

Abstract
Interleukin-6 (IL-6) is a multifunctional cytokine that activates the signaling pathways of Janus kinases-signal transducers and activators of transcription (STAT) and/or mitogen-activated protein kinases (MAPK) in various tumors. Thus, it modulates cell growth and apoptosis. IL-6 levels are elevated in tissues and sera from prostate cancer patients and IL-6 receptor expression has been detected in prostate cancer cell lines and clinical specimens. Continuous exposure of prostate cancer cells to IL-6 might alter their responsiveness to this cytokine. To gain more insight into the function of IL-6 in prostate carcinoma, we have inoculated LNCaP-IL-6+ cells, generated after prolonged treatment with IL-6, into nude mice (total n = 16, two independent experiments). Controls included animals bearing LNCaP-IL-6− cells, passaged at the same time as LNCaP-IL-6+ cells without supplementation of IL-6. LNCaP-IL-6+ tumor volumes were larger than those of their counterparts at all time points. There were no signs of cachexia in any of the experimental animals and all mice were free of metastases. To better understand the mechanisms responsible for accelerated growth of LNCaP-IL-6+ tumors, we have investigated the expression of cell-cycle regulatory molecules by Western blot analysis. The levels of cyclin-dependent kinase 2 were elevated in LNCaP-IL-6+ cells. There was a strong down-regulation of cyclins D1 and E in the LNCaP-IL-6+ subline. The cell-cycle inhibitor p27 was expressed at a low level in LNCaP-IL-6+ cells and could not be up-regulated by addition of IL-6. Most notably, LNCaP-IL-6+ cells exhibited a reduced expression of the hypophosphorylated form of the retinoblastoma protein (pRb). Accelerated tumor growth in our model system was also associated with alterations in IL-6-signaling pathways. The ability of IL-6 to induce tyrosine phosphorylation of STAT3 was abolished in the LNCaP-IL-6+ subline. In contrast, the levels of the MAPK extracellular signal-regulated kinases 1/2 increased in cells generated after long-term IL-6 treatment. The inhibitor of MAPK kinase PD 98059 retarded the proliferation of LNCaP-IL-6+ but not that of control cells. In summary, we show in the present study that chronic exposure of prostate cancer cells to IL-6 facilitates tumor growth in vivo by abolishment of the growth control by pRb and activation of the MAPK signaling pathway. These findings could be relevant to understand the role of IL-6 in prostate cancer progression.
Apart from regulation of immune responses, interleukin-6 (IL-6) influences growth of normal and tumor cells. The IL-6 receptor is composed of the ligand-binding subunit gp 80 and the signal-transducing subunit gp 130, which is shared by related cytokines. After binding of IL-6 to its receptor, Janus kinases (JAK) and/or ras-mediated signaling become activated. JAK phosphorylates signal transducers and activators of transcription (STAT) factors, which translocate to the nucleus and activate the transcription of genes containing the STAT response element. Among STAT factors, STAT3 is implicated in IL-6 action in most tissues. The ras-mediated cellular events ultimately lead to activation of the mitogen-activated protein kinase (MAPK)-signaling pathway. Because of different interactions with elements of these pathways in target tissues, IL-6 causes a variety of biological responses ranging from induction of growth arrest to suppression of apoptosis. A consistent growth-promoting effect of IL-6 has been demonstrated in renal cell carcinoma and myeloma. 1,2 In several other malignancies, both positive and negative regulation of proliferation of cancer cells were reported. Most interestingly, melanoma cell lines derived from early-stage lesions are inhibited by IL-6, whereas these cells lose sensitivity to IL-6 during tumor progression. 3
The initial observation that IL-6 is elevated in sera from patients with advanced carcinoma of the prostate has stimulated research on its expression and function in this most frequently diagnosed tumor in industrialized countries. 4 More recently, it was reported that the levels of IL-6 and its receptor increase in tissue extracts obtained from patients with clinically localized prostate cancer. 5 IL-6 elicits variable responses in established prostate cancer cell lines; in PC-3 cells derived from a bone metastasis, IL-6 acts as an autocrine growth factor. 6 For LNCaP prostate cancer cells, which respond to androgen and express the androgen receptor, contrasting effects of IL-6 on growth have been reported. Several researchers demonstrated that treatment of LNCaP with IL-6 retards the proliferation and increases the percentage of cells in the G1 phase of the cell cycle. 7-12 In contrast, other groups showed that the growth of LNCaP cells is stimulated after addition of IL-6. 5,13-15 In the authors’ laboratory, LNCaP cells have constantly been inhibited by IL-6. 16 LNCaP cells up-regulate the expression of prostate-specific antigen after treatment with IL-6 and this effect is antagonized by the nonsteroidal anti-androgen bicalutamide. 16,17 Thus, ligand-independent activation of the androgen receptor is associated with cellular differentiation in LNCaP cells.
We hypothesized that prolonged stimulation of prostate cancer cells by IL-6 may modify an initial response to this cytokine. For this reason, we have generated a new subline, LNCaP-IL-6+, which does not show a growth-inhibitory response to IL-6 and, in contrast to parental cells, expresses endogenous IL-6. 18 However, in LNCaP-IL-6+ cells, IL-6 was capable of stimulating androgen receptor-mediated reporter gene activity. 18 For the development of novel therapeutical approaches based on interference with IL-6 action, it is necessary to obtain more information about IL-6 regulation of prostate cancer in vivo. In the present study, we have investigated the growth of LNCaP-IL-6+ tumors in nude mice and analyzed alterations in the expression of key cell-cycle regulators after continuous treatment with IL-6.
Materials and Methods
Cell Culture
LNCaP-IL-6+ cells were maintained in culture as previously described. 18 High subline passages were used for in vivo experiments. LNCaP-IL-6− cells were passaged at the same time as LNCaP-IL-6+ cells in the absence of IL-6. 18 In terms of IL-6 responsiveness, they do not differ from parental LNCaP cells.
In Vivo Growth
Male nude mice (nu/nu) were purchased from Charles River Laboratories (Sulzfeld, Germany). To assess the growth of LNCaP-IL-6+ cells in vivo, two independent experiments were performed. Before transplantation, mice were supplemented with slow-release testosterone pellets (12.5 mg, 60-day release; Innovative Research of America, Sarasota, FL). In each experiment, eight animals were inoculated subcutaneously with LNCaP-IL-6+ and eight animals with LNCaP-IL-6− cells. The experimental protocol was approved by the Committee of the Austrian Federal Ministry of Education, Science, and Culture. Cells (1 × 106) were inoculated with Matrigel (Pharmingen Becton Dickinson, San Diego, CA). Animal weight and tumor size were monitored weekly. Tumor size was measured using a caliper and tumor volume was calculated according to the formula: length × width2 of a tumor area/2. After 6 weeks, nude animals were sacrificed by beheading and the tumors and lymph nodes were removed for pathohistological examination. The animals were X-rayed to detect metastatic spread. Blood samples were collected, frozen, and stored for IL-6 determination.
