

Abstract
Mutations in α-synuclein (αS) and parkin cause heritable forms of Parkinson disease (PD). We hypothesized that neuronal parkin, a known E3 ubiquitin ligase, facilitates the formation of Lewy bodies (LBs), a pathological hallmark of PD. Here, we report that affinity-purified parkin antibodies labeled classical LBs in substantia nigra sections from four related human disorders: sporadic PD, inherited αS-linked PD, dementia with LBs (DLB), and LB-positive, parkin-linked PD. Anti-parkin antibodies also detected LBs in entorhinal and cingulate cortices from DLB brain and αS inclusions in sympathetic gangliocytes from sporadic PD. Double labeling with confocal microscopy of DLB midbrain sections revealed that ∼90% of anti-αS-reactive LBs were also detected by a parkin antibody to amino acids 342 to 353. Accordingly, parkin proteins, including the 53-kd mature isoform, were present in affinity-isolated LBs from DLB cortex. Fluorescence resonance energy transfer and immunoelectron microscopy showed that αS and parkin co-localized within brainstem and cortical LBs. Biochemically, parkin appeared most enriched in cytosolic and postsynaptic fractions of adult rat brain, but also in purified, αS-rich presynaptic elements that additionally contained parkin’s E2-binding partner, UbcH7. We conclude that parkin and UbcH7 are present with αS in subcellular compartments of normal brain and that parkin frequently co-localizes with αS aggregates in the characteristic LB inclusions of PD and DLB. These results suggest that functional parkin proteins may be required during LB formation.
Two missense mutations in α-synuclein (αS), A30P and A53T, cause rare autosomal-dominant forms of Parkinson disease (PD), 1,2 and a variety of mutant parkin genotypes are linked to autosomal-recessive parkinsonism with predominantly juvenile or young adult onset. 3,4 Clinically, a subset of parkin-linked cases are virtually indistinguishable from sporadic PD. 4,5 Neuropathologically, sporadic and αS-linked PD share neuronal loss and Lewy body (LB) inclusions in selective brainstem nuclei. 6-8 The etiology of sporadic PD remains largely unknown, but mechanisms involving oxidative stress and mitochondrial dysfunction have been implicated. 9 In the related disorder, dementia with LBs (DLB), both brainstem and cortical neurons are affected by LB formation. 10-12 αS and ubiquitin (Ub) represent the principal known protein constituents of these inclusions. 13-15 In contrast, LBs are generally absent in parkin-linked PD brains, 16-20 with one recently reported exception. 21
The 465-amino acid parkin protein contains an N-terminal Ub-like domain linked to a C-terminal RING box 22 that contains two canonical RING-finger domains (C3H1C4) and an imperfect, third RING finger motif (C6H1C1), also referred to as “in-between-RING” 23 or “double-RING-finger-linked” domain 24 (Figure 1A) ▶ . As such, parkin is a likely member of a class of zinc-binding proteins that comprises several Ub ligases. 25,26 Indeed, parkin has been shown to act as an E3 Ub ligase in transfected cell cultures and in vitro assays, in which it principally recruits one of two E2 Ub-conjugating proteins at its RING box, UbcH7 22,27,28 or UbcH8. 29 In general, the transfer of more than four activated Ub molecules from an E2 protein onto a substrate, a process facilitated by an E3 Ub ligase, provides a signal for proteasomal degradation of the substrate. 30 In cell culture systems, parkin fusion proteins have also been shown to interact with the synaptic vesicle protein, CDC-rel1, 29 the αS-binding protein, synphilin-1, 31 and actin filaments. 32 Furthermore, parkin has been found to be up-regulated during the integrated cellular response to misfolded protein-induced stress. 27
Figure 1.

Characterization of affinity-purified antibodies to human parkin. A: Schematic diagram of human parkin and five peptide antibodies (amino acid numbering according to Kitada et al 3 ). B: Dot blot analysis of selected proteins and parkin peptides. Bovine serum albumin, His-tagged Ub, human αS (10 ng each), and synthetic peptides (50 ng) were diluted in water and loaded horizontally. Membranes were probed with antibodies (Ab) as indicated (abs., absorbed with cognate peptide; sister lanes were mock-absorbed with noncognate parkin peptide). C: PAGE and Western blotting (WB) of recombinant αS and Ub (20 ng), soluble extracts from control brains (CNS, 40 μg) and lysates of SH-SY5Y cells stably expressing mycParkin (cell, 10 μg). Membranes were probed with antibodies to Ub, αS (syn-1), or parkin (HP6A, HP1A, HP1A abs., HP2A, and HP2A abs.), as indicated. Black arrowheads indicate relative position of monomeric Ub; white arrowheads identify proteolytic fragments of parkin. Asterisk denotes polyubiquitinated, high Mr protein smear in cell lysates. Note, anti-parkin antibodies fail to detect recombinant or endogenous αS and Ub proteins.
Specific targets for parkin’s E3 Ub ligase activity in vivo include an O-glycosylated form of αS, αSp22, 28 and Pael-R, a Parkin-associated endothelin-like receptor. 33 The homozygous inheritance of exonic deletions in the parkin gene results in the accumulation of nonubiquitinated forms of these two substrates in the brain. 28,33 However, the extent of parkin’s physiological role in maintaining dopaminergic neuronal function and its pathophysiological role in LB-positive disorders, eg, PD and DLB, are not yet known. The general absence of detectable αS-inclusions in cases of parkin-linked PD led us to hypothesize that parkin promotes a critical step in LB formation by ubiquitinating αS and other unknown substrates. Therefore, we investigated the subcellular distribution of parkin in normal adult brain as it relates to αS and UbcH7, and analyzed LB-rich tissues from sporadic PD, inherited PD, and DLB for parkin immunoreactivity both morphologically and biochemically.
Materials and Methods
Antibody Production
Synthetic peptides of human parkin protein [HP6A (amino acids 6 to 15), HP7A (amino acids 51 to 62), HP1A (amino acids 84 to 98), HP2A (amino acids 342 to 353), and HP5A (amino acids 453 to 465); for numbering see Kitada et al 3 ] were purified by high pressure liquid chromatography, sequence-verified (Biopolymer Laboratory, Brigham and Women’s Hospital), coupled to keyhole limpet hemocyanin (KLH), and injected into rabbits. Sera were pooled, affinity-purified against their resin-coupled peptide antigens, and analyzed by enzyme-linked immunosorbent assay (Research Genetics Inc, AL). The purified antibodies were used at final dilutions of 1:75 to 1:100 for immunohistochemistry, at 1:500 to 1:1000 for Western blots, and at 1:1000 to 1:5000 for dot blots. Purified bovine Ub was from Sigma Chemical Co. (St. Louis, MO). Recombinant, bacterially expressed αS and a human αS-encoding cDNA were provided by P. Lansbury of Brigham and Women’s Hospital. 34 Anti-αS LB509 was from Zymed (South San Francisco, CA); anti-Ub from Molecular Biological Laboratories; anti-synaptophysin from Sigma Chemical Co., and anti-postsynaptic density protein (PSD)-95 from Chemicon (Temecula, CA). Anti-syn-1, anti-UbcH7, and lysates of Jurkat cells expressing UbcH7 were from Transduction Labs (Lexington, KY).
