

Abstract
Hypersynchronous neuronal firing is a hallmark of epilepsy, but the mechanisms underlying simultaneous activation of multiple neurons remains unknown. Epileptic discharges are in part initiated by a local depolarization shift that drives groups of neurons into synchronous bursting. In an attempt to define the cellular basis for hypersynchronous bursting activity, we studied the occurrence of paroxysmal depolarization shifts after suppressing synaptic activity by TTX and voltage-gated Ca2+ channel blockers. Here we report that paroxysmal depolarization shifts can be initiated by release of glutamate from extrasynaptic sources or by photolysis of caged Ca2+ in astrocytes. Two-photon imaging of live exposed cortex revealed that several anti-epileptics, including valproate, gabapentin and phenytoin, reduced the ability of astrocytes to transmit Ca2+ signaling. Our results reveal an unanticipated key role for astrocytes in seizure activity. As such, these findings identify astrocytes as a proximal target for the treatment of epileptic disorders.
Epilepsy is a neurological disorder in which normal brain function is disrupted as a consequence of intensive burst activity from groups of neurons1. Epilepsies result from long-lasting plastic changes in the brain affecting the expression of receptors and channels, and involve sprouting and reorganization of synapses, as well as reactive gliosis2,3. Several lines of evidence suggest a key role of glutamate in the pathogenesis of epilepsy. Local or systemic administration of glutamate agonists triggers excessive neuronal firing, whereas glutamate receptor (GluR) antagonists have anticonvulsant properties4. The observation that astrocytes release glutamate via a regulated Ca2+ dependent mechanism5-8 prompted us to hypothesize that glutamate released by astrocytes plays a causal role in synchronous firing of large populations of neurons.
Paroxysmal depolarization shifts (PDSs) are abnormal prolonged depolarizations with repetitive spiking and are reflected as interictal discharges in the electroencephalogram2,3. We report here that glutamate released by astrocytes can trigger PDSs in several models of experimental seizure. A unifying feature of seizure activity was its consistent association with antecedent astrocytic Ca2+ signaling. Oscillatory, TTX-insensitive increases in astrocytic Ca2+ preceded or occurred concomitantly with PDSs, and targeting astrocytes by photolysis of caged Ca2+ evoked PDSs. Furthermore, several anti-epileptic agents, including valproate, gabapentin, and phenytoin, potently reduced astrocytic Ca2+ signaling detected by 2-photon imaging in live animals. This suggests that pathologic activation of astrocytes may play a central role in the genesis of epilepsy, as well in the pathways targeted by current anti-epileptics.
RESULTS
PDSs can be triggered by an action potential- independent mechanism
To examine the cellular mechanism underlying PDSs, we patched CA1 pyramidal neurons in rat hippocampal slices exposed to 4-aminopyridine (4-AP). 4-AP is a K+ channel blocker that induces intense electrical discharges in slices9 and seizure activity in experimental animals10. All slices exposed to 4-AP (61 slices from 23 rats) exhibited epileptiform bursting activity expressed as transient episodes of neuronal depolarizations eliciting trains of action potentials (Fig. 1a). Bath application of TTX promptly eliminated neuronal firing (Fig. 1b). Unexpectedly, the paroxysmal neuronal depolarization events evoked by 4-AP were largely TTX-insensitive (Fig. 1b). Pyramidal neurons exposed to 4-AP continued to exhibit 10–30 mV depolarization shifts after addition of TTX, despite complete suppression of action potentials (Fig. 1b). To ensure that all synaptic activity was eliminated, we added a mixture of voltage-gated Ca2+ channel (VGCC) blockers, including nifedipine, mibefradil, omega-conotoxin MVIIC, omega-conotoxin GVIA, and SNX-48211. Notably, this cocktail of VGCC blockers did not suppress the expression of 4-AP-induced PDSs compared with TTX alone (Fig. 1b vs d). In contrast to neurons, voltage changes in astrocytes during PDSs were minor, 0.5–2 mV, in accordance with the non-excitable properties of astrocytic plasma membranes (Fig. 1e,f).
Figure 1.

Synaptic activity is not required for PDSs in hippocampal slices evoked by 4-AP. (a) Whole-cell recording of CA1 pyramidal neuron during epileptiform activity triggered by 4-AP (100 μM, upper trace) combined with field potential recording (lower trace). Spontaneous neuronal depolarization events elicit trains of action potentials, which are mirrored by negative deflections of the field potential. (b) Addition of TTX (1 μM) eliminated neuronal firing, but not the transient episodes of neuronal depolarizations and the drop in field potential. (c) 4-AP induced PDSs in a CA1 pyramidal neuron, (d) continue in presence of a cocktail of voltage-gated Ca2+ blockers, Nifedipine (L-type channel blocker, 10 μM); Mibefradil (T-type channel blocker, 10 μM); Omega-Conotoxin MVIIC (P/Q type Blocker, 1 μM); Omega-Conotoxin GVIA (N-type blocker, 1 μM); SNX-482 (R-type blocker, 0.1 μM) and TTX (1 μM). (e) Astrocytic membrane potential declined 0.5–1.0 mV during PDSs before, and (f) after addition of TTX. In all recordings, the field potential electrode was placed less than 30 μm from either the neuronal (a–d), or astrocytic cell body (e–f).
Combined, these experiments demonstrate that PDSs can be triggered by an action potential-independent mechanism. Neurons exhibited a 16 ± 5 mV (n = 24) depolarization shift, whereas astrocytes only display a modest change in membrane potential (0.5 ± 0.2 mV, n = 22) during PDSs in the presence of TTX.
Glutamate release mediates PDSs
To examine the role of glutamate released from action potential-independent sources in PDSs, we next quantified the occurrence of PDSs in the presence of TTX and GluR antagonists. The PDSs evoked by 4-AP resulted primarily from activation of ionotropic glutamate receptors, because APV and CNQX potently reduced both the frequency and the amplitude of the PDSs, in accordance with earlier studies (Fig. 2a-c)4. Washout of TTX, APV, and CNQX resulted in partial recovery of PDSs (Fig. 2a-c). Addition of VGCC blockers did not cause an additional decrease in the frequency and amplitude of PDSs compared with TTX alone, further supporting the notion that glutamate was released from an action potential-independent source (Fig. 2d). PDSs persisted in the presence of D,L-threo-beta-benzyloxyaspartate (TBOA, a glutamate transport inhibitor), and TBOA increased the frequency and amplitude of PDSs significantly suggesting that inverted transport of glutamate did not contribute to PDSs (Fig. 2e).
Figure 2.
