

Abstract
Chemokines are low molecular weight cytokines which act as chemoattractants for infiltrating cells bearing appropriate receptors (CCR) to sites of inflammation. It has been proposed that CCR2 on monocytes is responsible for their recruitment into the central nervous system (CNS) in experimental autoimmune encephalomyelitis (EAE), a model for multiple sclerosis, and two previous reports have described resistance of CCR2−/− mice to EAE. The present study examined three different mouse strains with CCR2 deletions for susceptibility to EAE. Animals were studied up to 4 months post-sensitization and were examined by neuropathology, RNase protection assay, in situ hybridization, and in vitro assays. All three strains were found to be susceptible to EAE: C57BL/6 × J129 and Balb c strains, 100%; and C57BL/6, 67%. Unusual in CNS lesions of CCR2−/− mice was an overabundance of neutrophils versus monocytes in wild-type animals. An attempt of the immune system to develop compensatory mechanisms for the lack of CCR2 was evidenced by a corresponding increase in mRNA for other chemokines and CCR. Inasmuch as neutrophils replaced monocytes and led to demyelination, our findings support the concept that promiscuity of chemokines and CCR was able to surmount the deletion of CCR2, still resulting in full expression of this autoimmune disease.
Experimental autoimmune encephalomyelitis (EAE) is a T cell-mediated inflammatory disease of the central nervous system (CNS) that serves as an animal model for multiple sclerosis (MS). 1,2 The disease can be induced by immunization with whole myelin or a variety of myelin antigens, their peptides, or T cells responsive to these antigens. 3 During induction of EAE, T cells sensitized to myelin antigens migrate across the blood-brain barrier into surrounding white matter, 4 re-encounter antigen and become activated to produce soluble mediators, including chemokines, for which there is compelling evidence for roles in lesion pathogenesis. 3 Chemokines are chemoattractants produced under pathological conditions by tissue elements and infiltrating leukocytes. 5,6 Two main groups of chemokines are recognized, α-chemokines, having two adjacent cysteines (C-C chemokines), and β-chemokines, with two cysteines separated by one amino acid, C-X-C chemokines. 7 Chemokines are involved in leukocyte maturation, lymphocyte trafficking, and renewal of circulating leukocytes. 8 The molecular regulation of leukocyte trafficking is complex and involves not only interactions between adhesion molecules and their receptors, but also between members of the entire superfamily of chemokines. 8 During EAE, involvement and up-regulation of several C-C chemokines, including macrophage inhibitory protein-1α (MIP-1α) and macrophage chemoattractant protein-1 (MCP-1), is well established. 9 MCP-1 is associated with monocyte/macrophage recruitment, 10,11 and its target is the chemokine receptor, CCR2, expressed by these cells. 12
Previous studies have reported that disruption of chemokine receptors (CCR) may lead to impaired monocyte function, including chemokine-directed chemotaxis, 13,14 and that manipulation of CCR might result in resistance to inflammatory disease. In the case of CCR2, recent communications 15,16 have reported that CCR2 knockout (ko) mice are resistant to EAE and that CCR2 is a key susceptibility factor in this disease. 15-17 Since we had access to three separate mouse strains with CCR2 deletions, we decided to extend a preliminary study on one strain, 18 to test further the susceptibility of CCR2−/− mice to EAE. Study of these three different strains with CCR2 disruption has revealed all to be highly susceptible. Interestingly, each strain displayed subtle differences, particularly in regard to the cellular make-up of the CNS infiltrate while the process of demyelination was not disrupted. As an approach toward mechanism, we have investigated cytokine, chemokine, and CCR profiles and found them to be in line with an increase in polymorphonuclear leukocytes, the cell type predominating in CCR2−/− EAE lesions. Therefore, CCR2 is not an all-important determining factor in EAE and it appears that multiple compensatory mechanisms exist in the expression of autoimmune demyelination.
Materials and Methods
Animals
Six- to eight-week-old wild-type (wt) and CCR2−/− mice on a C57BL/6 × J129 (C57/J129) background, a C57BL/6 × J129 background back-crossed × 8 onto C57BL/6 (C57BL/6), or a Balb c background (six back-crosses) were used. With the exception of wt Balb c mice, all mice were provided by W. Kuziel. Control wt female Balb c mice were purchased from The Jackson Laboratory, Bar Harbor, ME. CCR2−/− mice were generated by homologous recombination and all were bred under pathogen-free conditions. In addition, 20 mice (10 CCR2−/−, 10 wt) 12- to 24-weeks of age, were tested for the susceptibility of older animals to develop EAE. These animals were studied for clinical susceptibility only. Animal care and use was performed in accordance with National Institutes of Health guidelines and mice were housed in an American Association for Accreditation of Laboratory Animal Care (AAALAC) accredited facility. A total of 214 animals was used; 60 C57/J129, 140 C57BL/6, and 14 Balb c.
Antigens and Antibodies
Myelin was prepared from guinea pig spinal cord (GPSC) according to established procedures. 19 An encephalitogenic peptide corresponding to myelin oligodendrocyte glycoprotein (MOG) residue aa35–55 (MOG35–55) was synthesized by an in-house facility. Amino acid composition was verified by mass spectroscopy: purity was > 98%. Monoclonal antibodies (mAB) were used to CD4 T cells (L3T4) and CD8a T cells (Lyt-2; both from BD PharMingen, La Jolla, CA); to neutrophils (7/4), macrophages (F4/80), and B cells (CD19; Serotec, Kidlington, Oxford, United Kingdom); and to IL-4 and IL-10 (BD PharMingen). Polyclonal antibodies were used to IFN-γ and TNF-α (R&D Systems, Minneapolis, MN).
Sensitization for Active EAE and Assessment of Clinical Signs
On day 0 (d 0) and day 7 post-immunization (p.i.) C57/J129 and C57BL/6 mice were sensitized for active EAE by subcutaneous (s.c.) injection (two sites, dorsal flank) with a total of 600 μg of encephalitogenic MOG35–55 emulsified in incomplete Freund’s adjuvant (IFA; Difco, Detroit, MI), containing 70 μg Mycobacterium tuberculosis, H37Ra (Difco). On day 0 and day 2 p.i., each mouse also received 500 ng pertussis toxin (PTX; List Biological Laboratories, Campbell, CA) intravenously (i.v.), via a tail vein. Balb c mice were sensitized for active EAE with 700 μg GPSC homogenate emulsified in IFA containing 35 μg M. tuberculosis. 100 ng PTX was also given i.v. on day 0, day 2, and day 7 p.i. Of the 12- to 24-week-old group, 10 (5 CCR2−/−, 5 wt) were sensitized as above and 10 (5 CCR2−/− and 5 wt) were sensitized according to the technique of Izikson et al, 16 with 100 μg MOG in complete Freund’s adjuvant (CFA) and intraperitoneal administration of PTX, for comparison of methods of induction. Animals were assessed daily for clinical signs and evaluated according to the following scale: grade 0, no abnormalities; grade 1, weak tail; grade 2, limp tail and weakness in hind-limbs; grade 3, hind-limb paraparesis; grade 4, tetraplegia; grade 5, moribund or death.