Brain Tissue and Immunohistochemistry
All tissue was collected in accordance with Institutional Review Board-approved guidelines. Paraffin sections of brains and peripheral gangliocytes fixed in 10% neutral buffered formalin were obtained from the Neuropathology Service at Brigham and Women’s Hospital. The diagnoses of PD (n = 3), DLB (n = 2), αS-linked PD 7 [(n = 1); provided by L. Golbe and D. Dickson of Robert Wood Johnson Medical School, New Jersey and D. Dickson of Mayo Clinic, Jacksonville, FL] and normal cases (n = 2) were established by standard neuropathological criteria. Sections from a parkin-linked PD case were obtained as described. 21 Immunohistochemistry was performed with the use of antigen retrieval buffer (BioGenex, San Ramon, CA) and microwaving. 35 For preabsorptions, 5 μg of parkin or control peptides per μl of parkin antibody wereused. Peptides were suspended in 8% dimethyl sulfoxide/water and stored at 1 μg/μl.
Fluorescence Resonance Energy Transfer (FRET) Analysis
Fresh-frozen midbrains (n = 5) from DLB were obtained from the Harvard Brain Tissue Resource Center, fixed in 4% paraformaldehyde, and processed as described. 36,37 Anti-αS (H3C; gift of D. Clayton of University of Illinois) and anti-Ub (DAKO, Inc., Carpinteria, CA) were used for FRET analysis. BODIPY- and cy3-conjugated secondary antibodies (Molecular Probes, Eugene, OR) were used at a dilution of 1:200. HP2A, anti-αS, and anti-Ub reactivities were examined systematically under a ×40 objective. Images were obtained with a MRC-1024 Bio-Rad laser confocal imaging system (Bio-Rad, Richmond, CA) as described. 36,37 Relative distances between epitopes were calculated by FRET analysis, in which the extent of energy transfer is assessed by measuring the donor fluorescence before (DA) and after (D) photobleaching of the acceptor. 38 A positive FRET signal (the ratio of D/DA > 1.0 being proportional to spatial proximity) was compared to the null hypothesis value of 1.0 by one-group t-tests, as described. 36,37 The efficiency of energy transfer is dependent on the inverse sixth power of the distance between the two fluorophores. 36-38
Purification of Lewy Bodies
Cerebral cortical gray matter from DLB or normal brain was obtained from the South Australian National Brain Bank at Flinders University, homogenized, and processed by glass wool filtration and Percoll density gradient centrifugation to obtain Lewy inclusion-rich brain fractions. LBs were affinity-isolated from pooled Percoll density gradient fractions using sheep anti-human αS antibody and streptavidin-coated magnetic beads (50 nm). LB-rich fractions were solubilized in urea/sodium dodecyl sulfate-containing lysis buffer, boiled, and analyzed by polyacrylamide gel electrophoresis (PAGE) or subjected to immunoelectron microscopy. 39-41
Immunoelectron Microscopy
LB-positive samples were fixed in 0.25% glutaraldehyde/2% formaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) for 1 hour and 0.5% OsO4 in 0.1 mol/L phosphate buffer (pH 7.4) for 1 hour at 4°C, dehydrated in ice-cold alcohol, and embedded in LR White resin (London Resin, Basingstoke, UK). Ultrathin sections (80- to 100-nm thick) were incubated for 2 hours at room temperature with primary antibody that were detected by 12-nm gold-conjugated donkey anti-rabbit IgG (Jackson Laboratories, Bar Harbor, ME). Sections were stained for 10 minutes with 2% aqueous uranyl acetate and an additional 10 minutes with Reynolds lead citrate, and examined with a Jeol JEM-1200EX electron microscope at 80 kV. 41
Cloning, Cell Culture, and Transfection
Plasmids pmyc∼parkin and pΔUblparkin were obtained by subcloning an EcoRI-SalI fragment from pGEM-T-Easy parkin (containing wild-type human parkin cDNA 3 ) in frame and out of frame, respectively, into the mammalian expression vector pCMV-Tag3 (Stratagene, La Jolla, CA). The pΔUblparkin cDNA construct reinitiates translation at M80 after a stop codon following the in-frame translation of the myc tag (see Results). All constructs were confirmed by sequencing from both ends and expression in in vitro translation assays as per the manufacturer (T3; Promega, Madison, WI). Site-directed mutagenesis of the cDNA-encoding full-length mycParkin was performed as per the manufacturer’s instructions (Stratagene; the primer sequences are available on request). HEK293, COS, and SH-SY5Y cells were cultured in 10-cm dishes and transiently transfected with 10 μg of plasmid DNA using Lipofectamine 2000 (Life Technologies, Inc., Grand Island, NY). Cells were collected after 24 hours in STEN/0.2% Nonidet P-40 lysis buffer containing an ethylenediaminetetraacetic acid-free protease inhibitor cocktail (Boehringer Mannheim, Indianapolis, IN). Lysates of cells (and brain homogenates) were precleared at 13,000 × g and added to 2× sodium dodecyl sulfate-Tris glycine sample buffer (Novex) containing 10 mmol/L of dithiothreitol (Sigma Chemical Co.) or to 4× Laemmli buffer, 28 then boiled and cleared again. PAGE was performed on 10%, 8 to 16%, or 4 to 20% gradient Tris-glycine gels (Invitrogen, Carlsbad, CA). Proteins were transferred to polyvinylidene difluoride membranes (0.4 μm; Immobilon) and visualized by SuperSignal (Pierce, Rockford, IL) or enhanced chemiluminescence (New England Nuclear, Boston, MA). Stable clones expressing mycParkin–derived from transiently transfected SH-SY5Y cells were selected by the addition of G418 (0.2 μmol/L; Life Technologies, Inc., Los Angeles, CA) to the culture medium.
Subcellular Fractionation
Pooled homogenates of five adult Sprague-Dawley rat forebrains were processed as described 42,43 to generate purified pre- and postsynaptic elements. Briefly, after the removal of cytosol (100,000 × g supernatant) from pelleted crude synaptosomal preparations, the latter were separated from myelin-rich fractions on sucrose density gradient no. 1 to yield purified synaptosomes. These were then extracted in 0.5% Triton X-100 (Sigma Chemical Co.)-containing buffer and centrifuged at 28,000 × g. Triton supernatants were respun at 165,000 × g to yield highly purified presynaptic elements (PRE) in the supernatant. 42 The Triton pellet was subjected to discontinuous sucrose density gradient no. 2; the 1.5- to 2.1-mol/L sucrose interface (crude PSD preparation) was re-extracted in 0.5% Triton X-100 and subjected to sucrose density gradient no. 3. The 1.5- to 2.1-mol/L sucrose interface contained highly purified postsynaptic elements (PSD). 43
Results
To examine parkin’s pathophysiological role, we raised affinity-purified antisera to various peptides of human parkin (Figure 1A) ▶ and characterized their specificity. Parkin antisera were first tested by dot blotting under nonreducing, nondenaturing conditions (Figure 1B) ▶ , and subsequently by Western blotting under both reducing and denaturing conditions (Figure 1C) ▶ . In these assays, anti-parkin antisera recognized their cognate antigens in a specific manner and failed to cross-react with other peptides of human parkin, bovine serum albumin, recombinant αS, or Ub (Figure 1B) ▶ , as expected. Rather, they specifically reacted with the ∼53-kd parkin protein of human brain extracts and the ∼54-kd mycParkin fusion protein stably expressed in SH-SY5Y neuroblastoma cells, as shown for HP6A (to amino acids 6 to 15), HP1A (to amino acids 84 to 98), and HP2A (to amino acids 342 to 353) (Figure 1C) ▶ . These antibodies did not detect either recombinant or endogenous forms of αS and Ub from human brain and SH-SY5Y neuroblastoma cell extracts (Figure 1C) ▶ .