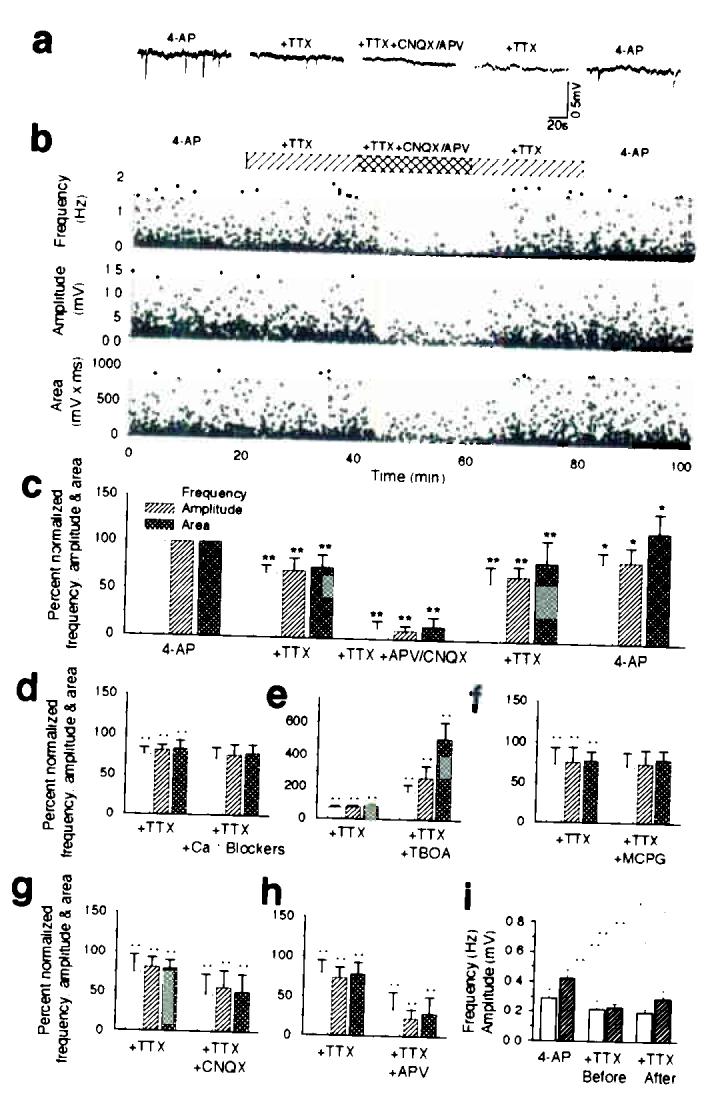
PDSs are mediated by release of glutamate from action potential-independent sources. (a) Representative traces of field potential recording in 4-AP; 4-AP and TTX; 4-AP, TTX, APV (50 μM), and CNQX (20 μM). (b) Frequency, amplitude and area (amplitude × duration) plotted as a function of time (n = 7). Spontaneous field potential events were observed in all slices exposed to 4-AP. The frequency and amplitude of PDSs were reduced by 20–35% by TTX and by 85–90% by APV and CNQX. (c) Normalized mean values of frequency, amplitude, and area (amplitude × duration) during exposure to 4-AP, 4-AP + TTX, 4-AP + TTX + APV/CNQX, during washout of APV/CNQX (4-AP + TTX), and during washout of TTX (4-AP) (n = 7). (d) The cocktail of VGCC blockers (Nifedipine, Mibefradil, Omega-Conotoxin MVIIC, Omega-Conotoxin GVIA, SNX-482, same concentration as in Fig. 1 and TTX did not decrease the frequency or amplitude of PDSs compared with TTX alone (n = 5). (e) D,L-threo-beta-benzyloxyaspartate (TBOA, a glutamate transport inhibitor, 100 μM) did not reduce the occurrence of PDSs, but increased the frequency, amplitude and area of PDSs significantly suggesting that inverted transport of glutamate did not contribute to PDSs (n = 6). (f) (S)-Alpha-methyl-4-carboxy-phenylglycine ((S)-MCPG, a non-selective mGluR antagonist, 1 mM) did not decrease the frequency or amplitude of PDSs compared with TTX alone (n = 7). (g) CNQX alone significantly reduced PDSs (n = 6). (h) APV alone highly significantly reduced PDSs (n = 6). (i) TTX added before (10–15 min) had no effect on frequency of PDSs, but significantly reduced the amplitude of PDSs compared with slices first exposed to TTX 20 min after addition of 4-AP (n = 7). *, P < 0.05; **, P < 0.001; student's t-test; mean ± s.d..
Addition of TTX and CNQX (no APV) resulted in a significant decrease in the occurrence of PDSs compared with TTX alone (Fig. 2g), whereas TTX and APV compared with TTX alone, displayed a highly significant reduction in both frequency and amplitude of PDSs (Fig. 2h). Thus, the larger fraction (57%) of TTX-insensitive PDSs is caused by activation of NMDA receptors, whereas activation of AMPA receptors plays less of a significant role in generation of PDSs (26%). (S)-Alpha-methyl-4-carboxyphenylglycine ((S)-MCPG, a non-selective mGluR antagonist)12 failed to reduce the frequency and amplitude of PDSs (Fig. 2f), indicating that the TTX-insensitive PDSs were not elicited by activation of mGluRs.
In all experiments thus far, TTX was first added after the hippocampal slices had been exposed for 20 min to 4-AP (Fig.1 and Fig 2a-h). To test the possibility that astrocytic activation was secondary to neuronal bursting activity triggered by 4-AP, we next added TTX (10–15 min) before exposing the slices to 4-AP (Fig. 2i). Interestingly, when TTX was added before 4-AP, the frequency and amplitude of 4-AP induced PDSs were only slightly decreased (Fig. 2i).
Combined, these observations show that TTX decreased the relative frequency of PDSs by 32 ± 8% (P = 0.001) compared with 4-AP alone (Fig. 2a-c). These observations suggest that 4-AP, in addition to its well known effects on neurons13, evoked a large number of paroxysmal depolarization events (approx 70% of total) which were triggered by release of glutamate from extrasynaptic sources.
PDSs in several acute seizure models
Seizures can be induced by a variety of inciting agents with apparently unrelated mechanisms of action. The traditionally defined mechanisms of epileptogenesis involve either the facilitation of excitatory synaptic activity, or the suppression of inhibitory transmission. To assess whether glutamate release from action potential-independent sources plays a role in experimental epilepsy, we analyzed the dependence of PDSs upon TTX and glutamate receptor antagonists in several seizure models. A common approach to induce hypersynchronous burst activity of large groups of neurons is to enhance excitatory synaptic activity by removing extracellular Mg2+. The epileptogenic action of Mg2+depletion has been attributed to the activation of NMDA receptors at the resting membrane potential14. In accordance with earlier reports, Mg2+-free solution triggered repeated PDSs (Fig. 3a). These, however, were sustained in the presence of TTX despite complete elimination of action potentials, whereas APV/CNQX blocked more than 80% of PDSs (Fig. 3a). Thus, PDSs evoked by low extracellular Mg2+ appeared to result from glutamate released from action potential-independent sources, in addition to the removal of the Mg2+ block on NMDA receptors. Bicuculline and penicillin are potent convulsants in slice preparations15 and in animal models16. The epileptogenic actions of bicuculline and penicillin have been ascribed to their antagonism of GABAA receptors15. Our recordings, however, suggested that both bicuculline and penicillin, similar to 4-AP and Mg2+-free solution, triggered TTX-insensitive depolarization shifts resulting from extrasynaptic glutamate release and reception (Fig. 3b,c). Simply lowering extracellular Ca2+ generates slow-wave and late-burst activity similar to seizures that occur in patients with hippocampal epilepsy17. As in the other seizure models, Ca2+-free solution induced repeated TTX-insensitive depolarization shifts, resulting from activation of neuronal glutamate receptors (Fig. 3d). We compared the effect of TTX added 10–15 min prior to inducing seizure activity vs addition of TTX 20 min later (when seizure activity was maximal). If TTX was added first, the frequency of PDSs were reduced in slices exposed to Mg2+-free solution, bicuculline, and penicillin, but not in slices incubated in Ca2+-free solution (Fig. 3). Similar to 4-AP (Fig. 2i), however, the major fraction of PDSs occurred independently of TTX addition before or after induction of seizure activity.