Histopathology and Immunohistochemistry
Light microscopy studies were performed on glutaraldehyde/osmium-fixed tissue from optic nerve, cerebrum, cerebellum, and spinal cord. The tissue was dehydrated and embedded in epoxy resin from which 1-μm sections were cut and stained with toluidine blue. Inflammation, demyelination, Wallerian degeneration (WD), and remyelination were scored on a scale of 0 to 5, as described previously. 20 Ultrastructural studies were conducted on thin sections contrasted with lead and uranium salts, carbon-coated, and scanned by electron microscopy (EM) in a Hitachi HS600 (Tokyo, Japan).
For immunohistochemistry, blocks of phosphate-buffered saline (PBS)-perfused lumbar spinal cord were snap-frozen in liquid nitrogen. Cryostat sections (10-μm) were fixed with cold acetone and methanol, and primary antibodies were applied overnight at 4°C. For evaluation of bound antibody, the Vectastain Elite kit (Vector, Burlingame, CA), was used. 3′, 3′ diaminobenzidine (KPL, Gaithersburg, MD), served as the substrate for horseradish peroxidase. Immunoreactive cells were counted in spinal cord sections from six to eight different areas at × 10 and × 63 magnification.
RNase Protection Assay
Spinal cords were collected from mice in each group and snap-frozen at different time-points after sensitization. Total RNA was prepared using standard procedures. RNA probes were used to detect chemokines and chemokine receptors, and cytokine mRNAs were generated by in vitro transcription. RNase protection assays (RPAs) were performed using RiboQuant template sets, mCK1, mCK3b, and mCR5 (BD Pharmingen), in combination with RPA II kit from Ambion (Austin, TX). Protected fragments were visualized by autoradiography and quantified by phosphoimaging the gels with a Storm 860 scanner and Image QuaNT V3.01 software (Molecular Dynamics, Sunnyvale, CA). For each sample, a ratio of the intensity of the cytokine, chemokine, or chemokine receptor band was obtained using the value of the band for the housekeeping genes, mL32 and GAPDH.
In Situ Hybridization
In situ hybridization (ISH) was performed on 10-μm frozen sections using 300- to 500-bp PCR products of different cytokines, chemokines, and chemokine receptors, labeled with digoxigenin by nick-translation (Roche, Indianapolis, IN), according to the manufacturer’s instructions. PCR products representing the following mRNA sequences were applied: CCR1, bp 309–650; CCR2, bp434–710; CCR5, bp706–1047; IL-8R, bp252–661; RANTES, bp199–491; MCP-1, bp163–501; MCP-5, bp78–378; MIP-1β, bp239–578; IL-8, bp118–651; TNF-α, bp1083–1434; IFN-γ, bp232–539; TGF-β, bp1327–1579; and IL-4, bp128–407. Negative controls for ISH consisted of hybridization solution without labeled probe.
Cell Culture
C57BL/6 mice were sensitized s.c. with a total of 600 μg MOG35–55/CFA on day 0 and day 7 p.i. On day 15 p.i., mice were anesthetized with ether and sampled, and popliteal and axillary lymph nodes and spleen excised. Single-cell suspensions from draining lymph nodes and spleen were made and activated by culturing at 37°C in 7% CO2 in RPMI medium supplemented with 1% mouse serum (Rockland, Gilbertsville, PA), 1 mmol/L glutamate, 1 mmol/L pyruvate, 100 units/ml penicillin, 100 μg/ml streptomycin (all from Invitrogen; Grand Island, NY), 0.02 mmol/L β-mercaptoethanol (Sigma, St. Louis, MI), to which were added different antigens, such as 30 μg/ml of MOG35–55, 30 μg/ml MBP, 2.5 μg/ml concanavalin A (ConA), and 500 ng/ml LPS (all from Sigma). To determine proliferation after 2 days, cells were treated with 25 μCi 3H-thymidine (NEN Life Science Prod, Pittsburgh, PA), for an additional 16 hours, before being harvested and assessed for radioactivity. 21
Statistical Analysis
Clinical and histological scores in all age groups were compared with the Mann-Whitney rank sum test, using Instat (Graph Pad). Results were expressed as mean ± SD.
Results
CCR2−/− Mice from All Three Strains Are Susceptible to EAE
In both C57 strains (C57/J129 and C57BL/6), CCR2−/− mice sensitized at 8 weeks of age developed clinical signs of MOG peptide-induced EAE (Figures 1, A and B) ▶ , which were characterized by a delay in onset of 3 to 5 days and reduced clinical severity in comparison to wt mice. In general, CCR2−/− mice developed a milder EAE, between grade 2 (C57/J129) and grade 1.5 (C57BL/6), versus 3.0 to 3.5 in wt mice. However, Balb c CCR2−/− mice showed a more typical response (grade 3) (Figure 1C) ▶ . As in both C57 strains, Balb c CCR2−/− mice displayed a delayed onset. Also noteworthy were differences in the course of disease; C57/J129 developed a relapsing EAE while C57BL/6 and Balb c mice showed a monophasic pattern. Significantly, 100% of 8-week-old sensitized C57/J129 and Balb c CCR2−/− mice displayed clinical signs, whereas in C57BL/6 CCR2−/− mice, the average incidence was 67%.
Figure 1.