To assess parkin immunoreactivity in LB-rich regions of the central nervous system, we first probed midbrain sections from sporadic PD, DLB, and normal brains by conventional immunohistochemistry. HP2A strongly labeled the cores of classical intracellular LBs in pigmented neurons of the substantia nigra in PD and DLB (Figure 2, a and b) ▶ . HP1A and HP7A (amino acids 51 to 62) strongly labeled cytoplasmic parkin in a granular pattern in cell bodies and proximal neurites of dopaminergic neurons in both diseased and normal brains (Figure 2, i and k) ▶ ; these two antibodies occasionally identified the cores of LBs within nigral neurons in PD and DLB (Figure 2; c to f) ▶ . Thus, the staining profile of HP1A and HP7A was very similar to that of a reported antiserum raised against human parkin amino acids 293 to 306 (referred to as M74 in Shimura et al 19 ). As expected, brainstem LBs were immunoreactive for αS (Figure 2, g and h) ▶ and Ub (not shown). The staining of LB inclusions, cytoplasm, and neuronal processes by anti-parkin antibodies was abolished by preabsorption with the respective antigen (Figure 2, l and m) ▶ . In the brainstem, perikarya of other neurons were also labeled by anti-parkin antibodies, sometimes more intensely than dopaminergic cells. These included neurons of cranial nerve nuclei III (Figure 2j) ▶ , V, VII, X, and XII; central raphe nuclei; locus coeruleus; peri-aqueductal gray; and inferior olive (data not shown). We did not observe staining of glial, ependymal, or meningothelial cells. There was no labeling of dystrophic Lewy neurites by anti-parkin under these conditions. Anti-parkin HP6A and HP5A (to amino acids 453 to 465) detected neuronal parkin weakly; their staining of LBs did not differ from that of the background neuropil (data not shown). When we probed midbrain sections from PD with a specific monoclonal antibody to UbcH7, parkin’s previously identified E2-binding partner in human brain, 28 we observed staining of neuronal cytoplasm and perinuclear structures, as expected, 24 and of the cores of LBs (Figure 2t) ▶ .
Figure 2.

Immunohistochemistry with antibodies to parkin, αS, and UbcH7 in sections of the central and peripheral nervous system. Brainstem Lewy bodies (LB, arrows) are detected by HP2A (a and b), HP1A (c and d), HP7A (e and f), LB509 (g and h), and UbcH7 (t) in substantia nigra sections from PD (a, c, e, g, l, and t) and DLB (b, d, f, h, and m) but not control brain (i, j, and k). Open arrowhead depicts granular staining of perikaryon in a dopaminergic neuron (i); cells of cranial nerve III (j) and neurites (arrowheads) in control nigra (k) stained by HP1A. Competition with respective antigen [HP2A abs. (l) and HP1A abs. (m)] abolishes LB staining but not the appearance of surrounding melanin granules. In entorhinal (n and o) and cingulate cortex (p) sections from DLB brain (n, o, and p), cortical LBs are detected by HP2A (n), HP1A (o), and LB509 (p). In sympathetic gangliocytes from PD (q, r, and s), peripheral LBs are stained by HP2A (q), HP1A (r), and LB509 (s). Scale bar, 20 μm.
To examine if parkin epitopes are present in LBs found elsewhere in the nervous system, we next probed sections of anterior cingulate and entorhinal cortices from cases of DLB brain. Both HP2A and HP1A recognized cortical LBs (Figure 2, n and o) ▶ in a distribution similar to that of anti-αS (Figure 2p) ▶ and detected neuronal parkin (eg, Figure 2n ▶ ). HP7A detected fewer cortical LBs than HP2A and HP1A (data not shown). In normal and DLB cortices, anti-parkin reactivity was associated with granular structures throughout the cytoplasm of many pyramidal neurons in layer IV and their apical dendrites. 32 Anti-parkin immunoreactivity in these cortical sections was completely abolished by absorption with cognate antigens (not shown).
To determine whether parkin immunoreactivity can also be detected in LBs outside the central nervous system, we probed serial sections of thoracic sympathetic ganglia from a sporadic PD case, a site routinely affected in PD. Anti-αS revealed numerous, intensely stained LB-like inclusions of variable sizes and forms in tyrosine hydroxylase-positive sympathetic gangliocytes (Figure 2s) ▶ . 8,44,45 In parallel sections, HP2A and HP1A specifically recognized the center of many club-shaped and elliptical inclusions (Figure 2, q and r) ▶ , and strongly reacted with parkin in the soma of tyrosine hydroxylase-positive cells. No inclusions were detected by anti-αS or anti-parkin in thoracic sympathetic ganglia from a normal patient, as expected. Cross sections of exiting nerve bundles of these thoracic ganglia revealed diffuse axonal parkin staining (not shown). We conclude from these immunohistochemical data collected from sections of sporadic PD, DLB, and control brains that parkin epitopes can be found in LBs at three distinct sites of the nervous system. Throughout these studies, we observed a distinct gradient of parkin immunoreactivity, with more intense central (core) than peripheral (rim) staining (eg, Figure 2; a to f ▶ ). This immunoreactivity within the LB core is similar to that observed for Ub, as reported in immunoelectron microscopic and immunofluorescence studies. 41
To determine whether parkin immunoreactivity could also be detected in the brainstem LBs of inherited PD cases, we first stained sections from the case of a patient who had been an affected member of the North American branch of the Contursi kindred (IX/56) 46 carrying a heterozygous mutation that resulted in an αSA53T substitution (Figure 3) ▶ . 1,47 In the substantia nigra, despite the extensive cell loss and reactive gliosis observed, rare cytoplasmic LBs and numerous nonsomatic inclusions (representing either axonal spheroids or large intraneuritic LBs) were stained by anti-αS (Figure 3, a and c) ▶ . Both of these neuropathological abnormalities were also detected by anti-parkin HP2A (Figure 3, b and d) ▶ . In an adjacent section, nonsomatic inclusions were also strongly reactive with HP1A, although no cytoplasmic LBs were present (not shown).
Figure 3.

Immunohistochemistry with antibodies to αS and parkin in sections of substantia nigra from an αSA53T PD brain. 46,47 Cytoplasmic Lewy bodies (a and b, arrows) and nonsomatic inclusions (c and d, arrowheads) are detected by LB509 (a and c) and HP2A (b and d). Scale bar, 20 μm.
Next, we examined substantia nigra sections of the only LB-positive parkin-linked PD case described to date (Figure 4) ▶ . The patient, referred to as index patient Pw3, had been a compound heterozygote at the parkin locus. 21 Genotyping revealed a parkinC924T missense mutation in exon 7 on one allele (resulting in a parkinR275W substitution) and a 40-bp deletion of exon 3 (that nevertheless preserved the reading frame) on the other allele, parkinΔ40 bp. 21 As concerns the mutant parkinR275W protein, the epitopes of all of our anti-parkin antibodies should be present (Figure 1A) ▶ , in the case of the shorter translation product of the parkinΔ40 bp allele, the epitope for HP1A should be absent. In sections of the substantia nigra (that showed severe cell loss and reactive gliosis), LBs were immunoreactive with anti-parkin HP2A (Figure 4, b and d) ▶ and faintly positive with anti-αS LB509 (Figure 4, c and e) ▶ . Both HP7A and HP2A demonstrated neuronal immunoreactivity, whereas HP1A stained neurons only weakly when compared with sections from sporadic PD processed in parallel. Anti-parkin HP2A-reactive LBs were also identified in sections of the locus coeruleus and nucleus basalis of Meynert from this compound heterozygous patient (data not shown). We conclude that parkin immunoreactivity occurs in nigral LBs of four related human disorders, sporadic PD, αS-linked PD, LB-positive parkin-linked PD, and DLB.