Figure 3.
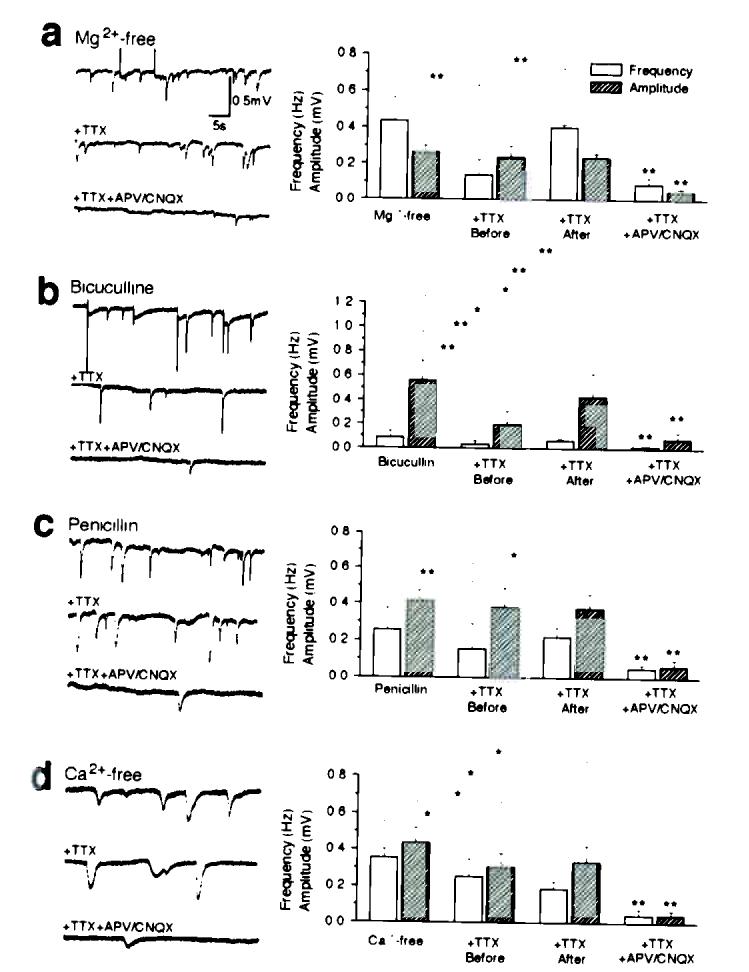
Spontaneous depolarization shifts in four experimental models of epilepsy. (a) Hippocampal slices were perfused with Mg2+-free solutions. Traces in left panel are representative field potential recordings in: Mg2+-free solution (upper); after addition of 1 μM TTX (middle), and after addition of TTX + 50 μM APV and 20 μM CNQX (lower). Left panel plots the frequency, amplitude, and area (amplitude × duration) of PDSs. (b–d) Similar sets of observations in hippocampal slices exposed to bicuculline (b, 30 μM), Penicillin (c, 2000 U/ml), and Ca2+-free solution (d, 1 mM EGTA). *, P < 0.05; **, P < 0.001; student's t-test; mean ± s.d.; n = 5–7.
Thus, in all experimental models of seizure analyzed, including exposure to 4-AP, Mg2+-free solution, bicuculline, penicillin, and removal of extracellular Ca2+, PDSs were largely insensitive to TTX. Depending upon the model, TTX (and VGCC blockers) reduced the frequency of PDSs to 70–90% of total, demonstrating that the majority of PDSs was evoked by action potential-independent pathways. Another key observation was that glutamate is the principal mediator of TTX-insensitive PDS, because combined exposure to APV/CNQX/MCPG decreased the frequency of PDSs to 5–20%. The TTX- and APV/CNQX/MCPG- insensitive PDS might be elicited by other action potential-independent mechanisms, including gap junctions17 and purinergic receptor activation possibly mediated by release of ATP by astrocytes18,19.
TTX-insensitive astrocytic Ca2+ signaling in experimental seizure models
Our recordings in hippocampal slices indicated that the cellular hallmark of epileptic discharge, PDSs, is caused by prolonged episodes (∼500 ms) of neuronal depolarization triggered by glutamate release from a non-synaptic source. Since a number of studies have documented that astrocytes can release glutamate in a Ca2+-dependent manner6-8, we asked whether activation of astrocytic Ca2+ signaling was a unifying feature of epileptogenesis. Hippocampal slices were loaded with the Ca2+ indicator, fluo-4/AM and viewed by two-photon laser scanning microscopy. The preferential loading of fluorescent acetoxymethyl esters indicators by astrocytes has been extensively reported20-22. Bath application of 4-AP potently initiated astrocytic Ca2+ signaling expressed as infrequent Ca2+ oscillations (Fig. 4a). TTX did not reduce either the frequency or the amplitude of astrocytic Ca2+ oscillations, indicating that astrocytic activation was not an indirect effect of transmitters released during neuronal firing, but resulted from a direct action of 4-AP on astrocytes (paired t-test, P = 0.4–0.8). In fact, 4-AP promptly induced Ca2+ signaling in cultured astrocytes, in the absence of co-cultured neurons, in accordance with previous publications (Fig. 4b)13. Frequent TTX-insensitive Ca2+ oscillations were also observed in hippocampal slices exposed to Mg2+-free solution, bicuculline, penicillin, and Ca2+-free solution. Thus, all paradigms of experimental seizure studied potently triggered Ca2+ signaling of astrocytes in hippocampal slices in the absence of action potentials. Astrocytic Ca2+ signaling was expressed as slow oscillatory elevations of cytosolic Ca2+ lasting 10–60 s in individual astrocytes, but small groups of neighboring astrocytes also frequently displayed synchronized increases in Ca2+. Similar results were obtained in cultures of astrocytes (Fig. 4b) with the exception that bicuculline only weakly induced Ca2+ signaling. We have no explanation for the difference response to bicuculline, but culturing of astrocytes is associated with major alterations of both morphology and receptor expression23. We next combined Ca2+ imaging with field potential recordings to establish the temporal connection between the two events (Fig. 4c). When the field electrode was placed in close proximity to the astrocytic cell body, at an average distance of 22 ± 2 μm (range 10 to 30 μm, n = 23, 7 slices), we found that oscillatory increases in astrocytic Ca2+ were linked to a negative shift in field potential (0.38 ± 0.06 mV, range 0.2 to 1.17 mV, 23 of a total of 45 spontaneous astrocytic Ca2+ increases in a total of 8 slices). Interestingly, the oscillatory increase in astrocytic Ca2+ always preceded the drop in field potential. The average delay between the onset of astrocytic Ca2+ increase to the onset of a decrease in field potential was 0.38 ± 0.06 s (range 0.05 to 1.69 s) (Fig. 4c).
Figure 4.