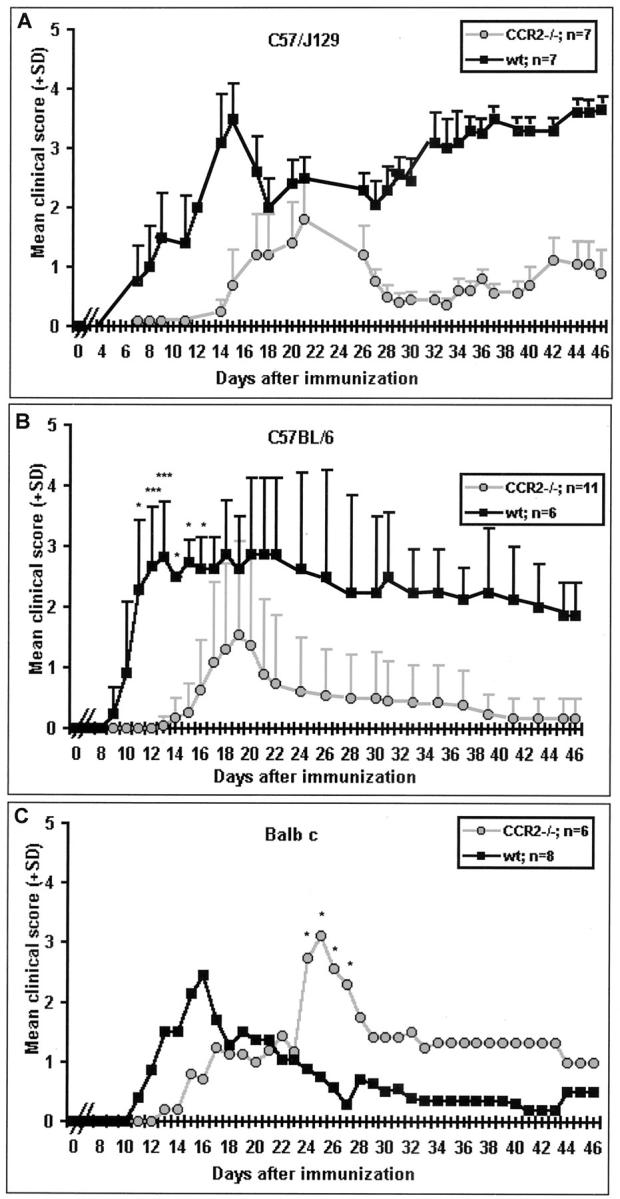
CCR2−/− mice from all three strains were susceptible to EAE. A: C57/J129; note the relapsing course of MOG35–55-induced EAE. B: C57/BL6; note the monophasic course in mice immunized with MOG35–55. C: Balb c; note monophasic course after immunization with guinea pig crude myelin. Both C57 strains showed delayed onset and reduced severity while Balb c displayed delayed onset with clinical scores comparable to wt mice. Clinical scores were assessed daily after immunization on day 0. Mean values and SD are indicated; P values refer to comparison between ko and wt mice. *, P < 0.001; **, P < 0.01; ***, P < 0.05.
Of the 12- to 24-week-old sensitized groups, four of five CCR2−/− mice from the group sensitized by the present technique developed signs of EAE (grades 1 to 3) versus two of five wt (grades 1 to 2), while of these sensitized according to Izikson et al, 16 one of five CCR2−/− (grade 1) and two of five wt (grade 2) developed disease.
EAE Lesions Differ among CCR2−/− Mice from Different Strains
In C57/J129 mice (CCR2−/− and wt) sensitized at 8 weeks of age, spinal cord lesions typical of acute EAE were seen (Figures 2 A–D ▶ and Figure 3 ▶ ). However, wt C57/J129 mice showed more severe pathology with widespread WD and a monocyte-rich infiltrate (Figure 2D) ▶ . In contrast, CNS lesions in C57/J129 CCR2−/− mice revealed a striking overabundance of infiltrating neutrophils and a paucity of monocytes (Figure 2C) ▶ . The lesion in CCR2−/− mice was less destructive and fewer axons showed damage. In C57BL/6 mice, comparable high numbers of neutrophils in CNS infiltrates and a similar degree of demyelination in both ko and wt (Figure 2, G–J) ▶ , were encountered. Similar to C57/J129 ko mice, CCR2−/− C57BL/6 mice displayed less WD in comparison to wt mice (Figure 2, I–J) ▶ .
Figure 2.
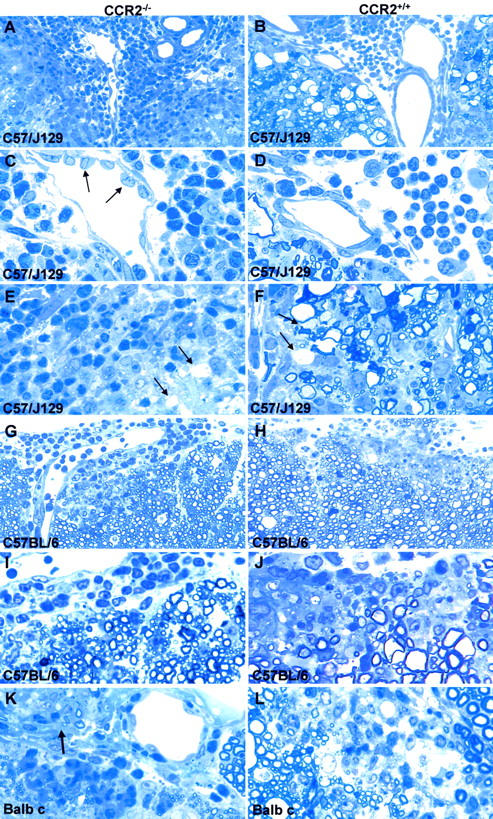
Typical acute EAE lesions in ko and wt. A–F: C57/J129 mice. Note that the inflammatory infiltrates are composed predominantly of neutrophils in CCR2−/− mice (A and C) versus monocytes in wt animals (B and D). Axonal damage is apparent as ballooned and condensed nerve fibers (WD), seen mainly in wt animals (B, D, F), and rarely in CCR2−/− (A, C, E). Arrows in C indicate high endothelial venules in ko animals. Demyelinated fibers were found in both CCR2−/− and wt animals (arrows in E and F). A: Magnification, ×250, B: Magnification, ×250, C: Magnification, ×625, D: Magnification, ×625, E: Magnification, ×625, F: Magnification, ×625. G–J: C57BL/6 mice displayed similar inflammatory infiltrates comprising neutrophils, more so in CCR2−/− (G, I), monocytes, macrophages, and lymphocytes. More severe demyelination was observed in the spinal cord of wt mice (H, J). G: Magnification, ×250, H: Magnification, ×250, I: Magnification, ×625, J: Magnification, ×625. K and L: Balb c CCR2−/− mice (K) mice displayed a preponderance of neutrophils, while wt lesions contained more monocytes (L). Note the affected root entry zone at arrow in (K). K: Magnification, ×625, L: Magnification, ×625.
Figure 3.