Figure 4.

Immunohistochemistry with antibodies to parkin (HP2A in a, b, and d) and αS (LB509 in c and e) in sections of substantia nigra from a compound heterozygous, parkin-linked PD brain. 21 Intracellular Lewy bodies (arrow) are seen at low power (a) and in adjacent, high power (original magnification, ×40). b to e are magnification images. Upper square in a denotes area of interest depicted in b and c, lower square images are depicted in d and e. Scale bar, 20 μm.
To compare the frequency of parkin and αS staining within compact cytoplasmic LBs, we first performed semiquantitative analysis of serial sections (10 μm) from six LB-positive midbrains that had been stained immunohistochemically with two antibodies, HP2A and LB509. HP2A was selected because of its robust detection of LBs, and its ability to distinguish LB-associated from normal neuronal parkin reactivity (eg, Figure 2 ▶ ). Review of these midbrain sections demonstrated a ratio for parkin:αS-reactive cytoplasmic LBs of 0.76 and 0.81 in sporadic PD and DLB, respectively (Table 1) ▶ . To calculate the co-labeling of classical LBs within the same plane, we used double-immunofluorescence and confocal-scanning laser microscopy to analyze paraformaldehyde-fixed, 40-μm-thick sections from five DLB midbrains, 36,37 which have more preserved dopaminergic neurons and higher numbers of LBs than do PD cases. Under these conditions, 90.6% of all anti-αS-positive nigral LBs were also stained by anti-parkin HP2A (Table 1) ▶ .
Table 1.
Detection of Cytoplasmic Lewy Bodies (LBs) with Anti-Parkin Antibody in Midbrains from LB Disorders
| A. Conventional immunohistochemistry of serial sections examined by light microscopy | ||||
|---|---|---|---|---|
| Case no. | Clinical diagnosis | LB detected by anti-parkin (hemi-midbrains counted) | LB detected by anti-αS (hemi-midbrains counted) | Ratio anti-parkin to anti-αS |
| 1 | Sporadic PD | 19 (2) | 22 (2) | 0.86 |
| 2 | Sporadic PD | 4 (1) | 12 (2) | 0.67 |
| 3 | DLB | 45 (4) | 43 (4) | 1.05 |
| 4 | DLB | 119 (2) | 208 (2) | 0.57 |
| 5 | αS-linked PD | 4 (4) | 1 (1) | 1.00 |
| 6 | parkin-linked PD | 3 (1) | 2 (1) | 1.50 |
| B. Double immunofluorescence labeling of single sections examined by confocal microscopy | |||||
|---|---|---|---|---|---|
| Case no. | Clinical diagnosis | Hemi-midbrains double labeled | LB detected by anti-parkin | LB detected by anti-αS | Percentage (%) of co-labeled LB |
| 1 | DLB | 2 | 60 | 68 | 88.2 |
| 2 | DLB | 2 | 38 | 39 | 97.4 |
| 3 | DLB | 2 | 7 | 8 | 87.5 |
| 4 | DLB | 2 | 9 | 11 | 81.8 |
| 5 | DLB | 2 | 12 | 13 | 92.3 |
Average ratio for sporadic Parkinson’s disease (PD): 0.76; average ratio for dementia with LB (DLB): 0.81.
Average percentage for dementia with LB (DLB): 90.6.
After establishing the co-localization of parkin epitopes with αS in LBs, we examined the spatial relation between the two proteins by performing FRET analysis and immunoelectron microscopy (see below). Two distinct fluorophore-labeled epitopes that generate a positive FRET signal (D/DA > 1.0) after photobleaching are spatially separated by <30 nm in situ, based on the average sizes of fluorophores and of antibodies. 36,37 Using FRET analysis, a tight intermolecular association has been reported between functionally related proteins, eg, dystrophin and actin. 38 We recently demonstrated strong FRET signals between αS- and Ub-epitopes within brainstem LBs, but not between αS and other LB constituents, such as the heat shock protein, αB crystallin. 36 In support of a close intermolecular association between parkin and αS in double-labeled, compact brainstem LBs, we obtained statistically significant FRET signals for the pairing of anti-parkin and anti-αS (D/DA = 1.38 ± 0.27, P < 0.005, n = 11). No double labeling of LBs was observed (and thus FRET analysis was not performed) in sections stained with pre-absorbed HP2A and anti-αS. In parallel experiments, a positive FRET signal was also obtained with anti-parkin and anti-Ub (D/DA = 1.29 ± 0.14, P < 0.005, n = 11). Thus, the human parkin sequence from amino acid 342 to 353 was found in proximity to both αS and Ub epitopes within classical LBs.
To extend the immunohistochemical and FRET evidence of parkin epitopes within LBs to the biochemical and ultrastructural levels, we used a previously published protocol to affinity-isolate cortical LBs. Homogenates of DLB or normal cortex were processed by glass wool filtration and Percoll density gradients, as described. 41 The resultant gradient fractions were analyzed individually (not shown), pooled, and anti-human αS IgG-coated magnetic beads were added to collect LB-positive material from DLB brain and the corresponding LB-negative material from normal brain (Figure 5A) ▶ . 40,41 After urea/sodium dodecyl sulfate solubilization of the particulate material that was isolated on the washed beads, LB-negative and LB-positive samples were immunoblotted with anti-αS, anti-Ub, and anti-parkin antibodies (Figure 5; B, C, and D) ▶ . Lysates of cells transiently transfected with pmyc∼parkin cDNA were analyzed as a positive control. In the LB-positive samples, we identified monomeric αS (16 kd), modified and/or oligomeric αS, and gel-excluded isoforms of αS (Figure 5B) ▶ , 15 faint monomeric Ub reactivity (∼8 kd), a strongly ubiquitinated protein band at ∼23 kd, an oligomeric smear, and gel-excluded isoforms of Ub (Figure 5C) ▶ . 40,48 Little immunoreactivity was detected in the LB-negative samples, consistent with the absence of Lewy inclusions by αS immunohistochemistry in this control brain (not shown). All three parkin antisera we tested, HP6A (Figure 5B) ▶ , HP1A (Figure 5C) ▶ , and HP2A (Figure 5D) ▶ , specifically labeled the 54-kd mycParkin fusion protein in cell lysates and the 53-kd endogenous neural parkin in the LB-positive but not in LB-negative extracts. In addition, insoluble gel-excluded, and soluble proteolytic fragments were detected by anti-parkin, including an 11- to 12-kd N-terminal fragment detected by HP6A and HP1A (Figure 5, B and C) ▶ . The high Mr, gel-excluded parkin immunoreactivity seen in the LB-positive samples was never observed in soluble or membrane-associated fractions processed from normal or DLB brain (not shown). Taken together with the data in Figure 1 ▶ , these results make a cross-reactivity of parkin antibodies with αS or Ub unlikely and suggest that parkin proteins occur within pathological αS aggregates.
Figure 5.