Epileptogenic agents evoke oscillatory increases in astrocytic cytosolic Ca2+ concentration, which precedes PDSs, and PDSs are spatially confined to small domains. (a) Upper panel: 2-photon imaging of astrocytic Ca2+ oscillations in stratum radiatum of the CA1 region in hippocampal slices exposed to 4-AP (100 μM) and TTX (1 μM). The frames were acquired with an interval of 8.2 s following 20 min exposure to 4-AP and TTX. White arrows indicate astrocytes with oscillatory increases in Ca2+. Scale bar, 50 μm. Lower panel: Histogram showing the frequency of Ca2+ oscillations in hippocampal slices exposed to 4-AP (100 μM), Mg2+-free solution, bicuculline (30 μM), penicillin (2000 U/ml), and Ca2+-free solution with and without TTX (1 μM) (mean ± s.d., n = 7). (b) Average increases in cytosolic Ca2+ in cultured astrocytes in response to 4-AP, Mg2+-free solution, bicuculline, penicillin, and Ca2+-free solution (mean ± s.d., n = 3). *, P < 0.05; ANOVA with Dunnett compared with vehicle. (c) Upper panel: 2-photon imaging of Ca2+ signaling combined with the field recordings in hippocampal slices exposed to 4-AP. The pipette solution contained 1 μM fluorescein-dextran to make the electrode visible during imaging (red in pseudocolor). White arrow indicates an astrocyte with a transient increase in cytosolic Ca2+. Scale bar, 30 μm. Middle panel: The rise in astrocytic Ca2+ concentration (upper tracing) preceded the negative deflection of the field potential (lower tracing). Numbers on the Ca2+ trace represent images in the upper panel. Lower panel: Histogram maps the latency between the onset of oscillatory increases in Ca2+ with the onset of drop in field potential. In all cases, astrocytic Ca2+ increment preceded the depolarization shift. (d) Both electrodes were placed in stratum radiatum of CA1. Representative tracings and summary histograms of dual field potential recordings with the electrodes placed at a distance of less than 100 μM (left panel); 100–200 μm (middle panel); and greater than 200 μm apart (right panel). (e) One electrode of the paired recordings was placed in stratum pyramidale of CA1 and the other one in stratum radiatum with a distance of less than 100 μM. Left panel: Representative tracings (top) and summary histograms (bottom) of dual field potential recordings. Central panel: Expanding recording traces (top) within the shadow area in top of left panel, the rise in astrocytic Ca2+ concentration (bottom) preceded the negative deflections of the field potentials. The numbers and letters are indicated in right panel. Right panel: The top photo is a DIC image which indicates the locations of the two electrodes. The other three photos are the 2-photon images of Ca2+ signaling in hippocampal slice exposed to 4-AP. White arrows indicate astrocytes with transient increases in cytosolic Ca2+. Scale bar, 20 μm.
Astrocytes within the cortex and hippocampus are organized in essentially non-overlapping microdomains with an average diameter of 40–70 μm, reviewed in24. Since Ca2+ oscillations are restricted to 1–3 neighboring astrocytes, it is expected that the PDSs are limited to small (< 50–200 μm) regions. To establish the spatial territories of PDSs, we first recorded with two field electrodes in the stratum radiatum of CA1 (Fig. 4d). Paired events were arbitrarily defined as negative shifts that occurred within a 5 s window at both electrode sites. If the electrodes were positioned < 100 μm apart, 56% of the paroxysmal depolarizations were temporally synchronized (307 of 544 events occurred within 100 ms of each other). When the electrodes were placed at a distance ranging from 100–200 μm, 4.8% of the paired events (24 of 505) occurred within the time window of 100 ms, whereas only 1.4% events (8 of 554) were synchronized if the electrodes were greater than 200 μm apart. Typically, the amplitude of the field potential deflections varied as a function of time and from event to event. Similarly, if the two electrodes were placed in the stratum radiatum and the stratum pyramidale, respectively, simultaneous depolarization events were observed in 356 of 583 pairs (61%) (Fig. 4e). Interestingly, the PDSs in the stratum pyramidale were preceded by Ca2+ increases in astrocytes, similar to PDSs recorded in the stratum radiatum (Fig. 4c).
Taken together, these observations demonstrate that astrocytic Ca2+ signaling is evoked in 5 different models of acute seizure. In all paradigms studied, astrocytic Ca2+ signaling was insensitive to TTX indicating direct stimulation of astrocytes, rather than a secondary response to neuronal bursting activity. Furthermore, PDSs were spatially restricted to a few hundred micrometers and increments in cytosolic Ca2+ of astrocytes always preceded PDSs by in the stratum radiatum.
Photolysis of caged Ca2+ in astrocytes triggers PDSs
To demonstrate that astrocytic activation is not only correlated with, but sufficient for generation of negative depolarization shifts, we photo released caged Ca2+ (NP-EGTA) in astrocytes (Fig. 5a)22. Increases in astrocytic Ca2+ evoked by uncaging of NP-EGTA triggered in 8 of 12 experiments a PDS, whereas UV-flash in an identical fashion of slices not loaded with NP-EGTA failed to increase astrocytic Ca2+ concentration or to evoke PDSs (n = 15) (Fig. 5a). Targeting neurons with the UV beam also failed to evoke PDSs (n = 7). This set of observations indicates that increases in astrocytic Ca2+ are sufficient to induce local depolarization shifts.
Figure 5.
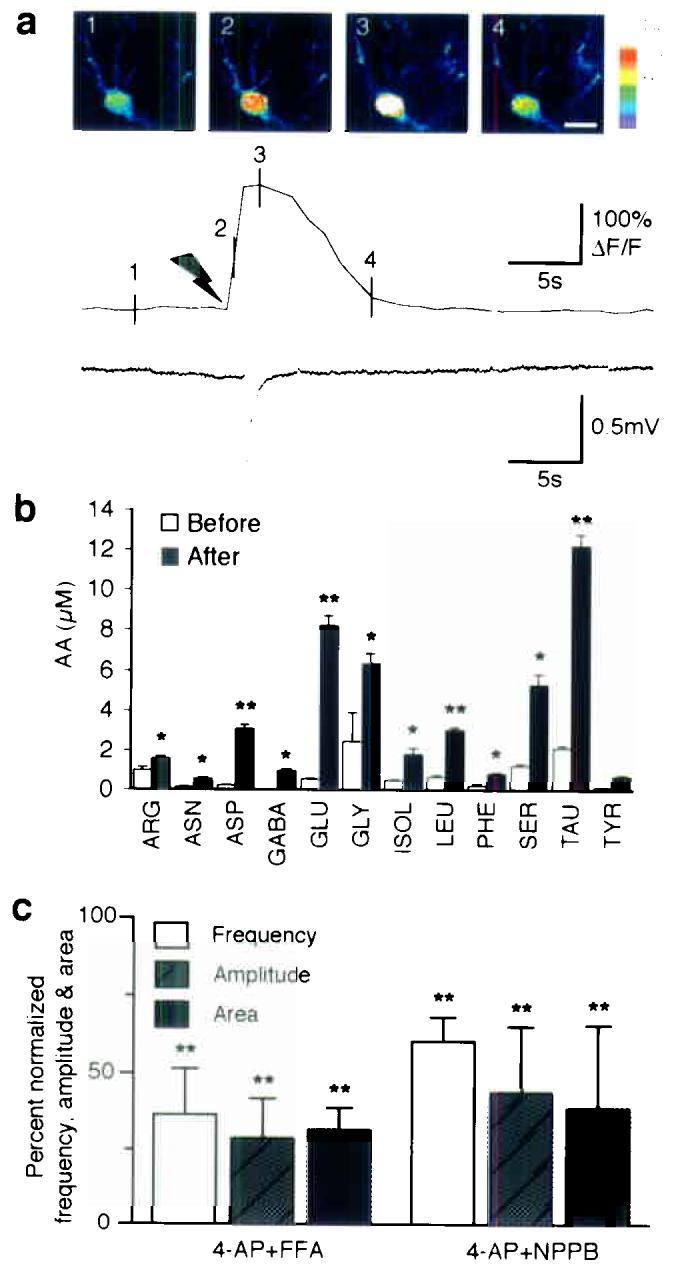
Astrocytes are the primary source of glutamate in experimental seizure. (a) Photolysis of caged Ca2+ (NP-EGTA) in an astrocyte elicits a local depolarization shift in the presence of 1 μM TTX. Upper panel: Sequence of pseudocolor images of an astrocyte loaded with NP-EGTA/AM and fluo-4/AM. Delivery of UV pulses targeting the astrocyte elevates cytosolic Ca2+ and triggers a spontaneous depolarization shift with a latency of 1.3 s. Scale bar, 10 μm. Lower panel: traces of astrocytic Ca2+ concentration and field potential. Black arrow represents the delivery of UV pulses. Numbers on the Ca2+ trace represent images in the upper panel. (b) Profile of amino acids released in an adult rat perfused with 4-AP (5 mM) and TTX (10 μM) through a microdialysis probe implanted in hippocampus. The histogram maps amino acid release before and after stimulation. *, P < 0.05; **, P < 0.001; paired student's test, mean ± s.d., n = 4. (c) Anion channel inhibitors, both 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB, 100 μM) and flufenamic acid (FFA, 100 μM), which reduce glutamate release from astrocytes, markedly decreased the frequency, amplitude and area of PDSs. **, P < 0.001 (Compared with 4-AP groups by paired student's t-test), mean ± s.d., n = 7.