Chemicals
For Western blot analyses, the following antibodies were purchased: monoclonal anti-cyclin-dependent kinase (cdk) 2 and cdk 4 and anti-phospho-Tyr1022/1023 JAK (pJAK) antibodies from Biosource International, Camarillo, CA. The monoclonal antibodies anti-retinoblastoma (pRb), which recognizes both hypo- and hyperphosphorylated forms of the protein, and anti-cyclin D1 were obtained from Lab Vision Neomarkers (Freemont, CA). Monoclonal anti-cyclin E, anti-p27, and anti-STAT3 antibodies were from Santa Cruz Biotechnologies (Santa Cruz, CA). The phospho-STAT3 (Y705) (pSTAT3) antibody was a product of Upstate Biotechnology (Lake Placid, NY) and the monoclonal antibody that reacts with both phosphorylated and nonphosphorylated forms of the extracellular signal-regulated kinases (ERK) 1/2 was from Zymed Laboratories (San Francisco, CA). The MAPK kinase inhibitor PD 98059 was purchased from VWR International (Darmstadt, Germany). Human recombinant IL-6 and IL-6 enzyme-linked immunosorbent assay were from R&D Systems (Minneapolis, MN). The secondary anti-mouse horseradish peroxidase-linked antibody was from Amersham Pharmacia Biotech (Freiburg, Germany) and the anti-rabbit secondary antibody from Santa Cruz Biotechnologies.
Western Blot Analyses
LNCaP-IL-6+ and LNCaP-IL-6− cells were maintained in culture in the presence or absence of IL-6, respectively. For detection of pJAK and pSTAT3, the cells were stimulated for 30 minutes before harvesting. Western blots for cdk, cyclins, p27, and pRb were performed after 48 hours of treatment. ERK 1/2 Western blots were performed after both 30 minutes and 48 hours of treatment. The experiments were performed in whole cell extracts with the exemption of p27 and STAT3 that were detected in nuclear preparations. Nuclear extracts were prepared from 2 × 106 cells using nuclear and cytoplasmatic extraction reagents (Pierce, Rockford, IL).
After the treatment, protein content in whole cell extracts was measured according to Bradford. 19 For this purpose, samples were lysed in a buffer containing 20 mmol/L NaH2PO4, pH 7.5, 1 mmol/L ethylenediaminetetraacetic acid, and 10% glycerol. For the electrophoresis, aliquots of the samples were then diluted in NuPAGE LDS sample buffer (Invitrogen, Leek, The Netherlands) according to the manufacturer’s instructions. Afterward, they were sonicated and boiled for 10 minutes at 70°C. The lysates were loaded onto 3 to 8% Tris-acetate gels and run for 1 hour at 150 V with 1× NuPAGE sodium dodecyl sulfate (for cdk and pRb Western blots) or 4 to 12% Bis-Tris gels and run with a buffer containing 3-(N-morpholino)-propanesulphonic acid buffer (Invitrogen) for 50 minutes to 1 hour for the other experiments. The proteins were then transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore, Bedford, MA) with help of the Xcell blot module for 1 hour at 30 V using NuPAGE transfer buffer. After the transfer procedure, the membranes were washed in Tris-buffered saline (TBS) once for 5 minutes and in TBS with 0.05% Tween-20 (TBST) three times for 5 minutes. Then the membranes were blocked with 5% skim milk in TBST (TBSTM) for 1 hour and incubated with the respective primary antibody overnight at 4°C. After two washes in TBS and five in TBST, the membranes were incubated with the respective secondary antibody. The antibodies were diluted as follows: anti-cdk 2 1:100, anti-cdk 4 1:50, both followed by a secondary anti-mouse antibody diluted 1:2000; the anti-cyclin D1 antibody was diluted 1:100 followed by the secondary anti-mouse antibody diluted 1:1000; the dilution of anti-cyclin E antibody was 1:100 and that of the secondary anti-mouse antibody 1:2000; the anti-p27 antibody was diluted 1:100 and the secondary anti-mouse antibody 1:1000; anti-pRb was diluted 1:100 and the secondary antibody 1:1000; anti pJAK was used diluted 1:1000 and the secondary anti-rabbit antibody 1:5000; anti pSTAT3 was applied diluted 1:500 and the secondary anti-rabbit antibody 1:8000; the dilution of anti-STAT3 was 1:2000 and that of the secondary anti-mouse antibody 1.8:4000; the anti-ERK 1/2 antibody was diluted 1:500 followed by the anti-mouse antibody diluted 1.8:4000. The membranes were washed as described above, with an additional final wash step with TBS. Western blots were developed by means of the ECL Plus+ substrate (Amersham). As a control for equal protein loading, Western blots for β-actin were performed.
Cell Proliferation Experiments
LNCaP-IL-6+ and LNCaP-IL-6− cells were grown on six-well plates (80,000 cells/well) in the absence or presence of the MAPK kinase inhibitor PD 98059 for 48 hours and counted afterward. The Mann-Whitney t-test was used for assessment of statistical significance.
Results
Growth of LNCaP-IL-6+ Tumors in Nude Mice
The growth of LNCaP-IL-6+ and LNCaP-IL-6− cells in host animals was evaluated in two independent experiments. Each group consisted of eight animals at the beginning of the experiment. In both experiments, all mice bearing LNCaP-IL-6+ tumors survived, whereas two animals inoculated with control cells died during the performance of the first experiment and one during the second experiment. There was no evidence of tumor in those dead mice. Tumor incidence was 100% after 3 weeks in the group bearing LNCaP-IL-6+ cells. In the control group, tumors were formed in five of six animals in the first and in six of seven mice in the second experiment (83.3% and 85.7%, respectively). However, in the control group two tumors appeared after 3 weeks, two after 4 weeks, and one after 6 weeks in the first experiment. In the second experiment, all control animals developed tumor lesions after 3 weeks. Most notably, differences in tumor volume were appreciable between the IL-6-producing tumors and the controls. The tumor volumes in the LNCaP-IL-6+ group were significantly larger at all time points (Figure 1) ▶ . At the time when the experiments were terminated, the mean tumor volume in the LNCaP-IL-6+ group was 0.93 cm3 in the first and 0.6 cm3 in the second experiment. In the control group, the volumes were 0.16 and 0.09 cm3, respectively. In one animal bearing LNCaP-IL-6+ tumor, skin ulceration was noted. The tumors were examined pathohistologically. In both groups, there were evident areas of necrosis. Lymph nodes removed from the vicinity of the tumors were tumor-free and X-ray imaging did not provide any evidence of the presence of bone metastases (data not shown). There was no statistically significant difference in weight between the animals inoculated with LNCaP-IL-6+ versus those inoculated with LNCaP-IL-6− cells. At the end of the first experiment, the mean weight of the mice bearing LNCaP-IL-6+ tumors was 31.5 g and that of their counterparts 31.6 g. The respective weights at the end of the second experiment were 26.6 g and 29.7 g. The ability of LNCaP-IL-6+ cells to secrete IL-6 in vivo was confirmed; serum IL-6 levels of 45.6 ± 5.5 pg/ml were measured in the first and 148.7 ± 35.7 pg/ml in the second experiment. IL-6 levels measured in sera from nude mice inoculated with LNCaP-IL-6− cells were below the detection limit.
