


Abstract
Recent genome-wide transcriptome analysis has identified diverse classes of non-coding RNAs (ncRNAs), some of which have been demonstrated to be functional, regulatory RNAs involved in various biological processes. Maturation of RNA molecules through various post-transcriptional processing events, including splicing, modification, editing and trimming of both ends, is required for correct folding and proper function of RNA molecules. To characterize post-transcriptional modifications and terminal chemical structures of fully processed native RNAs, it is necessary to isolate individual RNA species from a limited quantity and complex mixture of cellular RNAs. However, there have been no general and convenient strategies for isolation of individual RNAs. We describe here the first example of automated parallel isolation of individual ncRNAs using a novel method named ‘reciprocal circulating chromatography (RCC)’. RCC employs multiple tip-columns packed with solid-phase DNA probes to isolate multiple RNA species from a common sample of total RNAs. A pilot RCC instrument successfully isolated various ncRNAs from E. coli, yeast and mouse.
INTRODUCTION
The completion of the genome projects of human and other model organisms provides us with an initial step towards deep understanding of various biological processes. One of the striking findings from genome analyses is the presence of diverse classes of transcripts which do not encode protein products, the non-coding RNAs (ncRNAs) (1–4). In addition to classical ncRNAs, such as tRNA, rRNA, snRNA and snoRNA, newly identified ncRNAs include microRNAs (miRNAs), endogenous small interfering RNAs (siRNAs), natural anti-sense transcripts and mRNA-like ncRNAs. The miRNAs, a class of well characterized small RNAs found in both plants and animals, are involved primarily in development and cell differentiation and function as negative regulators of gene expression through translational repression and/or mRNA cleavage by a mechanism similar to RNA interference (RNAi) (5–7). Recently, germline-specific small RNAs were identified as another class of ncRNAs (8–10). Understanding the mechanisms by which ncRNAs function as regulatory elements will reveal a global picture of cellular and molecular networks connecting the transcriptome, proteome, interactome and epigenetics of the cell.
Post-transcriptional maturation of RNA molecules includes various processing events, such as capping, splicing, polyadenylation, modification, editing, 5′- and/or 3′-end processing, which take place in a spatiotemporal sequence. According to studies of classical ncRNAs, various post-transcriptional processing events are important for correct folding, interaction and assembly with proteins, and determination of subcellular localization, so as to execute the proper functions of the RNAs. Additionally, RNA modification (or editing) is a characteristic structural feature of ncRNAs. To date, more than 100 different RNA modifications have been reported in many RNA molecules from all types of living organisms (11). Most of these modifications have been found in abundant RNA molecules that have been isolated and analyzed, such as tRNAs, rRNAs or UsnRNAs. However, it has been reported that even miRNAs are modified. Plant miRNAs display 2′-O-methylation of the 3′-end nucleoside (12,13), which is required for normal miRNA maturation (14). A recent study revealed that drosophila rasiRNA contains an unidentified modification of the 3′-end (15). A fraction of mammalian miRNAs (∼6%) contain inosine (I) which is produced in pre-miRNAs by A-to-I editing (16–18). These observations indicate that RNA modifications are general and ubiquitous events during RNA maturation. Cellular RNAs can be detected by various hybridization-based techniques, including Northern hybridization, microarray technology and reverse transcription (RT)-PCR. Although RT-PCR is a highly sensitive and convenient technique to analyze quantitative aspects of RNA molecules, qualitative information, such as the type or position of modified nucleosides in an RNA molecule, is not readily available through PCR. To obtain information on the functional aspects of R2NA molecules, it is desirable to employ fully processed natural RNA molecules obtained from the cell. Isolation of individual RNA species, however, has required complex and technically challenging procedures, which are both time-consuming and labor intensive. In fact, most RNA research has no choice other than to ignore the precise characterization of RNA modifications. Thus, development of highly efficient and convenient methods for isolation of individual RNA species will provide a fundamental strategy to enable characterization of the functional aspects of large numbers of RNA molecules.