One of the characteristics of Ca2+-dependent astrocytic glutamate release is that other amino acids, including aspartate, glutamine, and taurine also are released25,26. These amino acids exit through volume sensitive channels (VSC) expressed by astrocytes, whereas other amino acids, including asparagine, isoleucine, leucine, phenylalanine and tyrosine, are released to a lesser extent. To test the idea that astrocytes release glutamate during epileptic seizures, a microdialysis probe with a built-in electrode for EEG recording27, was implanted in the hippocampus and perfused with artificial cerebrospinal fluid (ACSF) containing 4-AP. The basal extracellular concentration of glutamate was low in accordance with earlier reports (0.5–1.5 μM)28, but increased to 6–10 μM approximately 10 min after addition of 4-AP. Consistent with the idea that glutamate is released by astrocytes, a 3–8 fold increase in release of amino acid osmolytes, including glutamate, aspartate, glutamine, and taurine, was observed (Fig. 5b). This profile of amino acid release was very similar to the profile of amino acid release triggered by Ca2+ signaling in cultured astrocytes, with the exception that the concentration of non osmolyte amino acids doubled during seizure activity. The shrinkage of the extracellular space that occurs during seizure activity has previously been reported to cause an artificial increase in the concentration of compounds collected by microdialysis29.
Given that photolysis experiments and HPLC analysis indicated that astrocytes contribute to elevations in extrasynaptic glutamate in epileptic tissue, we predicted that compounds that reduce astrocytic glutamate release would suppress epileptiform activity. Based on culture experiments, it has been documented that anion channel inhibitors, including 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) and flufenamic acid (FFA), reduce glutamate release from astrocytes26. To evaluate the effect of anion channel inhibition upon epileptic discharges, FFA or NPPB were bath applied to hippocampal slices exhibiting 4-AP induced seizures. Both NPPB and FFA markedly reduced the frequency and amplitude of PDSs (Fig. 5c).
Combined, these observations indicate: 1) that targeting astrocytes by photolysis of caged Ca2+ triggered PDSs, whereas similar stimulation of neurons had no effect upon the field potential; 2) that the footprint of amino acids released during 4-AP induced seizures was similar to Ca2+-dependent amino acids released from cultured astrocytes, and; 3) that anion channel inhibitors reduce the frequency and amplitude of PDSs. Together, these findings support the idea that astrocytes contribute to action potential-independent glutamate release in 4-AP evoked seizures.
Suppression of astrocytic Ca2+ signaling by anti-epileptic drugs
To test the importance of astrocytic activation in generation of seizures in live animals, we next used two-photon imaging of Ca2+ signaling in the exposed cortex of adult mice. The primary somatosensory cortex was loaded with fluo-4/AM prior to imaging. In initial experiments, Fluo-4/AM was loaded concomitant with the astrocyte specific marker Sulforhodamine 10130. Fluo-4 and Sulforhodamine 101 were co-localized, indicating that fluo-4 is preferentially taken up by astrocytes in live exposed cortex as previously reported (Fig. 6a)31. 4-AP was delivered locally by an electrode used for recording of the field potential. Application of 4-AP triggered propagating Ca2+ waves and repeated oscillatory increases in Ca2+ (data not shown). In addition, astrocytes displayed Ca2+ signaling in conjunction with the spontaneous seizure activity that occurred 5–30 min after application of 4-AP (Fig. 6c). Only these late events were further analyzed. Interestingly, astrocytic Ca2+ signaling preceded bursting activity (Fig. 6c). We recorded a total of 31 epileptic activities in 5 animals, where 19 were preceded by astrocytic Ca2+ increases in the area of the recordings. The increases in astrocytic Ca2+ occurred 4.7 ± 2.8 sec (Mean ± SD, n = 19) prior to seizure activity and were characterized by a widespread increase in Ca2+ across multiple astrocytes. Only two episodes of spontaneous astrocytic Ca2+ increases (2 of 21 total events) were not linked to epileptiform activity. Opposite, 19 of 31 seizure-like neuronal burstings were preceded by astrocytic Ca2+ signaling, whereas 10 seizure-like events occurred without an increase in astrocytic Ca2+. Valproate reduced both the amplitude of neuronal discharges and astrocytic responses to 4-AP (Fig. 6d). 4-AP-induced Ca2+ signaling was reduced by 69.7% in animals treated with valproate, by 55.6% in animals with gabapentin, and by 45.5% in animals with phenytoin (Fig. 6g). Thus, three commonly used anti-epileptic drugs all depressed astrocytic Ca2+ signaling triggered by 4-AP. Since previous work has established that astrocytic Ca2+ signaling is activated during seizure activity31,32, the inhibition of astrocytic Ca2+ signaling might simply reflect that the anti-epileptics reduced the neuronal activity. To test the alternative idea, that valproate, gabapentin, and phenytoin, directly targeted astrocytes and by suppression of their ability to transmit Ca2+ signaling reduced epileptiform activity, we next evoked Ca2+ waves by iontophoretic application of ATP. In control animals, ATP triggered astrocytic Ca2+ waves, which propagated and spread beyond the field of view (Fig. 6e). In animals pretreated with valproate, gabapentin, and phenytoin, Ca2+ wave propagation was significantly decreased (Fig. 6f,h). Valproate depressed Ca2+ signaling by 64.9%, gabapentin by 53.8%, whereas phenytoin was least efficient (23.8%). Thus, all anti-epileptics tested directly suppressed astrocytic Ca2+ signaling evoked by purinergic receptor stimulation in control non-epileptic mice,
Figure 6.