Histopathological differences between CCR2−/− and wt mice with disease progression. A–C: Acute EAE. Balb c CCR2−/− mice revealed decreased pathology compared to wt. D–F: Remission. CCR2−/− mice of both C57 strains displayed less pathology (D and E), which was less prominent in Balb c ko animals (F). Interestingly, CCR2−/− mice with a C57/J129 background showed decreased axonal damage (D), in contrast to C57BL/6 (E) and Balb c CCR2 ko mice (F). G–I. Chronic EAE. Balb c mice showed less pathology, but increased demyelination and WD in CCR2−/− mice (I), in contrast to both CCR2−/− from C57 strains (G and H). Data represent average histological scores of three animals (C57 strains) or a single animal (Balb c), evaluated from semi-thin cross sections of lower spinal cord (four levels, eight slides). *, ± P < 0.001; **, P < 0.01; ***, P < 0.05
Balb c wt mice with acute EAE displayed widespread lesion activity with extensive cellular infiltration and demyelination (Figure 2L) ▶ . CCR2−/− Balb c mice, on the other hand, displayed small CNS lesions with only a few infiltrating cells, comprised mainly of neutrophils (Figure 2K) ▶ . Occasionally, in CCR2−/− animals, the CNS portion of the PNS in the anterior root entry zone was also affected. Also of interest in Balb c and C57/J129 CCR2−/− mice was the appearance of lymph node-like high endothelial venules in the CNS (Figure 2, C and K) ▶ , indicative of active trafficking and homing of lymphocytes, described previously in EAE. 4
In the remission phase, during which the most statistically significant (Mann-Whitney) differences were observed, C57/J129 CCR2−/− mice displayed fewer demyelinated fibers and less WD than wt (Figure 3) ▶ . Both C57 strains displayed less infiltration and WD in the CCR2−/− groups. In Balb c CCR2−/−, there were similar degrees of demyelination and remyelination and WD was less common than in wt, but WD was more extensive. During the chronic phase, except for some differences in the amount of WD, both ko and wt mice from all strains was comparable. As in MS, 3 with time, the plaque area in EAE animals was largely taken over by fibrous astrocytes and reactive microglia. This phenomenon was seen in ko and wt animals from C57BL/6 and Balb c (not shown). The same plaques were devoid of CNPase immunoreactivity due to depletion of oligodendrocytes and myelin and gliosis appeared more pronounced in C57BL/6 than Balb c mice, perhaps a reflection of the smaller CNS lesions in Balb c.
Process of Demyelination Is Intact in CCR2−/− Mice
Ultrastructural study confirmed the predominance of neutrophils in EAE lesions in CCR2−/− C57/J129 mice. Neutrophils and some eosinophils were found in both the leptomeningeal space and CNS compartment and were seen to traverse into the CNS between subpial astrocytic processes through gaps in the glia limitans (Figure 4A) ▶ . Occasionally, neutrophils were seen to flank nerve fibers with attenuated degenerating myelin sheaths (Figure 4B) ▶ . Significantly, EM study showed the process of demyelination to be intact in CCR2−/− mice. As is well documented in mice and other species, 3 disrupted myelin formed an extracellular vesicular network around affected axons, resulting in the formation of lamellar myelin droplets (Figure 4C) ▶ . These were then internalized by macrophages following their attachment to clathrin-coated pits on the macrophage surface (Figure 4D) ▶ , described in detail in EAE 22 and MS. 23 This process has been shown to involve immunoglobulin whereby opsonized myelin droplets bind to Fc receptors expressed in coated pits on the surface of macrophages (Figure 4B) ▶ . 24 This phenomenon, known as receptor-mediated phagocytosis of myelin, is peculiar to autoimmune demyelinating models and MS. 3 Presumably, immunoglobulin mediation in the process of demyelination was not disrupted by CCR2 deletion.
Figure 4.
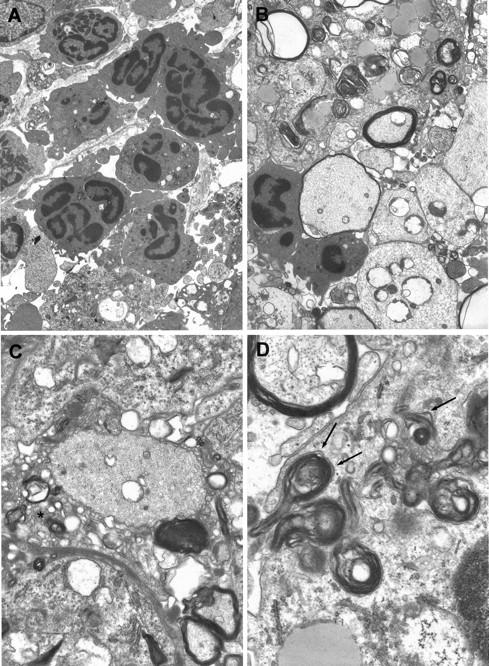
Ultrastructure of CCR2−/− mice showed features typical of wt acute EAE. Section of lumbar spinal cord from a CCR2−/− C57/J129 mouse (clinical score 3), day 4 after disease onset, is depicted. A: Neutrophils are seen within the leptomeningial compartment and the CNS parenchyma. One neutrophil can be seen traversing the glia limitans (arrows). Magnification, ×4400. B: Demyelinated axons (lower right) are shown and one nerve fiber displays an attenuated sheath (left center) flanked by two neutrophils. Elsewhere, macrophages contain myelin debris. Magnification, ×5300. C: The process of demyelination in CCR2−/− mice was intact. Disrupted myelin forms a myelin network around the demyelinated axon (center). Magnification, ×1300. D: Uptake of myelin by a macrophage occurs via its attachment to clathrin-coated pits (arrows) on the cell surface, a phenomenon known as receptor-mediated phagocytosis. Magnification, ×2500.
Cellular Composition of EAE Lesions Is Altered in CCR2−/− Mice
Immunohistochemical study of EAE lesions in C57BL/6 mice also confirmed the predominance of neutrophils compared to macrophages and T cells in both groups (data not shown). Among T cell populations, CD4+ lymphocytes were common in both CCR2−/− and wt mice. However, the number of macrophages and CD4+ T cells were increased in CCR2−/− mice, while the number of neutrophils was lower. Lesions in Balb c animals were comparable in ko and wt mice. Although CCR2−/− Balb c mice displayed lower numbers of neutrophils and F4/80+ macrophages, the numbers of CD4+ and CD8+ T cells were comparable in ko and wt. However, CD8+ cells were generally a minor component of the T cell population. CD19+ B cells were rarely seen during the acute phase of EAE, particularly in CCR2−/− C57BL/6 and Balb c mice.