For efficient isolation of individual RNAs, we previously developed the ‘chaplet column chromatography (CCC)’ method (19–22), which modified a technique for sequence-specific RNA/DNA isolation using solid-phase DNA probes (23). The CCC method employs a DNA oligo probe complementary to each target RNA. The DNA oligo probes are each immobilized on a resin and packed in columns which are then connected in series. A solution of total RNA is circulated through the serially connected columns to entrap each target RNA. Using this method, we successfully isolated various species of minor RNAs, such as mitochondrial tRNAs. The CCC method, however, is still a manual, labor-intensive approach that requires significant technical expertise. In addition, the high back-pressure developed by serially connecting the columns limits the degree to which this method can be scaled up. Considering these issues for RNA isolation, we describe here a new platform technology named ‘reciprocal circulating chromatography (RCC)’ which we developed for parallel and automated affinity purification of multiple species of RNA molecules. The most remarkable aspect of this method is that multiple RNAs are purified from a common pool of total RNA under the same conditions. In addition, the RCC method is suitable for automated operation, which provides readily controllable procedures and high reproducibility. Here, we demonstrate the automated isolation of various species of ncRNAs from E. coli, yeast and mouse using a pilot RCC instrument based on an eight-channel automatic pipetting device.
MATERIALS AND METHODS
Assembling the RCC instrument
The layout of the RCC instrument is shown in Figure 1B. A programmable liquid-handling device equipped with an 8-channel pipette (CHOT system, Nichiryo, Japan) was connected to a peristaltic pump (ADVANTEC) and a Windows PC which controls the RCC instrument. The NSS-mini editor (Nichiryo, Japan) was used to configure the instrument for immobilization of DNA probes and RCC. A customized heat block (Sakaguchi Giken, Japan) with 2 ml sample reservoir and two 96-well-type heating blocks (Sakaguchi Giken, Japan) where 1.1 ml of buffer in 8-strip tubes (Continental Lab Products, San Diego) can be incubated were placed in the working area of the instrument. An FMI pump (Yamazen, Japan) which is operated at a speed coordinated with the evaporation of the RNA solution during chromatography was connected to the sample reservoir to supply diluent (water).
Figure 1.

Schematic diagram illustrating the basic principles of reciprocal circulating chromatography. (A) Each DNA probe is immobilized in a separate tip-column. The total RNA solution in the common sample reservoir is circulated through all tip-columns by aspiration, dispensation and mixing at 66–70°C (reciprocal circulation step). After sufficient repetitions of the pipetting cycle, tip-columns are washed with washing buffer at 40°C (washing step), and bound RNAs are eluted at 68°C in the elution buffer (elution step). (B) Deck layout of the RCC instrument.
Preparation of affinity tip-columns
Streptavidin Sepharose HP (35 μl, GE Healthcare) was packed in a 300 μl pipette tip. Quartz wool (Wako, Japan) was used for filters caps for the top and bottom of the resin, and was fixed in place by pressing with a silicon tube.
Immobilization of DNA probes
All procedures were programmed into the RCC instrument. DNA probes were immobilized onto the tip-columns by the RCC instrument by pipetting 20 times in 240 μl immobilization buffer [400 mM NaCl, 10 mM HEPES-KOH (pH 7.5) and 5 mM EDTA] containing 66 μg of an individual 3′-biotinylated DNA oligonucleotide probe (Hokkaido System Science, Japan). The tip-columns were then washed in 400 μl immobilization buffer by pipetting three times. The same operation was repeated 10 times in a new tube to wash the columns. To prevent detachment of immobilized probes from the tip-columns during RCC operation, a round of RCC was performed with trace RNA. The tip-columns were independently washed in 400 μl binding buffer [1.2 M NaCl, 30 mM HEPES-KOH (pH 7.5), 15 mM EDTA and 0.5 mM DTT] containing 160 μg E. coli total RNA by pipetting 15 times at 68°C in parallel. Residual RNA in the tip-columns was washed away with 400 μl elution buffer [20 mM NaCl, 0.5 mM HEPES-KOH (pH 7.5), 0.25 mM EDTA] by pipetting three times and the same operations were repeated 11 times.