Experimental seizure in adult mice and the effect of anti-epileptic agents on astrocytic Ca2+ signaling. (a) The primary somatosensory cortex was exposed and loaded with fluo-4/AM and the astrocyte specific dye, sulforhodamine (SR101). Spacebar, 25 μm. (b) Normal EEG activity and stable astrocytic cytosolic Ca2+ levels under resting condition in an anesthetized mouse. Images were collected 130 μm below the pial surface. (c) 4-AP was delivered locally by an electrode and triggered delayed spontaneous episodes of high frequency, large amplitude discharges and astrocytic Ca2+ signaling. (d) In an animal receiving valproate (450 mg/kg i.p.), 4-AP induced seizure activity and astrocytic Ca2+ signaling were reduced. (e) Astrocytic Ca2+ wave induced by iontophoretic application of ATP during basal condition, and (f) in an animal treated with valpropate (450 mg/kg i.p.). Lower panels map changes in fluo-4 emission (ΔF/F) as a function of time. (g) Histogram summarizing the effect of valproate, gabapentin (200 mg/kg i.p.), and phenytoin (100 mg/kg i.p.) on 4-AP induced astrocytic Ca2+ signaling (5-30 min after delivery of 4-AP). (h) Histogram summarizing the effect of valproate (450 mg/kg i.p.), gabapentin (200 mg/kg i.p.), and phenytoin (100 mg/kg i.p.) on ATP-induced Ca2+ waves. *, P < 0.05; **, P < 0.001; student's t-test; mean ± s.d.; n = 5–7. Space bar, 50 μm.
DISCUSSION
Astrogliosis is a prominent feature of the epileptic brain, with autopsy and surgical resection specimens demonstrating that post-traumatic seizures and chronic temporal lobes epilepsy, may originate from gliotic scars32-34. In addition, astrocytes can modulate synaptic transmission through release of glutamate35. For example, spontaneous astrocytic Ca2+ oscillations drive NMDA-receptor-mediated neuronal excitation in the rat ventrobasal thalamus and activate groups of neurons in hippocampus7,8. These and other studies have pointed to glutamate as a key transmitter of bi-directional communication between astrocytes and neurons26,35. Nonetheless, experimental observations implicating astrocytes in initiation, maintenance, or spread of seizure activity, have not existed until now. Our study suggests that prolonged episodes of neuronal depolarization evoked by astrocytic glutamate release contribute to epileptiform discharges. Synchronized population spikes are key concomitants to seizure. Prior studies have demonstrated that multisynaptic excitatory pathways can trigger synchronized burst activity in picrotoxin-induced seizure activity36, whereas other evidence has been presented for roles of both recurrent inhibition and gap junction coupling17. Our observations suggest that an additional mechanism exists, that an action potential-independent source of glutamate can trigger local depolarization events and synchronized bursting activity. We cannot exclude that other cells, including neurons, contribute to extrasynaptic glutamate release, but several observations point to astrocytes as the primary source. First, the existence of a Ca2+-dependent mechanism of astrocytic glutamate release has been documented by several groups35. Second, photolysis of caged Ca2+ in astrocytes was sufficient to trigger PDSs (Fig. 5a). Third, astrocytic Ca2+ signaling was triggered in all models of seizure studied (Fig. 4a). Fourth, glutamate was not released in isolation, but was joined by the release of several amino acids present in the cytosol of astrocytes, including aspartate and taurine (Fig. 5b). Fifth, all conventional anti-epileptics tested suppressed astrocytic Ca2+ signaling following systemic administration (Fig. 6g).
Our study documented that 70–90% of PDSs were TTX-insensitive and that a non-synaptic mechanism thereby played a predominant role in generating seizure activity in the 5 models of experimental epilepsy studied. This observation does not exclude that astrocyte may play a role in seizure activity that originate in neurons. Astrocytes may amplify, maintain, and expand neurogenic seizure activity. Excessive neuronal firing is associated with marked alterations in the composition of the extracellular ions, most notably an increase in K+ and a reduction of Ca2+37. Lowering of extracellular Ca2+ potently elicits astrocytic Ca2+ signaling38 and glutamate release39, and secondary engagement of astrocytes may convert an otherwise self-limited episode of intense neuronal firing into a seizure focus. It is also possible that spillover of glutamate from excitatory synapses contributes to activation of astrocytic Ca2+ signaling by binding to mGluR21. Thus, astrocytes may initially be activated by excessive neuronal activity, but once activated, neuronal firing may no longer be required for continued activity of astrocytes, and thereby for maintenance and propagation of abnormal electrical activity. Similar action potential-independent mechanisms may underlie local expansion of a seizure focus. Lowering of extracellular Ca2+ triggers propagation of astrocytic Ca2+ waves that spread into adjacent tissue40. Long-distance astrocytic Ca2+ waves excite neurons along their path by release of glutamate26. In turn, neuronal activity lowers extracellular Ca2+ resulting in activation of astrocytes in increasing distances from the seizure focus41. Thus, a cascade of events in which astrocytic Ca2+ signaling plays a key role may cause conversion of normal brain tissue remote from the center of seizure initiation into an epileptic focus. The new observation reported here is that astrocytic activation directly can trigger seizure activity and that epilepsy thereby, at least in part, may originate in astrocytes.
We propose here that seizure activity may have an astrocytic basis, in addition to the well-established neurogenic mechanisms. The primary argument for existence of an astrocytic basis for seizure is that the larger fraction (70–90%) of PDSs was TTX-insensitive in five experimental models of seizure studied (Figs. 1-3). The new observation in our report is that astrocytic glutamate release constitutes a mechanism for generation of PDS, and thereby for hypersynchronous neuronal firing. Accepting that seizure activity can orginate from both astrocytes and neurons, it is also of importance to acknowledge that both astrocytes and neurons may contribute to the maintenance and spread of seizure activity. Even in gliogenic-induced seizures, excessive neuronal activity is associated with increases in interstitial K+, decreases in Ca2+, and additional glutamate release. High K+, low Ca2+, and glutamate21,38,42 are all potent triggers of astrocytic Ca2+ signaling and may be independent of etiology of the seizure due to secondary astrocytic activation. Synaptic mechanisms can on the other hand also amplify or generalize a local seizure focus4. Because trans-synaptic spread of seizure activity is driven by synaptic input, it is likely that TTX-sensitive seizures are not preceded by astrocytic Ca2+ increases or PDS. Consistent with this idea, 90% of Ca2+ increments in astrocytes (19 of 21) was followed by a seizure-like event in animals exposed to 4-AP, whereas only 61% (19 of 31) of the seizure events was preceded by increases in astrocytic Ca2+(Fig. 6c,g).
Existing drugs available for treatment of epilepsy fall into three categories. Na+ channel blockers attenuate high-frequency firing by reducing the amplitude and rate of rise of action potentials. GABA receptor agonists mimic the action of GABA , thereby increasing inhibitory synaptic transmission. Lastly, glutamate receptor antagonists block ionotopic glutamate receptors thereby reducing excitatory synaptic transmission43. The downside of these drugs is that the therapeutic mechanisms of action also suppress normal neural activity. We report here that valproate, gabapentin, and phenytoin all reduced astrocytic Ca2+ signaling in animals exposed to 4-AP. Even more intriguing, valproate, gabapentin, and phenytoin directly depressed astrocytic Ca2+ signaling evoked by purinergic receptor activation, demonstrating a direct effect on the ability of astrocytes to mobilize Ca2+ and/or transmit intercellular Ca2+ signaling. Thus, the anticonvulsive activity of valproate, gabapentin, and phenytoin, may be mediated by directly depressing astrocytic activity. Since our study suggests that epileptic discharges are secondary to glial pathology, astrocytes may represent a promising new target for epileptogenic interventions. Pharmacotherapy directed specifically at suppressing glial Ca2+ signaling or decreasing TTX-insensitive glutamate release may achieve seizure control, without the suppression of neural transmission associated with current treatment options.