Cytokine Expression Shows Minor Differences in CCR2−/− Mice
Lumbar spinal cord lesions in C57BL/6 CCR2−/− mice showed IFN-γ, TNF-α, and IL-6 protein and mRNA expression to be marginally decreased (from wt values). Infiltrating cells expressing anti-inflammatory cytokines, like IL-4 and IL-10, were found scattered around CNS blood vessels in both ko and wt mice. In Balb c CCR2−/− mice with EAE, proinflammatory cytokines, such as IFN-γ, TNF-α, and IL-6, were slightly elevated at both protein and mRNA levels, whereas IL-10 expression was lower. However, anti-inflammatory IL-4 (protein and mRNA) and TGF-β (mRNA), could not be detected in ko and wt animals (not shown).
mRNA Expression of Chemokines/Chemokine Receptors Differs in CCR2−/− Mice
RNA study using RPA in C57/J129 mice showed that in CNS tissue, message for CCR2 and CCR5 was decreased in CCR2−/− mice, whereas the mRNA level of CCR1 was comparable in ko and wt animals (Figure 5) ▶ . In addition, message levels of MIF and cytokines like TGF-β, TNF-α, and IFN-γ were also similar.
Figure 5.

RPA quantification of chemokine/chemokine receptor/cytokine mRNA expression in CNS tissue of CCR2−/− and wt C57/J129 mice with acute EAE. Intensity of the signal for protected bands was determined by phosphoimaging. Data are expressed as a ratio of the band of interest to the sum of signals for L32 and GAPDH. In ko animals, mRNA transcripts of CCR2 and CCR5 in CNS tissue were decreased (A). However, mRNA expression of MIF and cytokines, like IL-1, TGF-β, IFN-γ, and TNF-α, was comparable in CCR2−/− and wt mice (B).
ISH of CNS tissue from C57BL/6 revealed a visible increase in message for MCP-1, a ligand of CCR2, in CCR2−/− mice versus wt (Figure 6, A and B) ▶ . Unexpectedly, mRNA expression for CCR2 was seen in ko mice to the same extent as wt C57BL/6 (Figure 6, C and D) ▶ . This can be explained from the nature of the probe (see Discussion). However, expression of the CCR1-ligand, RANTES, was similar in ko and wt, as was that of the CCR5-ligand, MIP-1β (not shown). CCR1 and CCR5 mRNA-expression was slightly lower in CCR2−/− C57BL/6 mice. Interestingly, mRNA expression for IL-8, a chemokine involved in neutrophil recruitment, was elevated in CCR2−/− animals (Figure 6, E and F) ▶ , perhaps contributing to the unusual neutrophil population in the CNS. In general, lesions in Balb c CCR2−/− mice displayed higher numbers of cells expressing mRNA for chemokine receptors and their ligands, like CCR1/RANTES and CCR5/MIP-1β (Figure 6, G and H) ▶ . Moreover, loss of expression of CCR2 mRNA was reflected by reduced message for MCP-5, a CCR2 ligand. In contrast, transcripts for IL-8 receptor were increased in CCR2−/− mice. Negative controls showed a lack of detectable signal (not shown).
Figure 6.
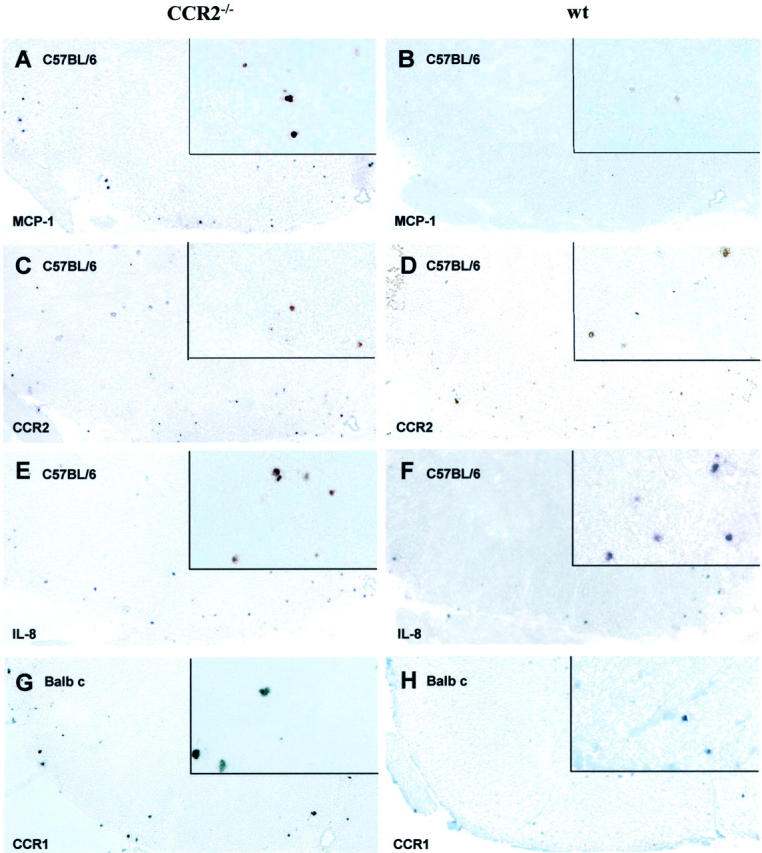
ISH for mRNA expression for chemokine receptors and their ligands in CCR2−/− and wt revealed increased MCP-1 mRNA expression in lumbar spinal cord of CCR2−/− (A) versus wt (B) C57BL/6 mice, while numbers of cells expressing CCR2 mRNA were comparable in both groups (C and D). The number of IL-8-expressing cells was higher in CCR2−/− (E) compared to wt (F) mice. Balb c CCR2 −/− mice displayed a higher mRNA expression for CCR1 (G), compared to corresponding wt mice (H). Magnification, ×100; insets: magnification, ×450.
Proliferative Response of Lymphocytes to MOG35–55 Is Not Altered in CCR2−/− Mice
In vitro proliferation assays on primary mixed lymphocyte cultures from ko and wt MOG35–55-primed C57BL/6 mice showed no differences. Lymphocytes and splenocytes from both groups displayed a high potential for MOG35–55-specific proliferation, whereas the response to MBP, an irrelevant myelin antigen, appeared inhibitory compared to controls (no antigen applied to cell culture), and proliferation indices of MOG35–55-specific lymphocyte populations from different lymphoid organs, lymph node and spleen, differed slightly (Table 1) ▶ . Therefore, at the level of lymphocyte responsiveness, no abnormalities were seen.
Table 1.