Figure 1.

Growth of LNCaP xenografts in nude animals. LNCaP-IL-6− and LNCaP-IL-6+ cells were injected with Matrigel subcutaneously in mice supplemented with slow-release testosterone pellets in two independent experiments. Tumor size was measured weekly using a caliper and tumor volume was calculated according to the formula: length × width2 of a tumor area/2. Top, first experiment; bottom, second experiment. **, P < 0.01, Mann-Whitney t-test. Bars, ±SE.
Expression of Cell-Cycle Regulatory Molecules
To gain more insight into the mechanisms responsible for accelerated growth of LNCaP-IL-6+ tumors, we first investigated the expression of cell-cycle regulatory molecules. Cdk 4-cyclin D complexes and cdk 2-cyclin E complexes govern the transition through the G1 phase of the cell cycle. In parental LNCaP cells, IL-6 down-regulates the expression and activity of cdk 2 and cdk 4 and up-regulates p27 expression. 11 In control cells, IL-6 treatment diminished the levels of both cdk (Figure 2) ▶ , consistent with the findings reported by Mori and associates. 11 As shown in Figure 2 ▶ , there were differences in the regulation of both kinases in LNCaP-IL-6+ cells. The expression of cdk 2 but not that of cdk 4 increased in untreated LNCaP-IL-6+ compared to LNCaP-IL-6− cells. In the LNCaP-IL-6+ subline, addition of IL-6 did not cause any effect on cdk expression. In parental LNCaP cells, there was a slightly reduced expression of cyclins D1 and E after IL-6 treatment. 11 In LNCaP-IL-6− control cells, we have observed a minor inhibitory effect of IL-6 on cyclin D1 levels. Interestingly, continuous IL-6 treatment yielded a considerably reduced expression of both cyclins D1 and E (Figure 3) ▶ . The cell-cycle inhibitor p27 was slightly induced by IL-6 in controls (Figure 4) ▶ . In contrast, its expression was negligible in the LNCaP-IL-6+ subline and it could not be increased by addition of IL-6 to culture media.
Figure 2.

Western analysis of cdk 2 and cdk 4 in LNCaP-IL-6− and LNCaP-IL-6+ cells. Western analyses with anti-cdk 2 and anti-cdk 4 monoclonal antibodies were performed after incubation of cells in the absence (lane 1) or presence of IL-6 (lane 2, 1 ng/ml; lane 3, 15 ng/ml).
Figure 3.
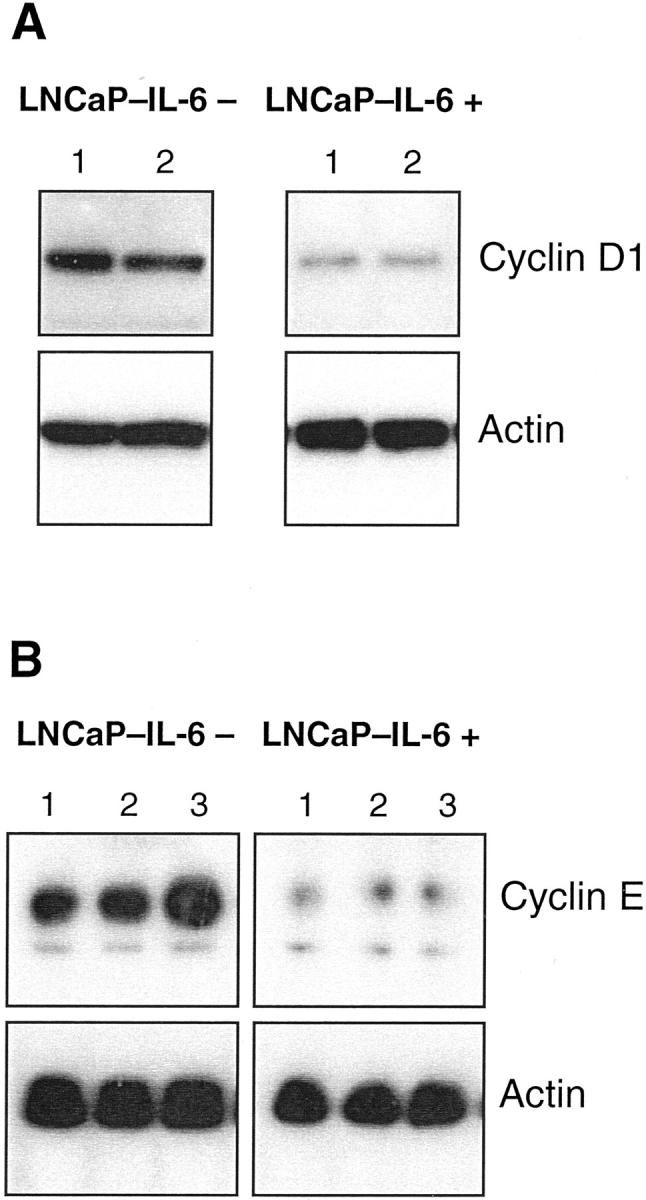
Western blot of cyclin D1 and cyclin E in controls and IL-6-resistant cells. Cyclins D1 and E were probed with monoclonal antibodies in extracts from LNCaP-IL-6− and LNCaP-IL-6+ cells that were cultured in the absence (lane 1) or presence of IL-6 (A: lane 2, 10 ng/ml; B: lane 2, 1 ng/ml; lane 3, 15 ng/ml).
Figure 4.

Western blot analysis of the cell-cycle inhibitor p27. Nuclear extracts were prepared from both LNCaP sublines cultured without (lane 1) or with IL-6 (lane 2, 10 ng/ml) and analyzed with a p27 monoclonal antibody.
The Rb gene product prevents unrestricted proliferation by binding to the transcription factor E2F. In untreated LNCaP-IL-6− cells, pRb was detected in its hypophosphorylated form, in concordance with the results published by Hofman and colleagues. 20 In our study, there were striking differences in the expression of the Rb protein; its levels were strongly down-regulated in LNCaP-IL-6+ versus control cells (Figure 5) ▶ .
Figure 5.

Presence of hypophosphorylated form of the tumor suppressor pRb in LNCaP-IL-6− and LNCaP-IL-6+ cells. Whole cell extracts obtained from LNCaP sublines were probed with a monoclonal antibody that recognizes both the hypophosphorylated and hyperphosphorylated forms of pRb. Only the hypophosphorylated form was detectable in both sublines. Lane 1, No treatment; lane 2, treatment with IL-6 (10 ng/ml).