RNA isolation using the RCC instrument
Total RNA (0.4–16 mg for E. coli) was dissolved in 2 ml binding buffer [1.2 M NaCl, 30 mM HEPES-KOH (pH 7.5), 15 mM EDTA and 0.5 mM DTT]. Affinity tip-columns were attached to the pipettor-head, and pipetted 40 times in the RNA mixture incubated in sample reservoir at 66°C. Aspiration volume for each tip-column was 200 μl. After each pipetting cycle, the RNA solution was moved by a peristaltic pump into a mixing reservoir and then pumped back into the sample reservoir and incubated for 1 min to maintain the desired temperature. Twelve 400 μl washing buffer [0.1 M NaCl, 2.5 mM HEPES-KOH (pH 7.5), 1.25 mM EDTA and 0.5 mM DTT] and six 400 μl elution buffer [20 mM NaCl, 0.5 mM HEPES-KOH (pH 7.5), 0.25 mM EDTA and 0.5 mM DTT] tubes were prepared for each tip-column, and the tip-columns were pipetted three times in each tube at 40°C for washing and 68°C for elution. The eluted RNAs were precipitated with 2-propanol. The total yield from each tip-column was calculated by measuring UV absorbance at 260 nm.
Validation of the RCC theoretical model
To validate the theoretical model of RCC, automatic purification of E. coli tRNAPro2 was performed. A small aliquot of the RNA solution was removed from the sample reservoir after every pipetting step during the isolation of E. coli tRNAPro2. The RNA solutions were diluted 1600-fold, and 2 µl of each solution was spotted on a Hybond-N+ membrane (GE Healthcare). Dot blot hybridization (24) was performed with a 5′-32P labeled DNA probe for E. coli tRNAPro2 (Table 1) to evaluate the equilibrium constant of E. coli tRNAPro2. The signals from hybridized DNA were analyzed with a BAS-5000 image analyzer (Fuji Photo Film). Purified E. coli tRNAPro2 was used as a standard control to quantify the amounts of tRNAPro2 in the RNA solution. The results were manually fitted to the theoretical equation by the least-squares method. Nmax was estimated from the eluted amount in an experiment using excess starting material. Nmax = 1.755 nmol and C0 = 0.462 µM were used.
Table 1.
List of probes and total yields for E. coli RNAs. Target RNA species, probe sequences, probe position for each target tRNA, amounts of immobilized probes and total yields of target RNAs are shown. Sequence of each RNA was obtained from the PEC database (http://www.shigen.nig.ac.jp/ecoli/pec/index.jsp). *ND: not determined, but detected by northern blotting
| RNA species | Probe sequence (5′ > 3′) | Probe position | Band intensity (%) | Yield of target RNA (μg) |
|---|---|---|---|---|
| Ala1B | TCCTGCGTGCAAAGCAGGCGCTCTCCCAGC | anticodon | 74 | 66 |
| Ala2 | TCTTGCATGCCATGCAAGCGCTCTCCCAGC | anticodon | 81 | 70 |
| Arg2 | TGCATCCGGGAGGATTCGAACCTCCGACCG | 3′ side | 60 | 38 |
| Arg3 | CGCGCCCGACAGGATTCGAACCTGAGACCT | 3′ side | 79 | 31 |
| Arg4 | CGCGCCCTGCAGGATTCGAACCTGCGGCCC | 3′ side | 51 | 10 |
| Arg5 | TGTCCCCTGCAGGAATCGAACCTGCAATTA | 3′ side | 96 | 11 |
| Asn | CTCCTCTGACTGGACTCGAACCAGTGACAT | 3′ side | 75 | 52 |
| Asp | CGGAACGGACGGGACTCGAACCCGCGACCC | 3′ side | 84 | 60 |
| Cys | AGGCGCGTTCCGGAGTCGAACCGGACTAGA | 3′ side | 94 | 78 |
| Gln1 | CTGGGGTACCTGGATTCGAACCAGGGAATG | 3′ side | 95 | 56 |
| Gln2 | CTGGGGTACGAGGATTCGAACCTCGGAATG | 3′ side | 100 | 69 |
| Glu2 | CGTCCCCTAGGGGATTCGAACCCCTGTTAC | 3′ side | 93 | 81 |
| Gly1 | AGCGGGCGAAGGGAATCGAACCCTCGTATA | 3′ side | 92 | 49 |
| Gly2 | AGCGGGCAGCGGGAATCGAACCCGCATCAT | 3′ side | 94 | 67 |
| Gly3 | AGCGGGAAACGAGACTCGAACTCGCGACCC | 3′ side | 79 | 83 |
| His | GGTGGCTAATGGGATTCGAACCCACGACAA | 3′ side | 89 | 81 |
| Ile1 | TAGGCCTGAGTGGACTTGAACCACCGACCT | 3′ side | 72 | 50 |
| Ile2, Ile2 variant | TATAAGTCGCCTGCTCTAACCACTGAGCTA | anticodon | 61 | 7 |
| Leu1(P), (Q, T, V) | TGCGAGGGGGGGGACTTGAACCCCCACGTC | 3′ side | 72 | 36 |
| Leu2 | TACCGAGGACGGGACTTGAACCCGTAAGCC | 3′ side | 17 | 8 |