Proliferative Response to MOG
| wt | CCR2−/− | |
|---|---|---|
| Lymphocytes | ||
| n | 6 | 10 |
| Mean | 3.27 | 2.15 |
| Median | 3.65 | 2.03 |
| SD | 1.4 | 1.22 |
| SE | 0.57 | 0.39 |
| Mann-Whitney | p < 0.09 | |
| Splenocytes | ||
| n | 6 | 4 |
| Mean | 1.5 | 1.97 |
| Median | 0.78 | 1.38 |
| SD | 0.32 | 0.69 |
| SE | 1.35 | 1.34 |
| Mann-Whitney | p < 0.76 |
Discussion
Previous reports have concluded that mice lacking CCR2 are resistant to the induction of EAE 16 or display much reduced disease. 15 The present study used three different strains of mice with CCR2 deletions and has shown mice from all three strains to be susceptible to EAE. Both previous studies used mice older (12 to 16 weeks) than the usual age for EAE induction (6 to 8 weeks). 25 In addition, procedures different from those usually used for EAE were used by one group, 16 ie, footpad injection of MOG35–55 followed by intraperitoneal challenge with PTX, versus s.c. immunization with MOG35–55 and i.v. PTX used here and by others. 26 A further critical parameter in the induction of EAE is amount of antigen. One of the studies, 16 used 100 μg MOG35–55, while the second used 200 μg MOG35–55, and the majority of studies, including the present on MOG-EAE, used 300 μg MOG35–55 or more. 26-28 Interestingly, both previous studies reported that protection from EAE within the CCR2−/− group was not 100% (93%), and that EAE in diseased CCR2−/− mice was characterized by delayed onset, the latter in accord with our own observations, clearly showing that these older animals displayed much reduced susceptibility. In attempt to reconcile these differences from the present results, we conducted a separate set of experiments using CCR2−/− and wt mice from older age groups (12 to 24 weeks), in which half were given the present high dose of antigen (300 μg MOG35–55), and half were given 100 μg MOG35–55 plus intraperitoneal PTX. 16 Animals were observed for 4 weeks. In view of the results, which showed that 80% of older ko mice sensitized by the present protocol developed EAE, while only 20% given the regimen of Izikson et al 16 displayed disease, it seems therefore, that the previously reported “resistance” to EAE in CCR2 ko mice may have been related to age and method of disease induction, rather than to gene deletion as claimed.
Chemokines are important mediators of inflammation, influencing migration of immunocompetent cells into damaged areas. 9,29-31 T cell and monocyte infiltration into the CNS are critical stages in the pathogenesis of demyelinating disease, as has been demonstrated by depletion studies. 17 Chemokine production by T cells, macrophages, and astrocytes has been shown to lead to infiltration of inflammatory cells into the CNS parenchyma during the acute phase of EAE. 32 Thus, CCR2, a receptor for C-C chemokines, is known to be a major regulator of macrophage, monocyte, and T lymphocyte trafficking in vivo. 33 Our results have shown consistently that all CCR2−/− groups from three different strains displayed delayed onset of disease. Thus, lack of CCR2 probably influences the function of C-C chemokines, like MCP-1, which is known to be expressed at high levels during early EAE, affecting migration of leukocytes into the target tissue. 34,10 We conclude that CCR2 contributes to but is not all-important for the initial recruitment of inflammatory cells into the CNS. The lack of CCR2 in C57/J129 mice was related to delayed onset, reduction of disease severity, and a failure of monocyte accumulation in the CNS. Apparently, other immune cell types, like neutrophils, were able to assume the role of the “blinded” monocyte. Neutrophils, which bear CCR1 and CXCR1 and CXCR2, are common but usually minor components in CNS inflammation in C57 mouse strains. 35 It is possible that these neutrophils, entering a CNS infiltrate depleted of monocytes in which neutrophil ligands are expressed at higher levels, may be capable of accumulation in greater numbers, compensating for the lack of CCR2. ISH analysis of CCR1, IL-8, and IL-8R showed an elevated expression that may account for the increased neutrophil recruitment. Interestingly, some CCR2−/− mice with EAE also displayed an increased number of eosinophils, cells known to be predominantly activated by C-C chemokines, such as RANTES, MIP-1α, MCP-2/-3/-4, and eotaxin. 35,36 In Balb c mice, neutrophils are normally common constituents of the infiltrate, 37 and might not serve to substitute for monocytes as much as may be the case in CCR2−/− C57/J129 mice. Inasmuch as increased numbers of neutrophils correlate with disease severity in other autoimmune diseases induced in CCR2−/− animals, 14 neutrophil invasion might be an event independent of the present interrupted MCP-1/CCR2 interaction.
One explanation for our observation of susceptibility to EAE in CCR2−/− mice may be that recruitment of leukocytes into tissue is a multi-step process in which chemokines participate but do not act alone. 38 Chemokines often act in concert with other chemokines and cytokines to effect tissue infiltration, 39 and in the present EAE lesion we found expression of MCPs [chemoattractants for monocytes and T lymphocytes 10 ], MIPs [CD8+ T cells 40 and neutrophils 41,42 ], and also IL-8 [neutrophils 43 ]. The latter chemokines, MIPs and IL-8, also contribute to the balance between Th1- and Th2-type responses. In this regard, it was reported that MIP-1α exposure up-regulated IFN-γ and skewed T cells in vitro toward a Th1-type profile. 34 In contrast, treatment with MCP-1 biased cells toward a Th2 phenotype by up-regulating IL-4. 34,44 Interestingly, CCR2 deletion in a Leishmania paradigm also caused a Th1 to Th2 shift, a phenomenon associated with a neutrophil-rich infiltrate. 45 Balb c mice, originally considered genetically resistant to EAE, 46 tended to mount a Th2-type immune response when challenged. 47 Thus, in Balb c, we expected the lack of CCR2 to have a greater impact on the induction and overall course of EAE. This was found not to be the case, since Balb c CCR2−/− mice were as susceptible as wt animals. However, only a minor influence of the lack of CCR2 on the Th1-type EAE in mice with a C57 background might have been predicted, since other chemokine receptors, like CCR1 and CCR5, also prominent in Th1-type responses, remained intact. This might suggest that not one, but several chemokine receptors cooperate during cellular infiltration, as previously suggested. 48 CCR1 has been implicated elsewhere in the selective recruitment of cells into inflammatory lesions during the development of EAE. 49 CCR1 has been identified on monocytes, neutrophils, and eosinophils, 50,51 as stated above, cell types over-represented in CNS lesions in CCR2−/− C57/J129 mice with EAE. Furthermore, CCR1 not only binds MIP-1α, RANTES, MCP-2, and MCP-3 (the latter two being CCR2 ligands), with high affinity, but also MIP-1β and MCP-1 with lower affinity. 52,53 This situation might be over-ridden by an increased level of MCP-1 such as that detected here in C57BL/6 CCR2−/− mice, perhaps due to lack of CCR2 binding sites. It should be noted that CCR2 and CCR1 share striking identity in amino acid sequence in mice and humans. 54 Previous studies have shown that monocyte chemotaxis can be maximally inhibited in the presence of neutralizing antibodies to both MCP-1 and MIP-1β. 55 Thus, inactivation of other chemokine receptors may be required to prevent exacerbation of EAE in the three strains investigated. The unexpected detection of CCR2 message in CCR2−/− mice might be explained by the ISH probe design, which contained a part of the CCR2 gene which was not disrupted and was still transcribed. 33 However, translation into functional CCR2 protein was impaired.