METHODS
Slice preparation, 2-photon laser scanning imaging, and photolysis
Hippocampal slices were prepared from Sprague-Dawley (SD) rats (P14–18) as previously described20-22. The slices were mounted in a perfusion chamber and viewed by a custom built laser scanning microscope (BX61WI, FV300, Olympus) attached to Mai Tai laser (SpectraPhysics, Inc.). For Ca2+ measurements, slices were loaded with the Ca2+ indicator, fluo-4/AM (10 μM, 1.5 h; Molecular Probes). For uncaging experiments, NP-EGTA/AM (200 μM; Molecular Probes) was co-incubated with fluo-4 AM. Photolysis was carried out by a 3 μm diameter UV pulse delivered as 10 trains (2 pulses with a duration of 10 ms and an interval of 50 ms; 100–500 μW) (DPSS lasers, Inc; 355 nm, 1.0 W).
Culture preparation and Ca2+ imaging
Cultured astrocytes were prepared from P1 rat pups as previously described 40. Confluent monolayer cultures were loaded with the Ca2+ indicator fluo-4 (5 μM for 1 h) and Ca2+ signaling monitored by confocal microscopy (BioRad, MRC1034)44. Maximum increase in fluo-4 intensity following stimulation occurred within 20–30 s and was normalized relative to baseline fluorescence.
Electrophysiology
Whole-cell recordings from CA1 pyramidal neurons and stratum radiatum astrocytes in hippocampal slices were performed as previously described22. The perfusion artificial cerebrospinal fluid (ACSF) contained (in mM): 125 NaCl, 5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 10 glucose and 25 NaHCO3, pH 7.4 when aerated with 95% O2, 5% CO245. Membrane potentials were filtered at 1 kHz, digitized at 5 kHz by using an Axopatch 200B amplifier, a pCLAMP 8.2 program and DigiData 1332A interface (Axon Instruments, Foster City, CA). Field potential recordings were made in stratum radiatum and stratum pyramidale of CA1 in hippocampal slices as previously described45. Recording signals were filtered at 1 kHz, digitized at 5 kHz. All experiments were performed at 32–34°C.
Microdialysis, EEG recordings, and HPLC analysis of amino acid release
Adult SD rats (220–250 g) were anesthetized by ketamine (60 mg/kg) and xylazine (10 mg/kg). Microdialysis probes with a built-in electrode for EEG recordings (Applied Neuroscience, London, UK) were stereotaxically implanted into the right dorsal hippocampus (from bregma: 3.0 mm rostral; 2.0 mm lateral; from dura: 3.5 mm vertical) and fixed to the skull using dental cement and perfused using a microinjection pump (Harvard Apparatus Inc. USA), at a rate of 2 μl/min28. Seizure activity was induced by delivering 4-AP (5 mM) through the microdialysis probe. The amino acid content was analyzed after reaction with ophthaldialdehyde utilizing fluorometric detection28. EEG (1–100 Hz) was recorded continuously by an amplifier (DP-311, Warner Instruments, Inc)46,47, a pCLAMP 9.2 program and DigiData 1332A interface with an interval of 200 μs.
In vivo two-photon imaging
Adult mice (25–30g) were anesthetized with ketamine (60 mg/kg) and xylazine (10 mg/kg) injection and a femoral artery catheterized. A custom made metal frame was glued to the skull with dental acrylic cement. A craniotomy (3 mm in diameter), centered 1–2 mm posterior to bregma and 2–3 mm from midline. Dura was removed and the exposed cortex loaded with fluo-4/am (2 mM, 1 hr) and in selected experiments, sulforhodamine 101 (100 μM, 10 min)30. Agarose (0.75%) in saline was poured into the craniotomy and a coverslip mounted. Valproate was administred i.p. 450 mg/kg, 30 min before imaging; gabapentin 200 mg/kg, 60 min before imaging; and Na+phenytoin, 100 mg/kg , 90 min before imaging48. A custom built microscope attached to Tsunami/Millinium laser (SpectraPhysics, Inc.) and a scanning box (FV300, Olympus) was utilized for two-photon imaging experiments. Electrodes filled with saline containing 100 mM 4-AP were inserted 100–150 μm from the pial surface for cortical EEG (CoEEG) recordings. CoEEG (1–100 Hz) was recorded continuously by an amplifier (700A, Axon Instruments Inc.)46,47, and a pCLAMP 9.2 program and DigiData 1332A interface with an interval of 200 μs. The seizure was induced by puffing 4-AP (5–10 pulses of 5–10 ms at 10 psi, Picospitzer). ATP (50 mM) was delivered iontophoretically (100 nA, 15 sec) with an electrode (100–150 μm from surface).
Animals were artificially ventilated with a ventilator (SAR-830, CWE) and blood gasses, pCO2(30–50 mm Hg), O2(100–150 mm Hg), and pH (7.25–7.45), monitored with a pH/blood gas analyzer (Rapidlab 248, Bayer, samples 40 μl). Body temperature was maintained at 37 °C by a homeothermic blanket system (Harvard Apparatus). All experiments were approved by the Institution Animal Care and Use Committee of University of Rochester.
ACKNOWLEDGEMENTS
We thank Steve Goldman, Steve Rothman, Edward Vates and Hermes Yeh for their comments. This work was supported in part by NS30007 and NS38073 (MN); NS39997 (JK); HD16396 (RZ).