| Leu3 | TGCGGGAGGCGAGACTTGAACTCGCACACC | 3′ side | 69 | 48 |
| Leu4 | TACCCGGAGCGGGACTTGAACCCGCACAGC | 3′ side | 37 | 15 |
| Leu5 | TGCCGAAGGCCGGACTCGAACCGGCACGTA | 3′ side | 71 | 16 |
| Lys | TGGGTCGTGCAGGATTCGAACCTGCGACCA | 3′ side | 42 | 18 |
| Met | TGGCTACGACGGGATTCGAACCTGTGACCC | 3′ side | 99 | 140 |
| fMet1, fMet2 | GTTATGAGCCCGACGAGCTACCAGGCTGCT | anticodon | 100 | 171 |
| Phe | TGCCCGGACTCGGAATCGAACCAAGGACAC | 3′ side | 73 | 68 |
| Pro1 | CCTTCGTCCCGAACGAAGTGCGCTACCAGG | anticodon | 100 | 133 |
| Pro2 | CCCGACACCCCATGACGGTGCGCTACCAGG | anticodon | 61 | 37 |
| Pro3 | CACTGGTCCCAAACCAGTTGCGCTACCAAG | anticodon | 59 | 45 |
| Sec | CGGAAGATCACAGGAGTCGAACCTGCCCGG | 3′ side | 65 | 29 |
| Ser1 | CGGAAGCGCAGAGATTCGAACTCTGGAACC | 3′ side | 78 | 53 |
| Ser2 | GTTGCCCCTACTCCGGTTTTCGAGACCGGT | anticodon | 58 | 14 |
| Ser3 | TTTGACCGCATACTCCCTTAGCAGGGGAGC | anticodon | 100 | 128 |
| Ser5 | CGTTGCCGTATACACACTTTCCAGGCGTGC | anticodon | 95 | 120 |
| Thr1 | TGCTGATACCCAGAGTCGAACTGGGGACCT | 3′ side | 95 | 61 |
| Thr2 | TGCCGATAATAGGAGTCGAACCTACGACCT | 3′ side | 96 | 49 |
| Thr3 | TGCTGATAGGCAGATTCGAACTGCCGACCT | 3′ side | 84 | 22 |
| Thr4 | TGCCGACTACCGGAATCGAACTGGTGACCT | 3′ side | 92 | 57 |
| Trp | CAGGGGCGGAGAGACTCGAACTCCCAACAC | 3′ side | 82 | 74 |
| Tyr1, Tyr2 | TGGTGGGGGAAGGATTCGAACCTTCGAAGT | 3′ side | 80 | 29 |
| Val1 | TGGGTGATGACGGGATCGAACCGCCGACCC | 3′ side | 52 | 35 |
| Val2A | TGCGTCCGAGTGGACTCGAACCACCGACCC | 3′ side | 45 | 13 |
| Val2B | TGCGTTCAATTGGACTCGAACCAACGACCC | 3′ side | 94 | 30 |
| 4.5S | GGGTGGGGGCCCTGCCAGCTACATCCCGGC | 3′ side | 22 | 6 |
| 6S | CTGGCCCGCTTGCGAACATCTCAGAGAAAT | 5′ side | 65 | 14 |
| DsrA | AATTCGTTACACCAGGAAATCTGATGTGTT | – | ND | ND |
| SraH | ACCGGGGTGCGCGAATACTGCGCCAACACC | – | ND | ND |
RESULTS
Concept and principle of reciprocal circulating chromatography
RCC is a method developed for parallel affinity isolation of multiple species of biological macromolecules, such as RNA, DNA or proteins. When solid-phase DNA probes are used as the affinity matrix, RCC enables the isolation of multiple species of RNA molecules from a common starting sample. The basic principle of RCC is shown schematically in Figure 1A. The detailed procedure is described in the Methods section. Pipette tips filled with an affinity matrix of streptavidin sepharose conjugated to a biotinylated DNA probe for each target RNA are attached to the head of a multi-channel pipettor. In this study, we used an 8-channel pipetting device. Each tip-column corresponds to a different species of target RNA. Total RNA extracted from cells is aspirated in parallel into the 8 tip-columns from a common solution under high salt, high temperature conditions. The solution is then dispensed back into the common reservoir. During this pipetting procedure, each target RNA hybridizes to its respective DNA probe in each tip-column. Since the concentration of each target RNA is decreased in the solution dispensed back into the reservoir, the RNA solution is mixed completely prior to the next aspiration step. Without this mixing step, the concentration of each target RNA develops a gradient in the solution, and reduces the efficiency of the yield. Sufficient repetitions of this cycle effectively circulate the entire RNA solution among all the tips. Since target RNAs are trapped on the affinity columns, minor species of RNA molecules that may be present at very low concentrations in the total RNA solution can be isolated effectively. In the washing step, non-specific RNAs in each tip-column are removed by pipetting using a buffer with an intermediate salt concentration. Finally, the target RNA bound to each tip-column is eluted into a separate tube using a low salt, high temperature buffer. Thus, several different RNAs can be isolated in parallel from the same crude RNA solution.