Taken in concert, the deletion of the chemokine receptor, CCR2, led to EAE with a different CNS infiltrate and chemokine/CCR environment from that usually encountered in EAE and a modified disease course with lesions in which the process of demyelination was not affected. These findings further underscore the plasticity of the immune system in situations where important molecules are deleted.
Acknowledgments
We thank Everett Swanson and Miriam Pakingan for technical expertise; and Patricia Cobban-Bond for administrative assistance.
Footnotes
Address reprint requests to Dr. Stefanie Gaupp, Department of Pathology, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461. E-mail: raine@aecom.yu.edu.
Supported in part by Health and Human Services (HHS) grants NS 11920, NS 08952, and NS 07098; and National Multiple Sclerosis Society grant, RG 1001-J-10. C.S.R. is the Wollowick Family Professor in Multiple Sclerosis Research.
Stefanie Gaupp and David Pitt contributed equally to this study.
References
- 1.Raine CS: Biology of disease. The analysis of autoimmune demyelination: its impact upon multiple sclerosis. Lab Invest 1984, 50:608-635 [PubMed] [Google Scholar]
- 2.Steinman L: Assessment to the utility of animal models for MS and demyelinating disease in the design of rational therapy. Neuron 1999, 24:511-514 [DOI] [PubMed] [Google Scholar]
- 3.Raine CS: The lesion in multiple sclerosis and chronic relapsing experimental allergic encephalomyelitis: a structural comparison. Raine CS McFarland HF Tourtellotte WW eds. Multiple Sclerosis: Clinical and Pathogenetic Basis. 1997:pp 243-286 Chapman and Hall, London
- 4.Cross AH, Cannella B, Brosnan CF, Raine CS: Homing to central nervous system vasculature by antigen-specific lymphocytes: I. localization of 14C-labeled cells during acute, chronic, and relapsing experimental allergic encephalomyelitis. Lab Invest 1990, 63:162-170 [PubMed] [Google Scholar]
- 5.Baggiolini M, Dahinden CA: CC chemokines in allergic inflammation. Immunol Today 1994, 15:127-133 [DOI] [PubMed] [Google Scholar]
- 6.Furie MB, Randolph GJ: Chemokines and tissue injury. Am J Pathol 1995, 146:1287-1301 [PMC free article] [PubMed] [Google Scholar]
- 7.Mantovani A: Chemokines: introduction and overview. Chem Immunol 1999, 72:1-6 [PubMed] [Google Scholar]
- 8.Baggiolini M: Chemokines and leukocyte traffic. Nature 1998, 392:565-568 [DOI] [PubMed] [Google Scholar]
- 9.Glabinski AR, Ransohoff RM: Chemokines and chemokine receptors in CNS pathology. J Neurovirol 1999, 5:3-12 [DOI] [PubMed] [Google Scholar]
- 10.Rollins BJ: Monocyte chemoattractant protein 1: a potential regulator of monocyte recruitment in inflammatory disease. Mol Med Today 1996, 2:198-204 [DOI] [PubMed] [Google Scholar]
- 11.Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, Bravo R, Lira SA: Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J Immunol 1995, 155:5769-5776 [PubMed] [Google Scholar]
- 12.Dzenko KA, Andjelkovic AV, Kuziel WA, Pachter JS: The chemokine receptor CCR2 mediates the binding and internalization of monocyte chemoattractant protein-1 along brain microvessels. J Neurosci 2001, 21:9214-9223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RVJ, Broxmeyer HE, Charo IF: Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest 1997, 100:2552-2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurihara TT, Warr G, Loy J, Bravo R: Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med 1997, 186:1757-1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ: CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med 2000, 192:899-905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD: Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med 2000, 192:1075-1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM: Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med 2001, 193:713-726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pitt D, Kuziel WA, Charles PA, Cannella B, Raine CS: Experimental autoimmune encephalomyelitis in chemokine receptor deficient mice. (Abstract) J Neuroimmunol 1998, 90:60 [Google Scholar]
- 19.Norton WT, Poduslo SE: Myelination in rat brain: method of myelin isolation. J Neurochem 1973, 21:749-757 [DOI] [PubMed] [Google Scholar]
- 20.Cannella B, Hoban CJ, Gao YL, Garcia-Arenas R, Lawson D, Marchionni M, Gwynne D, Raine CS: The neuregulin, glial growth factor 2, diminishes autoimmune demyelination and enhances remyelination in a chronic relapsing model for multiple sclerosis. Proc Natl Acad Sci USA 1998, 95:10100-10105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sung S, Rose CE, Fu SM: Intratracheal priming with ovalbumin- and ovalbumin 323–339 peptide-pulsed dendritic cells induces airway hyper-responsiveness, lung eosinophilia, goblet cell hyperplasia, and inflammation. J Immunol 2001, 166:1261-1271 [DOI] [PubMed] [Google Scholar]
- 22.Epstein LG, Prineas JW, Raine CS: Attachment of myelin to coated pits on macrophages in experimental allergic encephalomyelitis. J Neurol Sci 1983, 61:341-348 [DOI] [PubMed] [Google Scholar]
- 23.Prineas JW, Connell F: The fine structure of chronically active multiple sclerosis plaques. Neurology 1978, 28:68-75 [DOI] [PubMed] [Google Scholar]
- 24.Moore GR, Raine CS: Immunogold localization and analysis of IgG during immune-mediated demyelination. Lab Invest 1988, 59:641-648 [PubMed] [Google Scholar]
- 25.Smith ME, Eller NL, McFarland HF, Racke MK, Raine CS: Age dependence of clinical and pathological manifestations of autoimmune demyelination: implications for multiple sclerosis Am J Pathol 1999, 155:1147-1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyons JA, San M, Happ MP, Cross AH: B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol 1999, 29:3432-3439 [DOI] [PubMed] [Google Scholar]
- 27.Mendel I, Gur H, Kerlero DR, Ben-Nun A: Experimental autoimmune encephalomyelitis induced in B6.C-H-2bm12 mice by myelin oligodendrocyte glycoprotein: effect of MHC class II mutation on immunodominant epitope selection and fine epitope specificity of encephalitogenic T cells. J Neuroimmunol 1999, 96:9-20 [DOI] [PubMed] [Google Scholar]