Alterations in Activation of Signaling Pathways
The two major signaling pathways of IL-6 are the JAK-STAT and the MAPK pathway. Spiotto and Chung, 10,21 who investigated the regulation of cellular events in LNCaP cells by IL-6, found that the phosphorylation of STAT3 is associated with growth inhibition and neuroendocrine differentiation. We evaluated the effect of IL-6 on the levels of phosphorylated and total STAT3 in LNCaP-IL-6+ cells and in controls. In control cells, there was a strong induction of pSTAT3 detected after treatment with IL-6 (Figure 6) ▶ . In sharp contrast, there was no STAT3 phosphorylation observed in LNCaP-IL-6+ cells. The expression of total STAT3 was slightly lower in the LNCaP-IL-6+ than in the LNCaP-IL-6− subline. Consistent with these findings, there was no increase in the phosphorylation of the upstream kinase JAK1 at Tyr1022/1023 in LNCaP-IL-6+ cells (data not shown).
Figure 6.
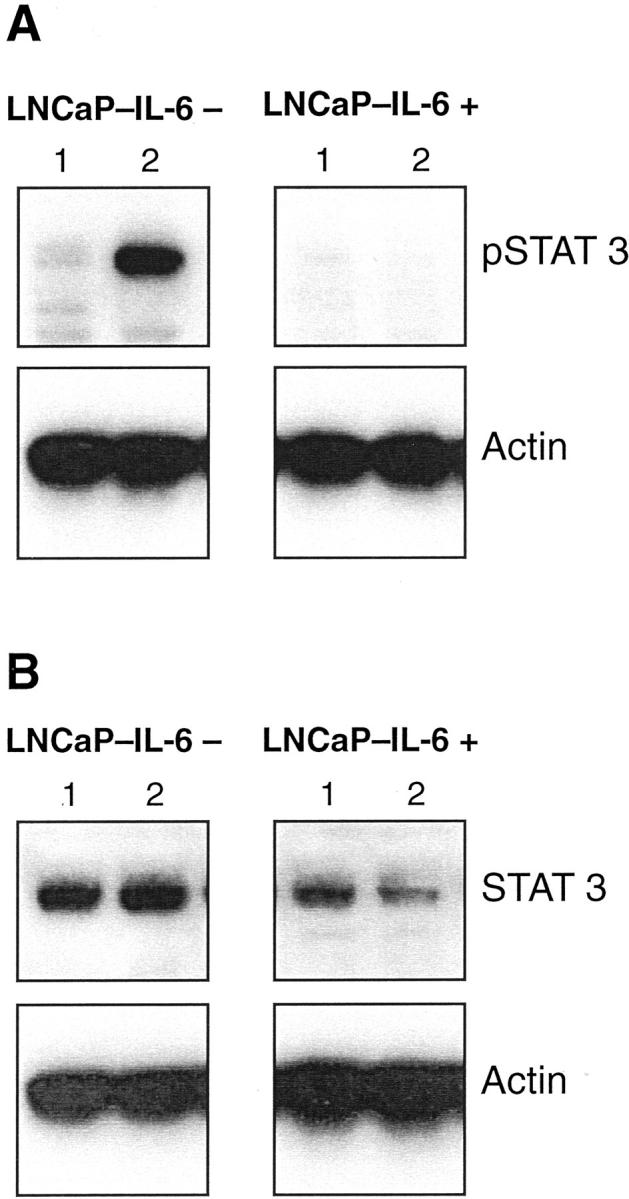
Phosphorylation of STAT3 in the LNCaP-IL-6− and LNCaP-IL-6+ sublines. LNCaP sublines were treated with IL-6 for 30 minutes (lane 2, 10 ng/ml) and the presence of pSTAT3 and total STAT3 was investigated in nuclear extracts with monoclonal antibodies by Western blot. Lane 1, Untreated cells.
The expression of the MAPK ERK 1/2 in LNCaP-IL-6+ cells was investigated using an antibody that reacts with both nonphosphorylated and phosphorylated kinases. Interestingly, MAPK expression was up-regulated in LNCaP-IL-6+ cells compared to controls (Figure 7) ▶ and the presence of IL-6 in culture either during 30 minutes (Figure 7A) ▶ or 48 hours (Figure 7B) ▶ did not cause a major effect on MAPK levels. As our antibody recognizes both MAPK forms, we asked whether elevated MAPK expression has a functional significance. For this purpose, we treated LNCaP-IL-6+ cells and controls with the MAPK kinase inhibitor PD 98059. As shown in Figure 8 ▶ , this compound had no effect on LNCaP-IL-6− cells. In contrast, PD 98059 caused a statistically significant inhibition of proliferation of LNCaP-IL-6+ cells.
Figure 7.
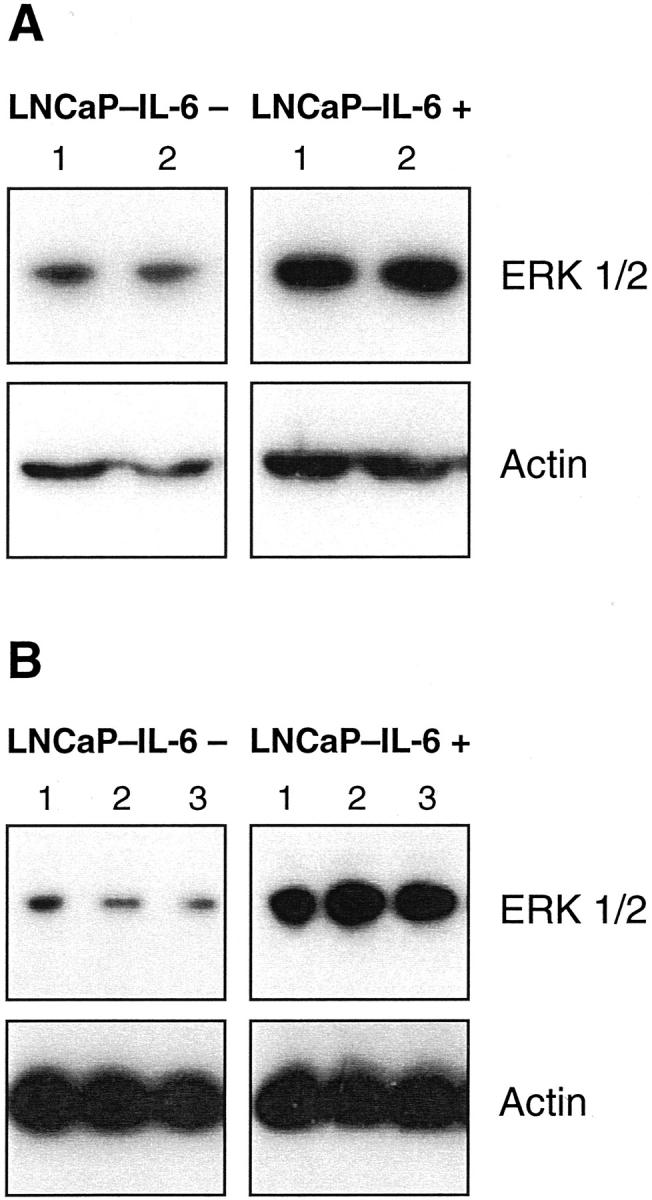
Detection of ERK 1/2 in LNCaP-IL-6− and LNCaP-IL-6+ cells. The sublines were cultured in the absence (lane 1) or presence of IL-6 (A: lane 2, 10 ng/ml; B: lane 2, 1 ng/ml; lane 3, 15 ng/ml). Western blot with a monoclonal antibody that reacts with both the phosphorylated and dephosphorylated forms of ERK was performed after either 30 minutes (A) or 48 hours (B) of treatment.