We assembled a prototype RCC instrument which realizes the concept of RCC. It consists of a liquid-handling instrument, customized heating blocks, a mixing pump, a water-feeding pump and a controller for the components. Each step of the RCC procedure was successfully programmed into this instrument and the experiments reported herein were performed using this prototype RCC instrument.
Theoretical model of RCC
In the RCC method, the number of cycles in the reciprocal circulation step is an important parameter governing maximum yield of the target RNAs. In order to estimate the requisite number of pipetting steps, we constructed a simplified theoretical model of RCC. As a first approximation, we assumed equilibrium binding based on the following reaction formula.
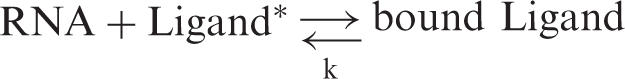 |
1 |
*Ligand stands for a DNA probe on the resin in this case.
The equilibrium constant, K was determined using the following Equation (2) where θn (0 ≤ θn ≤ 1) is the coverage of the affinity resin and expresses the ratio of bound ligand to the maximum binding capacity, and 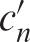 is the concentration of a target RNA in the solution dispensed from the tip-column during the nth pipetting cycle.
is the concentration of a target RNA in the solution dispensed from the tip-column during the nth pipetting cycle.
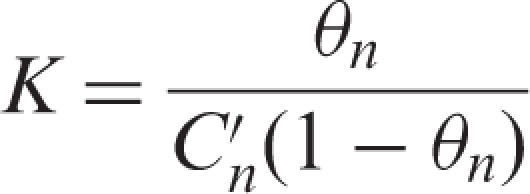 |
2 |
| 3 |
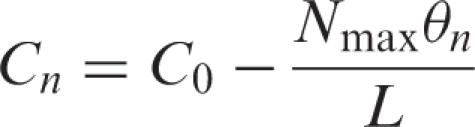 |
4 |
 |
5 |
Balance Equations (3) and (4) were substituted into equation (2) and finally, the following recurrence formula (equation (6)) was obtained by solving the quadratic equation for θn under the given initial conditions (equation (5)), where ℓ is the aspirating volume, L is the sample reservoir volume, Nmax is the maximal binding capacity of an affinity tip-column and Cn is the target RNA concentration in the sample reservoir after pipetting n times.
 |
6 |
Bn was defined as follows:
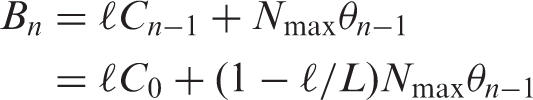 |
7 |
Bn shows the total target RNA trapped in the tip-column after the aspiration step of n pipetting cycles.