Characterization of affinity-isolated cortical Lewy bodies from DLB brain. A: Schematic diagram of Percoll density gradient fractions that contained LB-rich material (LB-positive) isolated by magnetic beads coupled to sheep anti-αS antibody. 41 B–D: PAGE and Western blotting (WB) of equal volumes of extracts from normal (LB-negative) and DLB (LB-positive) brain, and lysates (16 μg) of mycParkin-expressing transfected cells (293, CHO). Antibodies depicted are to the N-terminus of parkin (HP6A) and to αS (LB509, syn-1) (B); to parkin’s linker domain (HP1A) and to Ub (C); and to the in-between-RING domain of parkin (HP2A and HP2A abs.) (D). Note, in LB-positive material anti-parkin antibodies detect mature 53-kd parkin, an 11- to 12-kd fragment (open arrowheads), and gel-excluded high Mr parkin isoforms (asterisks). Anti-αS antibodies faintly recognize an ∼22-kd protein (black arrowhead). Brackets indicate oligomeric proteins.
Next, we subjected aliquots of the affinity-isolated cortical LBs to immunoelectron microscopy with anti-αS and anti-parkin, as described. 41 Anti-parkin HP2A (Figure 6c) ▶ , but not HP2A preabsorbed with its cognate antigen (Figure 6d) ▶ , specifically decorated filamentous material in a comparable but less frequent manner than the anti-αS antibody (Figure 6b) ▶ , as expected from the immunohistochemical staining (Figure 6a) ▶ . Thus, these biochemical and immunoelectron microscopic analyses of affinity-isolated αS-aggregates show that the 53-kd parkin and related isoforms occur in cortical LBs of DLB brain.
Figure 6.
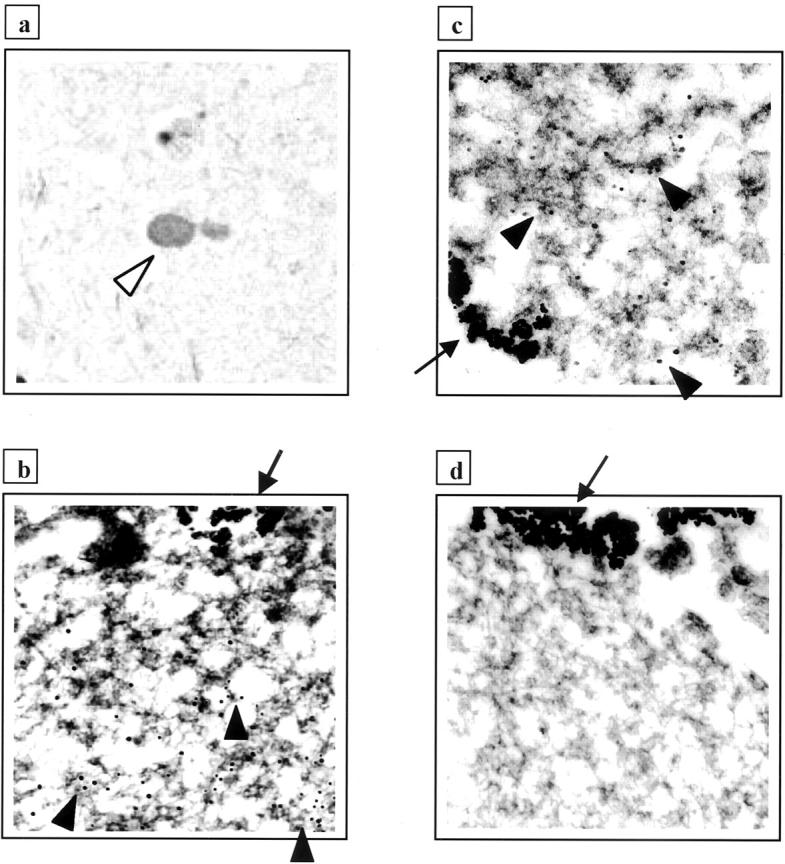
Light and electron microscopic localization of parkin protein in Lewy bodies (LB) from DLB cortex. a: LB staining (white arrowhead) in entorhinal cortex from DLB by immunohistochemistry with anti-parkin HP2A. b to d: LBs were isolated by magnetic beads (arrows) as in Figure 5A ▶ ; only segments of whole LBs are depicted. Antibody decoration of filamentous material by 12-nm gold particles (black arrowheads) using anti-αS (b) and anti-parkin HP2A (c), but not with preabsorbed HP2A (d).
The observation of an 11- to 12-kd, N-terminal parkin fragment in LB-positive extracts and HEK293 cells by two parkin antibodies, HP6A and HP1A (Figure 5, B and C) ▶ , and of a ∼42-kd C-terminal parkin fragment in brain lysates (Figure 1C) ▶ prompted us to investigate whether mature parkin (53 to 54 kd) is physiologically cleaved to release its Ub-like domain. In contrast to the integral type-2 Ub-like domains, type-1 Ub-like proteins (with Ub as the prototype) are activated by cleavage after a signature –GG motif and then covalently bound to protein substrates. 49 Intriguingly, we identified two –NAxGG- motifs near the end of the Ub-like domain of human parkin (Figure 7A) ▶ . 3 Therefore, we first engineered cDNAs encoding N-terminal parkin fragments with nascent C-termini of –GG84/85 or –GG93/94, and a C-terminal parkin protein lacking the Ub-like domain (Figure 7A) ▶ . When the constructs encoding wild-type mycParkin, mycUbl1–94, and ΔUblParkin were expressed in in vitro translation assays, we observed proteins of Mr 54, 12, and 42 kd, respectively (Figure 7B) ▶ , matching the Mr of the proteolytic parkin isoforms observed above. To determine whether the cleavage of mature mycParkin observed in HEK293 cells required intact residue(s) at G85 and/or G94, as would be expected for a type-1 Ub-like protein, 49 we engineered mycParkin cDNAs that encoded G85A or G94A substitutions or both (Figure 7A) ▶ . Transient expression of these constructs in HEK293 cells showed that cleavage of mycParkin was neither diminished in the single mutants nor abrogated in the double mutant (Figure 7C) ▶ . Thus, in transfected HEK293 cells mycParkin was apparently cleaved near the end of its Ub-like domain, a proteolysis that did not require intact -GG motifs at amino acids 84 and 85 or 93 and 94.
Figure 7.

Mutational analysis of parkin’s cleavage in HEK293 cells. A: Diagram of two –NAxGG- motifs (underlined) in the sequence of human parkin (numbering according to Kitada et al 3 ), and of cDNA constructs encoding parkin proteins, as indicated. B: PAGE and autoradiogram of in vitro-translated cDNA constructs encoding full-length and truncated parkin proteins. C: PAGE and Western blotting (WB) of HEK293 cell lysates transfected with cDNA constructs encoding wild-type and mutant parkin proteins, as indicated. Note, generation of the 42-kd C-terminal parkin fragment was not abolished in parkin mutants, as detected by anti-parkin HP5A. Identical results were obtained using anti-parkin HP2A. Asterisks in B and C denote nonspecific background bands.