Figure 8.

The effect of the MAPK kinase inhibitor PD 98059 on proliferation of LNCaP-IL-6− (open symbols) and LNCaP-IL-6+ (shaded symbols) cells. The cells were cultured in the absence or presence of the inhibitor for 48 hours and counted afterward (three independent experiments; *, P < 0.05, Mann-Whitney t-test); bars, ±SE.
Discussion
In a series of recent studies, IL-6 was identified as an important growth regulator in human prostate cancer. 5,7,8,10-15,18 For LNCaP cells, which express the androgen receptor and are frequently used in studies on hormonal effects in prostate carcinoma, either positive or negative growth regulation by IL-6 has been reported. One reason for these variances might be the fact that, in contrast to our approach, a LNCaP subline was generated by stable transfection of IL-6 cDNA. 15 In myeloma, basal cell carcinoma, and urothelial tumors, transfection of IL-6 cDNA leads to the acquisition of a growth advantage. 22-24 In these cases, the growth-controlling mechanisms could differ from those relevant to a paracrine growth regulation. Continuous treatment of the prostate cancer cell line LNCaP with IL-6 led to the establishment of a subline that confers a growth advantage in vitro, does not exhibit growth inhibition in response to exogenous IL-6, and expresses IL-6 mRNA and protein. 18 In fact, in our previous study we showed that there are three stages in the development of IL-6 resistance in prostate cancer: LNCaP cells are initially inhibited by IL-6, after continuous treatment the inhibitory effect is abolished, and in the next step there is a marked up-regulation of IL-6 expression and secretion. 18
In the present study, we show for the first time that cancer cells generated by long-term IL-6 treatment not only lose sensitivity to IL-6 inhibition in vitro but also grow rapidly in vivo. Thus, there is a correlation between our in vitro and in vivo findings with LNCaP-IL-6+ cells. The acquisition of a growth advantage of IL-6-producing cells was also observed in melanoma. 25 Cells derived from metastatic lesions of human malignant melanoma were growth-inhibited in vivo when transfected with IL-6 anti-sense cDNA. In contrast to LNCaP-IL-6+ cells, those cells were not generated by prolonged treatment with IL-6. However, in the case of melanoma cell lines there was no clear-cut correlation between the IL-6 growth regulation in vitro and in vivo. Interestingly, production of IL-6 by murine melanoma cells showed an inverse correlation with tumorigenicity. 26 Contrary to prostate cancer and melanoma, IL-6 acts in myeloma solely as a growth-promoting factor both in vitro and in vivo. 22 This points to the requirement of different signaling pathways of IL-6 in malignant tumors.
In prostate cancer, the role of IL-6 as a stimulatory growth factor in vivo has been documented in PC-3 tumors. 27 Treatment of PC-3 xenografts with an anti-IL-6 antibody led to tumor regression by induction of apoptosis. There is, however, a notable difference in the regulation of growth by IL-6 between LNCaP and PC-3 cells because only LNCaP cells undergo transition in the growth regulation. Alterations in apoptotic pathways such as increased activation of the phosphatidylinositol-3 kinase in PC-3 cells could occur after long-term IL-6 treatment and are being investigated in a separate study. 6
On pathohistological examination, necrotic areas were detectable in LNCaP-IL-6+ tumors and controls. The main differences between the two groups are that LNCaP-IL-6+ tumors appeared earlier and that the tumor volumes were larger, as revealed in two independent experiments. LNCaP-IL-6+ cells did not produce metastases in any animal. In previous studies, in which parental LNCaP cells were inoculated subcutaneously in nude animals, there was no evidence of metastasis formation. 28 Thus, although the cells generated after long-term IL-6 treatment grow more rapidly, they did not acquire metastatic potential during 6-week experiments. It could be speculated that an orthotopic injection of LNCaP-IL-6+ cells in host animals would result in the appearance of metastases. IL-6 is known to stimulate the expression of the matrix metalloproteinase promatrilysin that increases the invasive potential of prostate carcinoma cells. 29 However, the primary focus of our study was on the growth regulation of prostate tumors that express IL-6 in vivo rather than on their metastatic spread. In this context, it is worthwhile to note that in previous experiments in which LNCaP cells were inoculated orthotopically, dissimilar results on frequency of metastases were reported. 30,31 Interestingly, there were no signs of cachexia in animals bearing LNCaP-IL-6+ cells. In this respect, our prostate cancer model differs from a human uterine cervical carcinoma xenograft. 32 A neutralizing antibody against human IL-6 did not suppress uterine tumor growth but significantly improved body weight loss.