- 28.Suen WE, Bergman CM, Hjelmstrom P, Ruddle NH: A critical role for lymphotoxin in experimental allergic encephalomyelitis. J Exp Med 1997, 186:1233-1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ransohoff RM, Tani M, Glabinski AR, Chernosky A, Krivacic K, Peterson JW, Chien HF, Trapp BD: Chemokines and chemokine receptors in model neurological pathologies: molecular and immunocytochemical approaches. Methods Enzymol 1997, 287:319-348 [DOI] [PubMed] [Google Scholar]
- 30.Kennedy KJ, Strieter RM, Kunkel SL, Lukacs NW, Karpus WJ: Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1 α and monocyte chemotactic protein-1. J Neuroimmunol 1998, 92:98-108 [DOI] [PubMed] [Google Scholar]
- 31.Murphy PM: Blood, sweat, and chemotactic cytokines. J Leukoc Biol 1995, 57:438-439 [DOI] [PubMed] [Google Scholar]
- 32.Miyagishi R, Kikuchi S, Takayama C, Inoue Y, Tashiro K: Identification of cell types producing RANTES, MIP-1 α, and MIP-1 β in rat experimental autoimmune encephalomyelitis by in situ hybridization. J Neuroimmunol 1997, 77:17-26 [DOI] [PubMed] [Google Scholar]
- 33.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N: Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA 1997, 94:12053-12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karpus WJ, Kennedy KJ: MIP-1α and MCP-1 differentially regulate acute and relapsing autoimmune encephalomyelitis as well as Th1/Th2 lymphocyte differentiation. J Leukoc Biol 1997, 62:681-687 [PubMed] [Google Scholar]
- 35.Baggiolini M: Chemotactic and inflammatory cytokines: CXC and CC proteins. Adv Exp Med Biol 1993, 351:1-11 [DOI] [PubMed] [Google Scholar]
- 36.Kim Y, Sung S, Kuziel WA, Feldman S, Fu SM, Rose CEJ: Enhanced airway Th2 response after allergen challenge in mice deficient in CC chemokine receptor-2 (CCR2). J Immunol 2001, 166:5183-5192 [DOI] [PubMed] [Google Scholar]
- 37.Nygardas PT, Maatta JA, Hinkkanen AE: Chemokine expression by central nervous system resident cells and infiltrating neutrophils during experimental autoimmune encephalomyelitis in the BALB/c mouse. Eur J Immunol 2000, 30:1911-1918 [DOI] [PubMed] [Google Scholar]
- 38.Luster AD: Chemokines: chemotactic cytokines that mediate inflammation. N Engl J Med 1998, 338:436-445 [DOI] [PubMed] [Google Scholar]
- 39.Collins PD, Marleau S, Griffiths-Johnson DA, Jose PJ, Williams TJ: Cooperation between interleukin-5 and the chemokine eotaxin to induce eosinophil accumulation in vivo. J Exp Med 1995, 182:1169-1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taub DD, Conlon K, Lloyd AR, Oppenheim JJ, Kelvin DJ: Preferential migration of activated CD4+ and CD8+ T cells in response to MIP-1 α and MIP-1 β. Science 1993, 260:355-358 [DOI] [PubMed] [Google Scholar]
- 41.Davatelis G, Tekamp-Olson P, Wolpe SD, Hermsen K, Luedke C, Gallegos C, Coit D, Merryweather J, Cerami A: Cloning and characterization of a cDNA for murine macrophage inflammatory protein (MIP), a novel monokine with inflammatory and chemokinetic properties. J Exp Med 1988, 167:1939-1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sherry B, Tekamp-Olson P, Gallegos C, Bauer D, Davatelis G, Wolpe SD, Masiarz F, Coit D, Cerami A: Resolution of the two components of macrophage inflammatory protein 1, and cloning and characterization of one of those components, macrophage inflammatory protein 1 β. J Exp Med 1988, 168:2251-2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baggiolini M, Loetscher P, Moser B: Interleukin-8 and the chemokine family. Int J Immunopharmacol 1995, 17:103-108 [DOI] [PubMed] [Google Scholar]
- 44.Lukacs NW, Chensue SW, Karpus WJ, Lincoln P, Keefer C, Strieter RM, Kunkel SL: C-C chemokines differentially alter interleukin-4 production from lymphocytes. Am J Pathol 1997, 150:1861-1868 [PMC free article] [PubMed] [Google Scholar]
- 45.Sato N, Ahuja SK, Quinones M, Kostecki V, Reddick RL, Melby PC, Kuziel WA, Ahuja SS: CC chemokine receptor (CCR)2 is required for Langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells: absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, B cell outgrowth, and sustained neutrophilic inflammation. J Exp Med 2000, 192:205-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernard CC: Experimental autoimmune encephalomyelitis in mice: genetic control of susceptibility. J Immunogenet 1976, 3:263-274 [DOI] [PubMed] [Google Scholar]
- 47.Charles PC, Weber KS, Cipriani B, Brosnan CF: Cytokine, chemokine, and chemokine receptor mRNA expression in different strains of normal mice: implications for establishment of a Th1/Th2 bias. J Neuroimmunol 1999, 100:64-73 [DOI] [PubMed] [Google Scholar]
- 48.Adamus G, Manczak M, Machnicki M: Expression of CC chemokines and their receptors in the eye in autoimmune anterior uveitis associated with EAE. Invest Ophthalmol Vis Sci 2001, 42:2894-2903 [PubMed] [Google Scholar]
- 49.Rottman JB, Slavin AJ, Silva R, Weiner HL, Gerard CG, Hancock WW: Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur J Immunol 2000, 30:2372-2377 [DOI] [PubMed] [Google Scholar]
- 50.Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F: Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 1998, 187:129-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A: Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med 1998, 187:875-883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao JL, Kuhns DB, Tiffany HL, McDermott D, Li X, Francke U, Murphy PM: Structure and functional expression of the human macrophage inflammatory protein 1 α/RANTES receptor. J Exp Med 1993, 177:1421-1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franci C, Wong LM, Van Damme J, Proost P, Charo IF: Monocyte chemoattractant protein-3, but not monocyte chemoattractant protein-2, is a functional ligand of the human monocyte chemoattractant protein-1 receptor. J Immunol 1995, 154:6511-6517 [PubMed] [Google Scholar]
- 54.Murdoch C, Finn A: Chemokine receptors and their role in inflammation and infectious diseases. Blood 2000, 95:3032-3043 [PubMed] [Google Scholar]
- 55.Arenberg DA, Keane MP, DiGiovine B, Kunkel SL, Strom SR, Burdick MD, Iannettoni MD, Strieter RM: Macrophage infiltration in human non-small-cell lung cancer: the role of CC chemokines. Cancer Immunol Immunother 2000, 49:63-70 [DOI] [PMC free article] [PubMed] [Google Scholar]