To determine whether the E3 Ub ligase, parkin, its predominant E2-binding partner, UbcH7, and αS are present within the same subcellular compartment of the adult nervous system, we fractionated fresh rat brain homogenates. Synaptosomal preparations were prepared by a previously published protocol (avoiding a hypotonic lysis step of synaptosomes) 50 and processed to yield purified pre- and postsynaptic elements (see Materials and Methods). All fractions were first screened with antibodies to synaptophysin and postsynaptic density protein-95 (PSD-95) to determine their purity and then probed with αS, parkin, and UbcH7 antibodies (Figure 8) ▶ . The 53-kd HP2A-positive parkin protein localized predominantly to the cytosolic (CY) and postsynaptic (PSD) fractions (Figure 8, a and d) ▶ . Importantly, parkin was also found in the presynaptic (PRE) fractions (Figure 8a) ▶ 51 that contained synaptophysin and αS (Figure 8, b and c) ▶ , two membrane- and synaptic vesicle-associated marker proteins, respectively. 50,52 UbcH7 was detected in the cytosolic and presynaptic fractions, but was absent postsynaptically (Figure 8e) ▶ . The finding of mature parkin within the purified presynaptic terminal fraction (PRE) was not because of contamination with postsynaptic proteins because two abundant postsynaptic markers, PSD-95 (Figure 8d) ▶ 53 and δ-catenin were undetectable. Little parkin reactivity was detected in nuclear pellets and myelin-rich fractions. Preabsorption of HP2A abolished the 53-kd band (not shown). We conclude that neural parkin and its E2-binding partner, UbcH7, physiologically co-localize with αS in at least two subcellular compartments of adult brain, cytosol and presynaptic terminals.
Figure 8.

a–e: Subcellular fractionation of adult rat brain generated total homogenate (HO), cytosol (CY), crude synaptosomes (CS), purified synaptosomes (PS), crude (TS) and purified (PRE) presynaptic terminal fractions, crude (TP), and purified (PSD) postsynaptic terminal fractions. Sixteen μg of each sample were loaded for PAGE. Western blots were probed with antibodies to parkin (HP2A, a), synaptophysin (b), αS (syn-1, c), PSD-95 (d), and UbcH7 (e). Lysates of HEK293 cells (293) expressing both mycParkin and untagged αS (a–d) or of Jurkat cells expressing UbcH7 (e) were analyzed in parallel. Note, that parkin, αS, and UbcH7 are present in the highly purified (PSD-95-negative) presynaptic fraction, PRE (vertical arrow).
Discussion
In this study, we demonstrate a close association between the two principal proteins, parkin and αS, known to cause inherited forms of PD. A pathophysiological link between the two proteins is supported by several lines of evidence. First, parkin epitopes are present by immunohistochemistry within classical LBs of sporadic PD, inherited PD, and DLB at three distinct sites in the nervous system. Second, the 53-kd mature parkin protein is detected biochemically in affinity-isolated LBs from DLB brain. And third, parkin shows tight intermolecular association with the principal LB constituent, αS, by both FRET analysis and immunoelectron microscopy.
Recently, Huynh and colleagues 32 reported a lack of parkin immunoreactivity in LBs. This discrepancy could be explained by differences in the affinity and avidity of the parkin antibodies, the degree of unmasking of parkin antigens within the densely packed insoluble αS fibrils of LBs, or conformational changes and posttranslational modifications of neuronal parkin. For example, HP6A (to amino acids 6 to 15) (Figure 1C ▶ and Figure 5B ▶ ) 28 and HP5A (amino acids 453 to 465) (Figure 7C) ▶ show robust binding to human parkin isoforms on immunoblots but fail to label LBs in brain sections (pretreatment with formic acid did not change these results). HP2A (amino acids 342 to 353) and HP1A (amino acids 84 to 98) have similar reactivities on Western blots (Figure 1C ▶ and Figure 5 ▶ ) and by immunoprecipitation 28 yet differ in their neuronal staining patterns in tissue sections: HP1A recognizes more cytoplasmic parkin (as does HP7A), whereas HP2A is more selective for LB-associated parkin (Figures 2, 3, 4, and 6) ▶ ▶ ▶ . Consistent with this selectivity of HP2A, in our initial screen of human parkin epitopes the parkin-2 peptide sequence (amino acids 342 to 353) achieved the highest surface probability plot value among the five peptides we chose for antibody production (Figure 1A) ▶ (MGS and D Teplow, unpublished observation). Notably, HP2A’s antigen is located within the in-between-RING domain of parkin (Figure 1A) ▶ that is essential for the recruitment of UbcH7. 22,24,27
Our results demonstrate that the three proteins, parkin, UbcH7, and Ub, which have been found to interact functionally to ubiquitinate substrates in vivo and in vitro, 22,24,27,28 all localize predominantly to the core of brainstem LBs by immunohistochemistry (Figure 2) ▶ . 41 This observation supports our hypothesis of an initiating role for parkin (together with its binding partners and substrates) in the formation of LBs. Such a function would be lost in autosomal recessive PD brains that lack detectable parkin proteins 19,28 but would proceed in autosomal dominant, αS-linked PD (Figure 3) ▶ . We speculate that in parkin-linked PD cases with detectable mutant parkin proteins, 21 eg, patient Pw3, residual parkin function still promotes inclusion formation (Figure 4) ▶ but is inefficient in Ub ligation 31 and in maintaining dopaminergic neuronal integrity. In this regard, an inclusion-promoting role for another neuronal E3 Ub ligase, E6-AP, was recently demonstrated in vivo using a mouse model of the progressive neurodegenerative disorder, spinocerebellar ataxia, type-1. 54 Alternatively, parkin’s role in LB formation could be merely that of an E3 ligase that ubiquitinates already aggregated proteins, as shown in an inclusion body model of HEK293 cells that co-express αS and synphilin-1. 31 However, the absence of any detection of αS aggregates in fully parkin-deficient PD brains to date suggests a primary role for parkin in LB formation.
Our experiments identify the physiological co-localization of a small pool of parkin with UbcH7 and αS in cytosolic and presynaptic terminals of normal adult brain. These data are consistent with parkin’s detection at synaptic vesicle membranes by immunoelectron microscopy. 51 The localization of two mainly soluble proteins, parkin and UbcH7, to presynaptic elements (Figure 8, a and e) ▶ may be mediated by lipid modification 55 or by association with integral membrane or membrane-bound proteins. One candidate binding partner is CDC-rel1, a syntaxin-interacting protein 56 that was shown to associate with human parkin in HEK293 cells. 29 The abundance of parkin in postsynaptic fractions of adult rat brain (Figure 8a) ▶ suggests that binding partners (including Ub-conjugating enzymes other than UbcH7) and substrates also exist at this site. In accord, parkin’s C-terminus has recently been found to associate in rat brain with CASK, a postsynaptic protein in the NMDA receptor-signaling complex. 57 The dual distribution of parkin within both axonal (presynaptic) and dendritic (postsynaptic) compartments can also be observed in rat hippocampal neurons transfected with pmyc∼parkin cDNA (MGS and TO, unpublished data).