In LNCaP cells, IL-6 causes down-regulation of cdk 2 and cdk 4 in parallel with accumulation of the cell-cycle inhibitor p27. 11 IL-6 slightly inhibited the expression of cyclins D1 and E in LNCaP cells in that study. The effect on cyclin D1 was also observed in LNCaP-IL-6− cells under our experimental conditions. It is therefore possible that a continuous treatment with IL-6 is responsible for a strongly diminished expression of both cyclins in LNCaP-IL-6+ cells. The most prominent alterations in the expression of cell-cycle regulators in the LNCaP-IL-6+ subline are the dysregulated control of cdk 2 and the down-regulation of pRb and p27. The hypophosphorylated form of the Rb protein binds to the transcription factor E2F thus preventing premature entry into S phase. It is possible that, in the case of LNCaP-IL-6+ cells, the loss of pRb additionally contributes to the down-regulation of cyclin D1. It was demonstrated that the cyclin D1 gene promoter is under the control of pRb. 33 In LNCaP cells, the phosphorylation of pRb increases after addition of low doses of androgen that induce proliferation. 20 Hyperphosphorylated pRb was not detectable after treatment of LNCaP cells with higher androgen concentrations that are capable of retarding the proliferation of the cell line. Consistent with our observations in LNCaP-IL-6+ cells, the down-regulatory effect of IL-6 on hypophosphorylated pRb was observed in multiple myeloma cells. 34 Previously, it was also shown that the IL-6 gene promoter is repressed by pRb. 35 On the basis of our results and those previous data, we propose the following mechanism for the development of IL-6 resistance in prostate cancer: initially, long-term treatment with IL-6 diminished the expression of the hypophosphorylated form of pRb and, as a consequence, repression of the IL-6 gene promoter was abolished. In this context, it is apparent that in some fast proliferating cancers the expression of cyclin D1 decreases in parallel with that of pRb. 36 Both the loss of pRb and the presence of IL-6 were reported in human prostate cancer. 37,38 Diminished expression of p27 in prostate cancer has been associated with tumor aggressiveness or reduced survival. 39,40 In the prostate cancer xenograft CWR22, tumor progression is characterized as a release from cell-cycle arrest. 41 Taken together with data reported by Agus and associates 41 and clinical studies on altered expression of pRb and p27 in advanced prostate cancer, our results might be of clinical relevance. 37,40
However, the expression of molecules that govern cell-cycle progression differs between IL-6-resistant cell lines. The levels of cyclin E in the IL-6-resistant melanoma cell line WM 9 were not considerably altered when compared to those in the IL-6-sensitive WM 35 cells. 42 Another important difference between IL-6-resistant melanoma and prostate cancer cells is the fact that cdk 2 expression was reduced in WM 9 cells. In these two tumor cell lines, IL-6 regulates different cell-cycle inhibitors. In melanoma, induction of the cell-cycle inhibitor p21 by IL-6 was lost during tumor progression. 43 In the present study, we show that IL-6 up-regulates p27 in control but not in LNCaP-IL-6+ cells.
We have observed the induction of STAT3 phosphorylation by IL-6 only in the LNCaP-IL-6− cell subline. In contrast, phosphorylated STAT3 did not increase at all after IL-6 treatment in LNCaP-IL-6+ cells. Diminished but not completely absent induction of STAT3 tyrosine phosphorylation was observed in IL-6-resistant melanoma cells WM 9. 42 In the case of LNCaP cells, STAT3 activation was reported by researchers who found either stimulation or inhibition of proliferation by IL-6. 5,10,15 One possibility to explain the different results on association of STAT3 activation with growth regulation is that the interactions between IL-6 and serum compounds critically influence the outcome of individual experiments. Several recent reports linked activated STAT3 to IL-6-induced carcinogenesis. 44-46 In contrast, it has been recognized that defective JAK-STAT signaling is associated with the lack of inhibition of growth of melanoma cells by interferon-α. 47 M1 acute myeloid leukemia cells undergo growth arrest, terminal differentiation, and apoptosis after IL-6 treatment and STAT3 was required for both induction of growth arrest and terminal differentiation. 48 We hypothesize that the absence of STAT3 activation in LNCaP-IL-6+ tumors is at least in part because of the direct inhibitory effect of MAPK. It was demonstrated that the stimulation of MAPK by basic fibroblast growth factor or a constitutively active mutant of Raf leads to a down-regulation of STAT activity. 49 In two clinical studies, increased activation of STAT3 was observed in prostate cancers compared to nonmalignant glands. 50,51 However, there was no indication that those samples were obtained from patients with advanced prostate cancer. Our hypothesis that the loss of phosphorylated STAT3 occurs in late stages of prostate cancer requires further investigation.
Our Western analyses of ERK 1/2 and the experiments with the MAPK kinase inhibitor PD 98059 suggest that the functional MAPK pathway is at least partly responsible for enhanced growth of cells generated after long-term IL-6 treatment. In parental LNCaP cells, in concordance with our data, PD 98059 did not significantly affect tumor cell growth in the absence of epidermal growth factor. 52 Those findings were further supported by results showing that in LNCaP cells there is no constitutive MAPK activity. 53,54 IL-6-refractory WM 9 melanoma cells, in contrast to our model, do not show elevated MAPK activity. 42 Taken together, all these observations implicate that the development of IL-6 resistance in various systems is not a uniform process but could be rather associated with different alterations in the expression and function of molecules involved in IL-6 signaling.
In summary, in the present study we show that continuous exposure of prostate cancer cells to IL-6 facilitates tumor growth in vivo by abolishment of the growth control by pRb and activation of the MAPK signaling pathway. These findings could have clinical implications because of the existence of IL-6 paracrine and autocrine loops in prostate tissue. 5,38 A better understanding of the molecular mechanisms underlying different IL-6 responses in prostate cancer cells is of importance for the design of novel therapeutic strategies based on the interference with IL-6 production or signaling.
Acknowledgments
We thank Dr. H. Dietrich (Central Laboratory Animal Facilities, University of Innsbruck) for advice; Drs. J. Hoffmann, D. Zopf (Schering AG, Berlin, Germany), W. Doppler (Department of Medical Chemistry and Biochemistry, University of Innsbruck), and Mr. M. Leidecker (Schering) for helpful suggestions; G. Sierek, M. Kofler, and H. Hübl for expert technical assistance; and Dr. H. Recheis and R. Schober for editorial assistance.
Footnotes
Address reprint requests to Dr. Zoran Culig, Department of Urology, University of Innsbruck, Anichstrasse 35, A-6020 Innsbruck, Austria. E-mail: zoran.culig@uibk.ac.at.
Supported by the University Clinic Innsbruck Research Support Fund (MFF) and the Austrian Research Fund (grant SFB 002 F203).
