, Jensen PH: In situ and in vitro study of colocalization and segregation of α-synuclein, ubiquitin, and lipids in Lewy bodies. Exp Neurol 2000, 166:324-333 [DOI] [PubMed] [Google Scholar]
- 47.El-Agnaf OMA, Curran MD, Wallace A, Middleton D, Murgatroyd C, Curtis A, Perry R, Jaros E: Mutation screening in exons 3 and 4 of α-synuclein in sporadic Parkinson’s and sporadic and familial dementia with Lewy bodies cases. NeuroReport 1998, 9:3925-3927 [DOI] [PubMed] [Google Scholar]
- 48.Wood SJ, Wypych J, Steavenson S, Louis J-C, Citron M, Biere AL: α-Synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J Biol Chem 1999, 274:19509-19512 [DOI] [PubMed] [Google Scholar]
- 49.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT: Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 2000, 3:1301-1306 [DOI] [PubMed] [Google Scholar]
- 50.Giasson BI, Duda JE, Murray IVJ, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM-Y: Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290:985-989 [DOI] [PubMed] [Google Scholar]
- 51.Münch G, Lüth HJ, Wong A, Arendt T, Hirsch E, Ravid R, Riederer P: Crosslinking of α-synuclein by advanced glycation endproducts—an early pathophysiological step in Lewy body formation? J Chem Neuroanat 2000, 20:253-257 [DOI] [PubMed] [Google Scholar]
- 52.Giasson BI, Murray IVJ, Trojanowski JQ, Lee VM-Y: A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem 2001, 276:2380-2386 [DOI] [PubMed] [Google Scholar]
- 53.Davidson WS, Jonas A, Clayton DF, George JM: Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 1998, 273:9443-9449 [DOI] [PubMed] [Google Scholar]
- 54.Jo E, McLaurin J, Yip CM, St. George-Hyslop P, Fraser PE: α-synuclein membrane interactions and lipid specificity. J Biol Chem 2000, 275:34328-34334 [DOI] [PubMed] [Google Scholar]