The apparent cleavage of a subset of mature parkin proteins just after their Ub-like domain (Figures 1 and 5) ▶ ▶ led us to examine whether parkin’s released N-terminal fragment was able to covalently modify protein substrates, analogous to classical type-1 Ub-like proteins (eg, Ub, SUMO-1), 49 and thus contribute to LB formation. However, our mutational analysis of mycParkin in HEK293 cells (Figure 7C) ▶ and the absence of any detectable conjugation of other proteins in nonneural cell cultures by the expression of Ubl1–94 or Ubl1–85 parkin fusion proteins (MGS and MM, unpublished data) suggest that parkin’s N-terminus acts in accordance with type-2 Ub-like domains (eg, Rad23, elongin B). 49 As such, its role would mainly involve protein-protein interaction, as has been recently demonstrated in in vitro experiments with heterologous parkin proteins. 28,58
In summary, our data identify parkin as the second protein that is both genetically linked to the PD phenotype in several pedigrees 3,4,21,59 and a constituent of its hallmark neuronal inclusions. This finding is consistent with our recent demonstration that an O-glycosylated form of αS, αSp22, is a substrate for parkin’s Ub ligase activity from human brain and that this interaction is absent in autosomal-recessive PD brains that lack detectable parkin proteins. 28 Intriguingly, we describe here a small amount of an ∼22-kd anti-αS-reactive protein in affinity-isolated LBs (Figure 5B) ▶ . Generation of glycosylation-specific antibodies to αS that do not react with the nascent 16-kd isoform may determine whether the interaction of parkin and αSp22 helps promote LB formation in human brain and constitutes a common biochemical pathway in PD and related synucleinopathies. Our results do not preclude an association of parkin with other cellular elements that help mediate its interaction with αS, eg, CDCrel-1, 29 synphilin-1, 31,60 lipid molecules, 41,61 or actin filaments, 32 because functional E3 Ub ligase activity in vivo frequently requires one or more adaptor molecules. 26
Acknowledgments
We thank D. Teplow, M. Baker, and C. Lemere for valuable advice; M. Farrer for his comments; L. Golbe and D. Dickson for tissue donation and thoughtful discussions of the clinical and neuropathological findings; and S. Mansourian and B. Sudarsky for technical assistance.
Footnotes
Address reprint requests to Michael Schlossmacher, M.D., Center for Neurologic Diseases, Brigham and Women’s Hospital, 77 Ave. Louis Pasteur, Boston, MA 02115. E-mail: schloss@cnd.bwh.harvard.edu.
Supported by grants from the Lefler Foundation, the Grass Foundation, the National Institutes of Health (NS02127 to M. G. S.), the P. Beeson Award (to M. P. F.), the National Health & Medical Research Council of Australia (to W. P. G.), the Veterans’ Administration Medical Research Program (to L. F.), and the Morris R. Udall Centers (NS38372 to B. T. H. and NS38375 to M. P. F., D. J. S., and K. S. K.).
D. J. S. and K. S. K. both contributed equally to this work.
References
- 1.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Diiorio G, Golbe LI, Nussbaum RL: Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276:2045-2047 [DOI] [PubMed] [Google Scholar]
- 2.Krüger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O: Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998, 18:106-108 [DOI] [PubMed] [Google Scholar]
- 3.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392:605-608 [DOI] [PubMed] [Google Scholar]
- 4.Lücking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A: Association between early-onset Parkinson’s disease and mutations in the parkin gene. French Parkinson’s Disease Genetics Study Group. N Engl J Med 2000, 342:1560-1567 [DOI] [PubMed] [Google Scholar]
- 5.Ujike H, Yamamoto M, Yamaguchi K, Kanzaki A, Takagi M, Kuroda S: Two cases of sporadic juvenile Parkinson’s disease caused by homozygous deletion of Parkin gene. No To Shinkei 1999, 51:1061-1064 [PubMed] [Google Scholar]
- 6.Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F: Brain dopamine and the syndromes of Parkinson and Huntington. J Neurol Sci 1973, 20:415-455 [DOI] [PubMed] [Google Scholar]
- 7.Golbe LI, Di Iorio G, Sanges G, Lazzarini AM, La Sala S, Bonavita V, Duvoisin RC: Clinical genetic analysis of Parkinson’s disease in the Contursi kindred. Ann Neurol 1996, 40:767-775 [DOI] [PubMed] [Google Scholar]
- 8.Forno LS, DeLanney LE, Irwin I, Langston JW: Electron microscopy of Lewy bodies in the amygdala-parahippocampal region. Comparison with inclusion bodies in the MPTP-treated squirrel monkey. Adv Neurol 1996, 69:217-228 [PubMed] [Google Scholar]
- 9.Dawson TM: New animal models for Parkinson’s disease. Cell 2000, 101:115-118 [DOI] [PubMed] [Google Scholar]
- 10.Kosaka K, Oyanagi S, Matsushita M, Hori A: Presenile dementia with Alzheimer-, Pck- and Lewy body changes. Acta Neuropathol (Berl) 1976, 36:221-233 [DOI] [PubMed] [Google Scholar]
- 11.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH: Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. Neurology 1996, 47:1113-1124 [DOI] [PubMed] [Google Scholar]
- 12.Litvan I, MacIntyre A, Goetz CG, Wenning GK, Jellinger K, Verny M, Bartko JJ, Jankovic J, McKee A, Brandel JP, Chaudhuri KR, Lai EC, D’Olhaberriague L, Pearce RK, Agid Y: Accuracy of the clinical diagnoses of Lewy body disease, Parkinson disease, and dementia with Lewy bodies: a clinicopathologic study. Arch Neurol 1998, 55:969-978 [DOI] [PubMed] [Google Scholar]
- 13.Spillantini MG, Schmidt ML, Lee VM, Trojanowski LQ, Jakes R, Goedert M: Alpha-synuclein in Lewy bodies. Nature 1997, 388:839-840 [DOI] [PubMed] [Google Scholar]
- 14.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M: Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 1998, 95:6469-6473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T: Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 1998, 152:879-884 [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi H, Ohama E, Suzuki S, Horikawa Y, Ishikawa A, Morita T, Tsuji S, Ikuta F: Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology 1994, 44:437-441 [DOI] [PubMed] [Google Scholar]
- 17.Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y: Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 1998, 51:890-892 [DOI] [PubMed] [Google Scholar]
- 18.Hayashi S, Wakabayashi K, Ishikawa A, Nagai H, Saito M, Maruyama M, Takahashi T, Ozawa T, Tsuji S, Takahashi H: An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord 2000, 15:884-888 [DOI] [PubMed] [Google Scholar]
- 19.Shimura H, Hattori N, Kubo S, Yoshikawa M, Kitada T, Matsumine H, Asakawa S, Minoshima S, Yamamura Y, Shimizu N, Mizuno Y: Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann Neurol 1999, 45:668-672 [DOI] [PubMed] [Google Scholar]
- 20.van de Warrenburg BP, Lammens M, Lucking CB, Denefle P, Wesseling P, Booij J, Praamstra P, Quinn N, Brice A, Horstink MW: Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 2001, 56:555-557 [DOI] [PubMed] [Google Scholar]
- 21.Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, Singleton A, Tanner C, Hardy J, Langston JW: Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 2001, 50:293-300 [DOI] [PubMed] [Google Scholar]
- 22.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T: Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 2000, 25:302-305 [DOI] [PubMed] [Google Scholar]
- 23.Morett E, Bork P: A novel transactivation domain in parkin. Trends Biochem Sci 1999, 24:229-231 [DOI] [PubMed] [Google Scholar]