Validation of the theoretical equation
To validate the theoretical model of RCC, we simulated an isolation profile of E. coli tRNAPro2 from total RNA solution, using a 30 mer 3′-biotinylated DNA probe complementary to the anticodon region of tRNAPro2. The exact concentration of E. coli tRNAPro2 (C0) in a 1.6 mg/ml E. coli total RNA solution was determined to be 0.462 µM by dot blot hybridization using highly purified E. coli tRNAPro2 as a standard control. The approximate value of Nmax was determined to be 1.755 nmol from the yield of E. coli tRNAPro2 isolated from an excess (8 mg/ml) of E. coli total RNA. Using these initial conditions for the recurrence formula (Equation (6)), the yield of E. coli tRNAPro2 versus number of pipetting cycles was plotted using various equilibrium constants (K ) as shown in Figure 2A. The number of pipetting cycles at which the yield reaches a plateau varies drastically depending on the K values, suggesting that the total yield depends on the strength of the ligand–target affinity. In addition, the number of pipetting cycles sufficient for the efficient isolation of a target RNA can be predicted by this graph.
Figure 2.

Fitting to the theoretical model for RCC. (A) Isolation profile of E. coli tRNAPro2 simulated by the recurrence equation (6) using the initial conditions C0 = 0.462 µM and Nmax = 1.755 nmol. Yields of E. coli tRNAPro2 after n pipetting cycles with various equilibrium constants (K) were plotted (solid lines). Dashed line represents a plot when K is the infinite value. Kapp = 11 µM−1 is an experimental value fitted to the theoretical model for RCC (dotted line). (B) Experimental values of E. coli tRNAPro2 concentration (Cn) in the sample reservoir were quantified by dot blot hybridization and fitted to the theoretical equation. Kapp was determined to be approximately 1.1 × 107 M−1 with the coefficient of determination (R2) 0.888. (C) Predicted yields calculated from the theoretical equation with Kapp = 1.1 × 107 M−1 were compared with actual experimental yields quantified by northern hybridization. Initial conditions are described below the graph.
To determine the apparent equilibrium constant (Kapp) for E. coli tRNAPro2 and its corresponding DNA probe, the concentration of E. coli tRNAPro2 (Cn) at every reciprocal circulating step during RCC isolation of E. coli tRNAPro2 was measured. A small aliquot was taken from the RNA solution after every pipetting step, and the amount of tRNAPro2 was quantified by dot blot hybridization. The data were manually fitted to the theoretical Equation (6), and the Kapp was determined to be ∼1.1 × 107 M−1 with R2 = 0.888 (Figure 2B). The isolation profile of E. coli tRNAPro2 with this Kapp value is shown in Figure 1A. The maximum yield after 40 RCC cycles was approximately 80%, and 20 cycles seems to be sufficient for efficient isolation.
Next, we predicted the total yields of tRNAPro2 under various initial conditions using the determined Kapp value. The predicted yield and actual yield of tRNAPro2 were compared when the initial amount of total RNA was varied, as shown in Figure 2C. The results indicate that the predicted yields were consistent with the experimental values; nevertheless, the theoretical model of RCC includes some approximation. It is noteworthy that the percent yield of an individual RNA species was greater when the initial amount of total RNA was limited, suggesting that RCC is suitable for isolation of minor RNAs. Percent yield of target RNA is roughly predictable if the Kapp value is available.
Advantage of parallel isolation
In conventional methods to isolate multiple RNA species, total RNA solutions are often divided into aliquots, and each RNA target is purified from a separate aliquot. In the RCC method, multiple targets are purified from the same solution in a common reservoir, which leads to more effective utilization of a limited sample. To estimate the advantage of parallel isolation by the RCC method, total yields of eight different tRNAs were compared between RCC isolated preparations and separate aliquot purifications. In this experiment, the RCC instrument was equipped with 8 tip-columns containing probes corresponding to E. coli tRNAs for Met, fMet, Phe, Pro1, Pro2, Pro3, Sec and Trp, respectively (Table 1). RCC was performed with 2 ml of E. coli total RNA (8 mg/ml) in a single reservoir. In contrast, the separate aliquot purifications were performed with the same probes, but each tip-column was used to pipette from a separate reservoir, containing 250 μl of E. coli total RNA (8 mg/ml). Thus, the amount of E. coli total RNA used for each experiment was identical. Yields from each purification scheme are summarized in Figure 3. As expected, yields of tRNAs from the RCC method were substantially higher than those from the separate aliquot purifications because the entire RNA solution can participate in the RCC method, but only one-eighth of the total RNA is available for each of the separate aliquot purifications.
Figure 3.