A. H. and Z. C. are joint senior authors of the manuscript.
References
- 1.Angelo LS, Talpaz M, Kurzrock R: Autocrine interleukin-6 production in renal cell carcinoma: evidence for the involvement of p53. Cancer Res 2002, 62:932-940 [PubMed] [Google Scholar]
- 2.Frassanito MA, Cusmai A, Iodice G, Dammacco F: Autocrine interleukin-6 production and highly malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood 2001, 97:483-489 [DOI] [PubMed] [Google Scholar]
- 3.Lu C, Vickers MF, Kerbel RS: Interleukin-6: a fibroblast-derived growth inhibitor of human melanoma cells from early but not advanced stages of tumor progression. Proc Natl Acad Sci USA 1992, 89:9215-9219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Twillie DA, Eisenberger MA, Carducci MA, Hseih W-S, Kim WY, Simons JW: Interleukin-6: a candidate mediator of human prostate cancer morbidity. Urology 1995, 45:542-549 [DOI] [PubMed] [Google Scholar]
- 5.Giri D, Ozen M, Ittmann M: Interleukin-6 is an autocrine growth factor in human prostate cancer. Am J Pathol 2001, 159:159-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung TD, Yu JJ, Kong TA, Spiotto MT, Lin JM: Interleukin-6 activates phosphatidylinositol-3 kinase, which inhibits apoptosis in human prostate cancer cell lines. Prostate 2000, 42:1-7 [DOI] [PubMed] [Google Scholar]
- 7.Degeorges A, Tatoud R, Fauvel Lafeve F, Podgorniak MP, Millot G, de Cremoux P, Calvo F: Stromal cells from human benign prostate hyperplasia produce a growth-inhibitory factor for LNCaP prostate cancer cells, identified as interleukin-6. Int J Cancer 1996, 68:207-214 [DOI] [PubMed] [Google Scholar]
- 8.Ritchie CK, Andrews LR, Thomas KG, Tindall DJ, Fitzpatrick LA: The effects of growth factors associated with osteoblasts on prostate carcinoma proliferation and chemotaxis: implications for the development of metastatic disease. Endocrinology 1997, 138:1145-1150 [DOI] [PubMed] [Google Scholar]
- 9.Levesque E, Beaulieu M, Guillemette C, Hum DW, Belanger A: Effect of interleukins on UGT2B15 and UGT2B17 steroid uridine diphosphate-glucuronosyltransferase expression and activity in the LNCaP cell line. Endocrinology 1998, 139:2375-2381 [DOI] [PubMed] [Google Scholar]
- 10.Spiotto MT, Chung TD: STAT3 mediates IL-6-induced growth inhibition in the human prostate cancer cell line LNCaP. Prostate 2000, 42:88-98 [DOI] [PubMed] [Google Scholar]
- 11.Mori S, Murakami-Mori K, Bonavida B: Interleukin-6 induces G1 arrest through induction of p27 (Kip1), a cyclin-dependent kinase inhibitor, and neuron-like morphology in LNCaP prostate tumor cells. Biochem Biophys Res Commun 1999, 257:609-614 [DOI] [PubMed] [Google Scholar]
- 12.Deeble PD, Murphy DJ, Parsons SJ, Cox ME: Interleukin-6 and cyclic-AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol Cell Biol 2001, 21:8471-8482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu Y, Ravi L, Kung H-J: Requirement of ErbB2 for signalling by interleukin-6 in prostate carcinoma cells. Nature 1998, 393:83-85 [DOI] [PubMed] [Google Scholar]
- 14.Okamoto M, Lee C, Oyasu R: Interleukin-6 as a paracrine and autocrine growth factor in human prostatic carcinoma cells in vitro. Cancer Res 1997, 57:141-146 [PubMed] [Google Scholar]
- 15.Lou W, Ni Z, Dyer K, Tweardy DJ, Gao AC: Interleukin-6 induces prostate cancer cell growth accompanied by activation of stat3 signaling pathway. Prostate 2000, 42:239-242 [DOI] [PubMed] [Google Scholar]
- 16.Hobisch A, Eder IE, Putz T, Horninger W, Bartsch G, Klocker H, Culig Z: Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res 1998, 58:4640-4645 [PubMed] [Google Scholar]
- 17.Lin DL, Whitney MC, Yao Z, Keller ET: Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression. Clin Cancer Res 2001, 7:1773-1781 [PubMed] [Google Scholar]
- 18.Hobisch A, Ramoner R, Fuchs D, Godoy-Tundidor S, Bartsch G, Klocker H, Culig Z: Prostate cancer cells (LNCaP) generated after long-term interleukin-6 treatment express interleukin-6 and acquire an interleukin-6-partially resistant phenotype. Clin Cancer Res 2001, 7:2941-2948 [PubMed] [Google Scholar]
- 19.Bradford M: A rapid and sensitive method for the quantitation of microgram quantities of protein using the principle of protein-dye binding. Anal Biochem 1978, 72:248-254 [DOI] [PubMed] [Google Scholar]
- 20.Hofman K, Swinnen JV, Verhoeven G, Heyns W: E2F activity is biphasically regulated by androgens in LNCaP cells. Biochem Biophys Res Commun 2001, 283:97-101 [DOI] [PubMed] [Google Scholar]
- 21.Spiotto MT, Chung TD: STAT3 mediates IL-6-induced neuroendocrine differentiation in prostate cancer cells. Prostate 2000, 42:186-195 [DOI] [PubMed] [Google Scholar]
- 22.Okuno Y, Takahashi T, Suzuki A, Fukumoto M, Nakamura K, Fukui H, Koishihara Y, Ohsugi Y, Imura H: Acquisition of growth autonomy and tumorigenicity by an interleukin 6-dependent human myeloma cell line transfected with interleukin 6 cDNA. Exp Hematol 1992, 20:395-400 [PubMed] [Google Scholar]
- 23.Jee SH, Shen SC, Chiu HC, Tsai WL, Kuo ML: Overexpression of interleukin-6 in human basal cell carcinoma cell lines increases anti-apoptotic activity and tumorigenic potency. Oncogene 2001, 20:198-208 [DOI] [PubMed] [Google Scholar]
- 24.Okamoto M, Oyasu R: Effect of transfected interleukin-6 in non-tumorigenic and tumorigenic rat urothelial cell lines. Int J Cancer 1996, 68:616-621 [DOI] [PubMed] [Google Scholar]
- 25.Lu C, Sheehan C, Rak JW, Chambers CA, Hozumi N, Kerbel RS: Endogenous interleukin-6 can function as an in vivo growth-stimulatory factor for advanced-stage human melanoma cells. Clin Cancer Res 1996, 2:1417-1425 [PubMed] [Google Scholar]
- 26.Armstrong CA, Murray N, Kennedy M, Koppula SV, Tara D, Ansel JC: Melanoma-derived interleukin 6 inhibits in vivo melanoma growth. J Invest Dermatol 1994, 102:278-284 [DOI] [PubMed] [Google Scholar]
- 27.Smith PC, Keller ET: Anti-interleukin-6 monoclonal antibody induces regression of human prostate cancer xenografts in nude mice. Prostate 2001, 48:47-53 [DOI] [PubMed] [Google Scholar]
- 28.Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, Pathak S, von Eschenbach AC, Chung LW: Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res 1994, 54:2577-2581 [PubMed] [Google Scholar]
- 29.Stratton MS, Sirvent H, Udayakumar TS, Nagle RB, Bowden GT: Expression of the matrix metalloproteinase promatrilysin in coculture of prostate carcinoma cell lines. Prostate 2001, 48:206-209 [DOI] [PubMed] [Google Scholar]