- 24.Ardley HC, Tan NG, Rose SA, Markham AF, Robinson PA: Features of the parkin/ariadne-like ubiquitin ligase, HHARI, that regulate its interaction with the ubiquitin-conjugating enzyme, Ubch7. J Biol Chem 2001, 276:19640-19647 [DOI] [PubMed] [Google Scholar]
- 25.Hartwig A: Zinc finger proteins as potential targets for toxic metal ions: differential effects on structure and function. Antioxid Redox Signal 2001, 3:625-634 [DOI] [PubMed] [Google Scholar]
- 26.Jackson PK, Eldridge AG, Freed E, Furstenthal L, Hsu JY, Kaiser BK, Reimann JD: The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol 2000, 10:429-439 [DOI] [PubMed] [Google Scholar]
- 27.Imai Y, Soda M, Takahashi R: Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem 2000, 275:35661-35664 [DOI] [PubMed] [Google Scholar]
- 28.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ: Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 2001, 293:263-269 [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM: Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA 2000, 97:13354-13359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanaka K, Suzuki T, Chiba T, Shimura H, Hattori N, Mizuno Y: Parkin is linked to the ubiquitin pathway. J Mol Med 2001, 79:482-494 [DOI] [PubMed] [Google Scholar]
- 31.Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM: Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med 2001, 7:1144-1150 [DOI] [PubMed] [Google Scholar]
- 32.Huynh DP, Scoles DR, Ho TH, Del Bigio MR, Pulst SM: Parkin is associated with actin filaments in neuronal and nonneural cells. Ann Neurol 2000, 48:737-744 [PubMed] [Google Scholar]
- 33.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R: An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 2001, 105:891-902 [DOI] [PubMed] [Google Scholar]
- 34.Conway KA, Harper JD, Lansbury PT: Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med 1998, 4:1318-1320 [DOI] [PubMed] [Google Scholar]
- 35.Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ, Lemere CA: Temporal accrual of complement proteins in amyloid plaques in Down’s syndrome with Alzheimer’s disease. Am J Pathol 2000, 156:489-499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma N, McLean PJ, Kawamata H, Irizarry MC, Hyman BT: Alpha-synuclein has an altered conformation and shows a tight intermolecular interaction with ubiquitin in Lewy bodies. Acta Neuropathol (Berl) 2001, 102:329-334 [DOI] [PubMed] [Google Scholar]
- 37.Sharma N, Hewett J, Ozelius LJ, Ramesh V, McLean PJ, Breakefield XO, Hyman BT: A close association of torsinA and alpha-synuclein in Lewy bodies: a fluorescence resonance energy transfer study. Am J Pathol 2001, 159:339-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Root DD: In situ molecular association of dystrophin with actin revealed by sensitized emission immuno-resonance energy transfer. Proc Natl Acad Sci USA 1997, 94:5685-5690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gai W, Power J, Blumbergs P, Blessing W: Multiple-system atrophy: a new alpha-synuclein disease? Lancet 1998, 352:547-548 [DOI] [PubMed] [Google Scholar]
- 40.Jensen PH, Islam K, Kenney J, Nielsen MS, Power J, Gai WP: Microtubule-associated protein 1B is a component of cortical Lewy bodies and binds alpha-synuclein filaments. J Biol Chem 2000, 275:21500-21507 [DOI] [PubMed] [Google Scholar]
- 41.Gai WP, Yuan HX, Li XQ, Power JT, Blumbergs PC, Jensen PH: In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp Neurol 2000, 166:324-333 [DOI] [PubMed] [Google Scholar]
- 42.Huttner WB, Schiebler W, Greengard P, De Camilli P: Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 1983, 96:1374-1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ochiishi T, Sugiura H, Yamauchi T: Characterization and autophosphorylation of Ca2+/calmodulin-dependent protein kinase in the postsynaptic density of the rat forebrain. Brain Res 1993, 610:97-107 [DOI] [PubMed] [Google Scholar]
- 44.Iwanaga K, Wakabayashi K, Yoshimoto M, Tomita I, Satoh H, Takashima H, Satoh A, Seto M, Tsujihata M, Takahashi H: Lewy body-type degeneration in cardiac plexus in Parkinson’s and incidental Lewy body diseases. Neurology 1999, 52:1269-1271 [DOI] [PubMed] [Google Scholar]
- 45.Kaufmann H, Hague K, Perl D: Accumulation of alpha-synuclein in autonomic nerves in pure autonomic failure. Neurology 2001, 56:980-981 [DOI] [PubMed] [Google Scholar]
- 46.Golbe L, DiIorio G, Sanges G, Lazzarini A, LaSala S, Bonavita V, Duvoisin R: Clinical genetic analysis of Parkinson’s disease in the Contursi kindred. Ann Neurol 1996, 40:767-775 [DOI] [PubMed] [Google Scholar]
- 47.Golbe LI, Di Iorio G, Bonavita V, Miller DC, Duvoisin RC: A large kindred with autosomal dominant Parkinson’s disease. Ann Neurol 1990, 27:276-282 [DOI] [PubMed] [Google Scholar]
- 48.Trojanowski JQ, Goedert M, Iwatsubo T, Lee VM: Fatal attractions: abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Differ 1998, 5:832-837 [DOI] [PubMed] [Google Scholar]
- 49.Jentsch S, Pyrowolakis G: Ubiquitin and its kin: how close are the family ties? Trends Cell Biol 2000, 10:335-342 [DOI] [PubMed] [Google Scholar]
- 50.Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C: Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J Neurosci 2000, 20:6365-6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kubo SI, Kitami T, Noda S, Shimura H, Uchiyama Y, Asakawa S, Minoshima S, Shimizu N, Mizuno Y, Hattori N: Parkin is associated with cellular vesicles. J Neurochem 2001, 78:42-54 [DOI] [PubMed] [Google Scholar]
- 52.McLean PJ, Ribich S, Hyman BT: Subcellular localization of alpha-synuclein in primary neuronal cultures: effect of missense mutations. J Neural Transm (Suppl) 2000, 58:53-63 [DOI] [PubMed] [Google Scholar]
- 53.El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS: PSD-95 involvement in maturation of excitatory synapses. Science 2000, 290:1364-1368 [PubMed] [Google Scholar]
- 54.Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY: Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 1999, 24:879-892 [DOI] [PubMed] [Google Scholar]
- 55.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y: A ubiquitin-like system mediates protein lipidation. Nature 2000, 408:488-492 [DOI] [PubMed] [Google Scholar]
- 56.Beites CL, Xie H, Bowser R, Trimble WS: The septin CDCrel-1 binds syntaxin and inhibits exocytosis. Nat Neurosci 1999, 2:434-439 [DOI] [PubMed] [Google Scholar]
- 57.Fallon L, Moreau F, Croft BG, Labib N, Gu WJ, Fon EA: Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. J Biol Chem 2002, 277:486-491 [DOI] [PubMed] [Google Scholar]
- 58.Tsai Y, Thakor N, Fishman P, Oyler G: Parkin facilitates degradation of misfolded proteins. Soc Neurosci 2001, 27:607A [Google Scholar]
- 59.Hattori N, Shimura H, Kubo S, Kitada T, Wang M, Asakawa S, Minashima S, Shimizu N, Suzuki T, Tanaka K, Mizuno Y: Autosomal recessive juvenile parkinsonism: a key to understanding nigral degeneration in sporadic Parkinson’s disease. Neuropathology 2000, 20(Suppl):S85-S90 [DOI] [PubMed] [Google Scholar]
- 60.Wakabayashi K, Engelender S, Tanaka Y, Yashimoto M, Mori F, Tsuji S, Ross CA, Takahashi H: Immunocytochemical localization of synphilin-1, an alpha-synuclein-associated protein, in neurodegenerative disorders. Acta Neuropathol (Berl) 2002, 103:209-214 [DOI] [PubMed] [Google Scholar]
- 61.Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ: Alpha-synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci USA 2001, 98:9110-9115 [DOI] [PMC free article] [PubMed] [Google Scholar]