Comparison between the RCC method and isolation from separate aliquots. RCC was performed with 2 ml of the E. coli RNA mixture (8 mg/ml) in a common reservoir. For separate aliquot isolation, the pipetting cycles for each tip-column used separate reservoirs, each of which contained 250 μl of the E. coli RNA mixture (8 mg/ml). Filled and open bars represent yields of RCC and separate aliquot isolation, respectively.
Isolation of total species of E. coli tRNAs and four sRNAs
To verify the applicability of RCC to high throughput methods, we conducted an automated isolation procedure for all the species of E. coli tRNA and four sRNAs using the RCC instrument. For sRNA targets, 4.5S and 6S RNAs were selected as abundant traditional sRNAs. DsrA (25) and SraH (RyhA) (26) were used as representatives of newly discovered sRNAs (27). We designed 30 mer 3′-biotinylated DNA probes complementary to the 3′ side or the anticodon region of each RNA depending on the sequence specificity. Targets and their probe sequences are shown in Table 1. Four pairs of tRNA species shared the same probes since their sequence similarity is quite high. Thus, of the 48 probes used in this experiment, 44 were probes for tRNAs and 4 were probes for sRNAs. The probes were divided into six groups, and RCC was performed with each set of eight probes. Approximately 50 μg (52 ± 4.2 μg) of each probe was immobilized on 35 μl streptavidin resin in each tip-column using the RCC instrument (data not shown).
Using a 16 mg sample of total RNA in a common reservoir as the starting material, 6 runs were conducted in which 8 different tip-columns were attached to the head of the RCC instrument for purification of the ncRNAs. Pipetting with the RNA solution in the reciprocal circulation was repeated 40 times at 66°C. Then, columns were sequentially washed and eluted by pipetting three times in 12 sets of washing tubes and 6 sets of eluting tubes at 40 and 68°C, respectively. Sufficient washing was confirmed by measuring the optical densities at 260 nm of washed fractions (not over 0.01 A260 in last tubes). Isolated RNAs collected from the eluting tubes were electrophoresed in 10% denaturing polyacrylamide gels (Figure 4A). All tRNA species were visible on the gel as a single band or as major bands with some contamination. We confirmed isolation of some minor tRNAs, such as tRNASec. Doublet bands of several tRNAs were found to be conformers originating from the same tRNA. Lower bands were degraded fragments. From 16 mg total RNA, total yields of isolated RNAs were ∼20–120 μg (Table 1). The binding capacities of the tip-columns limited the total yield of some abundant tRNAs. Half of the target RNAs showed over 80% purity, although purities of several RNAs were low due to inappropriate probe designs (Table 1) which were improved by redesigning probe sequences (data not shown)
Figure 4.

Automated parallel isolation of all species of E. coli tRNA and four sRNAs. (A) Polyacrylamide gel electrophoresis of E. coli tRNAs, 4.5S and 6S RNAs isolated by the RCC instrument. RNAs were visualized by ethidium bromide staining. Several tRNAs migrated as doublet bands, which were conformers of the same tRNAs. Species of tRNAs are shown as single letter abbreviations for the corresponding amino acid and a number extension in the case of multiple tRNAs for the same amino acid. E. coli total RNA (asterisk) was used as a marker. (B) Northern blot analysis of DsrA and SraH sRNA. Strong signals were observed in the eluted fraction (elution), weak signals were visible in the starting sample (load), and no signals were found in the flow through fraction (FT). Ethidium bromide (EtBr) staining is also shown. Most of the target RNAs were highly concentrated in the eluted fractions.
To confirm the RCC isolation, each tRNA was purified from the major band on the gel. The purified tRNAs were digested by RNase T1, and then analyzed by LC/MS using iontrap mass spectrometry. For all tRNAs, and the 4.5S and 6S RNAs, most of the RNase T1-digested fragments could be assigned to known sequences in the target RNAs with their modifications (Supplementary Figure 1). Detailed analyses of the purified tRNAs will be reported separately.
DsrA and SraH were detected by northern blot analysis due to their low abundance in the cell (Figure 4B). In E. coli total RNA, weak signals by northern blot for both sRNAs could be detected, which then disappeared in the flow through fraction. Strong signals were observed in the eluted fraction, nevertheless no bands corresponding to the northern signals were visible on ethidium bromide stained gels. This result demonstrated that a large fraction of the target sRNAs were efficiently trapped onto the tip-columns, and highly concentrated in the eluted solution. Thus, if a large amount of total RNAs is used for RCC purification, these bands should also be visible on ethidium bromide stained gels.