- 30.Stephenson RA, Dinney CP, Gohji K, Ordonez NG, Killion JJ, Fidler IJ: Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst 1992, 84:951-957 [DOI] [PubMed] [Google Scholar]
- 31.Rembrink K, Romijn JC, van der Kwast TH, Rubben H, Schroder FH: Orthotopic implantation of human prostate cancer cell lines: a clinically relevant animal model for metastatic prostate cancer. Prostate 1997, 31:168-174 [DOI] [PubMed] [Google Scholar]
- 32.Tamura S, Ouchi KF, Mori K, Endo M, Matsumoto T, Eda H, Tanaka Y, Ishitsuka H, Tokita H, Yamaguchi K: Involvement of human interleukin 6 in experimental cachexia induced by a human uterine cervical carcinoma xenograft. Clin Cancer Res 1995, 1:1353-1358 [PubMed] [Google Scholar]
- 33.Müller H, Lukas J, Schneider A, Warthoe P, Bartek J, Eilers M, Strauss M: Cyclin D1 expression is regulated by the retinoblastoma protein. Proc Natl Acad Sci USA 1994, 91:2945-2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Urashima M, Ogata A, Chauhan D, Vidriales MB, Teoh G, Hoshi Y, Schlossman RL, DeCaprio JA, Anderson KC: Interleukin-6 promotes multiple myeloma cell growth via phosphorylation of retinoblastoma protein. Blood 1996, 88:2219-2227 [PubMed] [Google Scholar]
- 35.Santhanam U, Ray A, Sehgal PB: Repression of the interleukin 6 gene promoter by p53 and the retinoblastoma susceptibility gene product. Proc Natl Acad Sci USA 1991, 88:7605-7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartek J, Lukas J, Bartkova J: Perspective: defects in cell cycle control and cancer. J Pathol 1999, 187:95-99 [DOI] [PubMed] [Google Scholar]
- 37.Phillips SM, Barton CM, Lee SJ, Morton DG, Wallace DM, Lemoine NR, Neoptolemos JP: Loss of the retinoblastoma susceptibility gene (RB1) is a frequent and early event in prostatic tumorigenesis. Br J Cancer 1994, 70:1252-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hobisch A, Rogatsch H, Hittmair A, Fuchs D, Bartsch GJ, Klocker H, Bartsch G, Culig Z: Immunohistochemical localization of interleukin-6 and its receptor in benign, premalignant and malignant prostate tissue. J Pathol 2000, 191:239-244 [DOI] [PubMed] [Google Scholar]
- 39.Cordon-Cardo C, Koff A, Drobnjak M, Capodieci P, Osman I, Millard SS, Gaudin PB, Fazzari M, Zhang ZF, Massague JF, Scher HI: Distinct altered patterns of p27kip1 gene expression in benign prostate hyperplasia and prostate carcinoma. J Natl Cancer Inst 1998, 90:1284-1291 [DOI] [PubMed] [Google Scholar]
- 40.Cheng L, Lloyd RV, Weaver AL, Pisansky TM, Cheville JC, Ramnani DM, Leibovich BC, Blute ML, Zincke H, Bostwick DG: The cell cycle inhibitors p21WAF1 and p27KIP1 are associated with survival in patients treated by salvage prostatectomy after radiation therapy. Clin Cancer Res 2000, 6:1896-1899 [PubMed] [Google Scholar]
- 41.Agus DB, Cordon-Cardo C, Fox W, Drobnjak M, Koff A, Golde DW, Scher HI: Prostate cancer cell cycle regulators: response to androgen withdrawal and development of androgen independence. J Natl Cancer Inst 1999, 91:1869-1876 [DOI] [PubMed] [Google Scholar]
- 42.Böhm M, Schulte U, Funk JO, Raghunath M, Behrmann I, Kortylewski M, Heinrich PC, Kues T, Luger TA, Schwarz T: Interleukin-6-resistant melanoma cells exhibit reduced activation of STAT3 and lack of inhibition of cyclin E-associated kinase activity. J Invest Dermatol 2001, 117:132-140 [DOI] [PubMed] [Google Scholar]
- 43.Florenes VA, Lu C, Bhattacharya N, Rak J, Sheehan C, Slingerland JM, Kerbel RS: Interleukin-6 dependent induction of the cyclin dependent kinase inhibitor p21WAF1/CIP1 is lost during progression of human malignant melanoma. Oncogene 1999, 18:1023-1032 [DOI] [PubMed] [Google Scholar]
- 44.Yu C, Wang L, Khaletskiy A, Farrar WL, Larner A, Colburn NH, Li JJ: STAT3 activation is required for interleukin-6 induced transformation in tumor-promotion sensitive mouse skin epithelial cells. Oncogene 2002, 21:3949-3960 [DOI] [PubMed] [Google Scholar]
- 45.Syed V, Ulinski G, Mok SC, Ho SM: Reproductive hormone-induced, STAT3-mediated interleukin-6 action in normal and malignant human ovarian surface epithelial cells J Natl Cancer Inst 2002, 94:617-629 [DOI] [PubMed] [Google Scholar]
- 46.Horiguchi A, Oya M, Marumo K, Murai M: STAT3, but not ERKs, mediates the IL-6-induced proliferation of renal cancer cells, ACHN and 769P. Kidney Int 2002, 61:926-938 [DOI] [PubMed] [Google Scholar]
- 47.Pansky A, Hildebrand P, Fasler-Kan E, Baselgia L, Ketterer S, Beglinger C, Heim MH: Defective Jak-STAT signal transduction pathway in melanoma cells resistant to growth inhibition by interferon-alpha. Int J Cancer 2000, 85:720-725 [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, Shen B, Li Y, Sun Y: STAT3 exerts two-way regulation in the biological effects of IL-6 in M1 leukemia cells. Leuk Res 2001, 25:463-472 [DOI] [PubMed] [Google Scholar]
- 49.Terstegen L, Gatsios P, Bode JG, Schaper F, Heinrich PC, Graeve L: The inhibition of interleukin-6-dependent STAT activation by mitogen-activated protein kinases depends on tyrosine 759 in the cytoplasmic tail of glycoprotein 130. J Biol Chem 2000, 275:18810-18817 [DOI] [PubMed] [Google Scholar]
- 50.Campbell CL, Jiang Z, Savarese DM, Savarese TM: Increased expression of the interleukin-11 receptor and evidence of STAT3 activation in prostate carcinoma. Am J Pathol 2001, 158:25-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dhir R, Ni Z, Lou W, DeMiguel F, Grandis JR, Gao AC: Stat3 activation in prostatic carcinomas. Prostate 2002, 51:241-246 [DOI] [PubMed] [Google Scholar]
- 52.Guo C, Luttrell LM, Price DT: Mitogenic signaling in androgen sensitive and insensitive prostate cancer cell lines. J Urol 2000, 163:1027-1032 [PubMed] [Google Scholar]
- 53.Chen T, Cho RW, Stork PJ, Weber MJ: Elevation of cyclic adenosine 3′5′-monophosphate potentiates activation of mitogen-activated protein kinase by growth factors in LNCaP prostate cancer cells. Cancer Res 1999, 59:213-218 [PubMed] [Google Scholar]
- 54.Putz T, Culig Z, Eder IE, Nessler-Menardi C, Bartsch G, Grunicke H, Überall F, Klocker H: Epidermal growth factor receptor blockade inhibits the action of EGF, insulin-like growth factor I, and a protein kinase A activator on the mitogen-activated protein kinase pathway in prostate cancer cell lines. Cancer Res 1999, 59:227-233 [PubMed] [Google Scholar]