REFERENCES
- 1.Wyllie E. The treatment of epilepsy Principles and Practice. lippincot, Williams, and Wilkins; New York: 2001. [Google Scholar]
- 2.Heinemann U, Gabriel S, Schuchmann S, Eder C. Contribution of astrocytes to seizure activity. Adv. Neurol. 1999;79:583–590. [PubMed] [Google Scholar]
- 3.Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- 4.Meldrum BS. Update on the mechanism of action of antiepileptic drugs. Epilepsia. 1996;37(Suppl. 6):S4–11. doi: 10.1111/j.1528-1157.1996.tb06038.x. [DOI] [PubMed] [Google Scholar]
- 5.Parpura V, et al. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 6.Bezzi P, et al. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- 7.Fellin T, et al. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J. Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luhmann HJ, Dzhala VI, Ben-Ari Y. Generation and propagation of 4-AP-induced epileptiform activity in neonatal intact limbic structures in vitro. Eur. J. Neurosci. 2000;12:2757–2768. doi: 10.1046/j.1460-9568.2000.00156.x. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi S, Rogawski MA. Effects of anticonvulsant drugs on 4-aminopyridine-induced seizures in mice. Epilepsy Res. 1992;11:9–16. doi: 10.1016/0920-1211(92)90016-m. [DOI] [PubMed] [Google Scholar]
- 11.Elmslie KS. Neurotransmitter modulation of neuronal calcium channels. J. Bioenerg. Biomembr. 2003;35:477–489. doi: 10.1023/b:jobb.0000008021.55853.18. [DOI] [PubMed] [Google Scholar]
- 12.Drew GM, Vaughan CW. Multiple metabotropic glutamate receptor subtypes modulate GABAergic neurotransmission in rat periaqueductal grey neurons in vitro. Neuropharmacology. 2004;46:927–934. doi: 10.1016/j.neuropharm.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Grimaldi M, Atzori M, Ray P, Alkon DL. Mobilization of calcium from intracellular stores, potentiation of neurotransmitter-induced calcium transients, and capacitative calcium entry by 4-aminopyridine. J. Neurosci. 2001;21:3135–3143. doi: 10.1523/JNEUROSCI.21-09-03135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuchmann S, Albrecht D, Heinemann U, von Bohlen und Halbach O. Nitric oxide modulates low-Mg2+-induced epileptiform activity in rat hippocampal-entorhinal cortex slices. Neurobiol. Dis. 2002;11:96–105. doi: 10.1006/nbdi.2002.0533. [DOI] [PubMed] [Google Scholar]
- 15.Schneiderman JH. The role of long-term potentiation in persistent epileptiform burst-induced hyperexcitability following GABAA receptor blockade. Neuroscience. 1997;81:1111–1122. doi: 10.1016/s0306-4522(97)00209-1. [DOI] [PubMed] [Google Scholar]
- 16.Jones MS, Barth DS. Effects of bicuculline methiodide on fast (>200 Hz) electrical oscillations in rat somatosensory cortex. J. Neurophysiol. 2002;88:1016–1125. doi: 10.1152/jn.2002.88.2.1016. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Velazquez JL, Valiante TA, Carlen PL. Modulation of gap junctional mechanisms during calcium-free induced field burst activity: a possible role for electrotonic coupling in epileptogenesis. J. Neurosci. 1994;14:4308–4317. doi: 10.1523/JNEUROSCI.14-07-04308.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cotrina ML, et al. Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. USA. 1998;95:15735–15740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cotrina ML, Lin JH, Lopez-Garcia JC, Naus CC, Nedergaard M. ATP-mediated glia signaling. J. Neurosci. 2000;20:2835–2844. doi: 10.1523/JNEUROSCI.20-08-02835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang J, Jiang L, Goldman S, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- 21.Zonta M, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- 22.Liu QS, Xu Q, Arcuino G, Kang J, Nedergaard M. Astrocyte-mediated activation of neuronal kainate receptors. Proc. Natl. Acad. Sci. USA. 2004;101:3172–3177. doi: 10.1073/pnas.0306731101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ransom B, Behar T, Nedergaard M. New roles for astrocytes (stars at last) Trends Neurosci. 2003;26:520–522. doi: 10.1016/j.tins.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Jeremic A, Jeftinija K, Stevanovic J, Glavaski A, Jeftinija S. ATP stimulates calcium-dependent glutamate release from cultured astrocytes. J. Neurochem. 2001;77:664–675. doi: 10.1046/j.1471-4159.2001.00272.x. [DOI] [PubMed] [Google Scholar]
- 26.Nedergaard M, Takano T, Hansen AJ. Beyond the role of glutamate as a neurotransmitter. Nat. Rev. Neurosci. 2002;3:748–755. doi: 10.1038/nrn916. [DOI] [PubMed] [Google Scholar]
- 27.Obrenovitch TP, Urenjak J, Zilkha E. Evidence disputing the link between seizure activity and high extracellular glutamate. J. Neurochem. 1996;66:2446–2454. doi: 10.1046/j.1471-4159.1996.66062446.x. [DOI] [PubMed] [Google Scholar]
- 28.Mena FV, Baab PJ, Zielke CL, Zielke HR. In vivo glutamine hydrolysis in the formation of extracellular glutamate in the injured rat brain. J. Neurosci. Res. 2000;60:632–641. doi: 10.1002/(SICI)1097-4547(20000601)60:5<632::AID-JNR8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 29.Benveniste H, Huttemeier PC. Microdialysis--theory and application. Prog. Neurobiol. 1990;35:195–215. doi: 10.1016/0301-0082(90)90027-e. [DOI] [PubMed] [Google Scholar]
- 30.Nimmerjahn A, Kirchhoff F, Kerr J, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nature Methods. 2004;1:1–7. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- 31.Hirase H, Creso J, Buzsaki G. Capillary level imaging of local cerebral blood flow in bicuculline-induced epileptic foci. Neuroscience. 2004;128:209–216. doi: 10.1016/j.neuroscience.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Tashiro A, Goldberg J, Yuste R. Calcium oscillations in neocortical astrocytes under epileptiform conditions. J. Neurobiol. 2002;50:45–55. doi: 10.1002/neu.10019. [DOI] [PubMed] [Google Scholar]
- 33.Rothstein JD, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 34.Duffy S, MacVicar B. Modulation of Neuronal Excitability by Astrocytes. In: Delgado-Escueta A, wilson W, Olsen R, Porter R, editors. Jasper's Basic Mechanisms of Epilepsies. Thrird. Vol. 79. Lippincott Williams & Wilkins; Philadelphia: 1999. Advances in Neurology. [Google Scholar]
- 35.Haydon PG. GLIA: listening and talking to the synapse. Nat. Rev. Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- 36.Miles R, Wong RK. Single neurones can initiate synchronized population discharge in the hippocampus. Nature. 1983;306:371–373. doi: 10.1038/306371a0. [DOI] [PubMed] [Google Scholar]
- 37.Heinemann U, Konnerth A, Pumain R, Wadman WJ. Extracellular calcium and potassium concentration changes in chronic epileptic brain tissue. Adv. Neurol. 1986;44:641–461. [PubMed] [Google Scholar]
- 38.Stout C, Charles A. Modulation of intercellular calcium signaling in astrocytes by extracellular calcium and magnesium. Glia. 2003;43:265–273. doi: 10.1002/glia.10257. [DOI] [PubMed] [Google Scholar]
- 39.Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arcuino G, et al. Intercellular calcium signaling mediated by point-source burst release of ATP. Proc. Natl. Acad. Sci. USA. 2002;99:9840–9845. doi: 10.1073/pnas.152588599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bikson M, Ghai RS, Baraban SC, Durand DM. Modulation of burst frequency, duration, and amplitude in the zero-Ca(2+) model of epileptiform activity. J. Neurophysiol. 1999;82:2262–2270. doi: 10.1152/jn.1999.82.5.2262. [DOI] [PubMed] [Google Scholar]
- 42.Carmignoto G, Pasti L, Pozzan T. On the role of voltage-dependent calcium channels in calcium signaling of astrocytes in situ. J. Neurosci. 1998;18:4637–4645. doi: 10.1523/JNEUROSCI.18-12-04637.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat. Med. 2004;10:685–692. doi: 10.1038/nm1074. [DOI] [PubMed] [Google Scholar]
- 44.Takano T, et al. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001;7:1010–1015. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- 45.Valiante TA, Perez Velazquez JL, Jahromi SS, Carlen PL. Coupling potentials in CA1 neurons during calcium-free-induced field burst activity. J. Neurosci. 1995;15:6946–6956. doi: 10.1523/JNEUROSCI.15-10-06946.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ayala GX, Tapia R. Expression of heat shock protein 70 induced by 4-aminopyridine through glutamate-mediated excitotoxic stress in rat hippocampus in vivo. Neuropharmacology. 2003;45:649–660. doi: 10.1016/s0028-3908(03)00230-2. [DOI] [PubMed] [Google Scholar]
- 47.Urenjak J, Obrenovitch TP. Kynurenine 3-hydroxylase inhibition in rats: effects on extracellular kynurenic acid concentration and N-methyl-Daspartate-induced depolarisation in the striatum. J. Neurochem. 2000;75:2427–2433. doi: 10.1046/j.1471-4159.2000.0752427.x. [DOI] [PubMed] [Google Scholar]
- 48.Boothe DM. Anticonvulsant therapy in small animals. Vet. Clin. North. Am. Small Anim. Pract. 1998;28:411–448. doi: 10.1016/s0195-5616(98)82011-1. [DOI] [PubMed] [Google Scholar]