Isolation of several ncRNAs from yeast and mouse
Eukaryotic ncRNAs were chosen for the next validation of the RCC method. In Saccharomyces cerevisiae, 8 probes were designed for U4 and U6 snRNAs, SRP RNA (SCR1), 4 species of snoRNAs and mitochondrial tRNAMet (Table 2). As shown in Figure 5, all target RNAs, including the long RNA SCR1 (522 nt), were successfully isolated by this method. From 40 mg total RNA, the yields of isolated RNAs were ∼0.16–3.24 μg (Table 2). Seven mouse ncRNAs targets were selected: 7SK, SRP, RNase P, RNase MRP, Y1, Vault and U5 (Table 2). As shown in Figure 5B, each target RNA was isolated by RCC and the yields of isolated RNAs were ∼0.08–2.56 μg, from a starting sample of 4 mg total RNA (Table 2).
Table 2.
List of probes and total yields for ncRNAs from yeast and mouse. Target RNA species, their length, probe sequences, amounts of immobilized probes and total yields of target RNAs are shown. The sequence of each ncRNA was obtained from the Rfam database (http://www.sanger.ac.uk/Software/Rfam/) (31). U4, U5 and U6 snRNAs are spliceosomal RNAs. SCR1 is an ncRNA in the signal recognition particle. SNR5, 9, 128 and 190 are snoRNAs. #Length of the human 7SL RNA is listed
| RNA species | Length | Probe sequence (5′ > 3′) | Yield of target RNA (μg) |
|---|---|---|---|
| Yeast | |||
| U4 snRNA | 162 | CACTGATATGCGTATTTCCCGTGCATAAGG | 0.72 |
| U6 snRNA | 112 | CATCCTTATGCAGGGGAACTGCTGATCATC | 1.84 |
| SCR1 | 522 | ACGCTGGATAAAACTCCCCTAACAGCGGTG | 3.24 |
| SNR5 | 197 | TATAGACATATGGAGGCGTGATGTCTTAAG | 0.44 |
| SNR9 | 187 | GACTAATGATAGGTGGGTCAGGATATCAGC | 1.12 |
| SNR128 | 126 | CCGTGGAAACTGCGAATGTTAAGGAACCAG | 0.16 |
| SNR190 | 190 | GCTCAGATCTGCATGTGTTGTATAACACTG | 0.68 |
| mt tRNAMet | 76 | TTATTTATTTATGAGACAAATGTTTTAACC | 0.24 |
| Mouse | |||
| 7SK RNA | 331 | CTCATTTGGATGTGTCTGGAGTCTTGGAAG | 0.32 |
| 7SL RNA | 303# | CCAGGCTGGAGTGCAGTGGCTATTCACAGG | 1.60 |
| RNase P (H1) | 310 | GGCCCGGGAGGTGCCTCACCTCAGCCATTG | 0.24 |
| RNase MRP | 275 | GCACGCCGCTCAGCTCGCCCCGGAGGGGTC | 0.08 |
| Y1 RNA | 111 | GTCAAGTGCAGTAGTGAGAAGGGGGGAAAG | 0.80 |
| Vault RNA | 141 | GGGCCAGGGAGCGCCCGCCGGTCTCGAACC | 0.40 |
| U5 snRNA | 117 | CAAAAAATTGGTTTAAGACTCAGAGTTGTT | 2.56 |
Figure 5.

Automated parallel isolation of several ncRNAs from yeast and mouse. (A) Polyacrylamide gel electrophoresis of U4 and U6 snRNAs, SCR1 (SRP RNA), 4 snoRNAs (SNR5, SNR9, SNR128 and SNR190) and mitochondrial tRNAMet, which were isolated by RCC. The gel is stained with SYBR Green II (Invitrogen). Target RNAs are indicated by arrow heads. (B) Isolated mouse ncRNAs: 7SK RNA, SRP RNA, RNaseP RNA, RNaseMRP RNA, Y1 RNA, Vault RNA and U5 snRNA. Target RNAs are indicated by arrow heads.
DISCUSSION
We have described a novel method and instrument, RCC, as a new platform technology for RNA isolation. Isolation of all species of E. coli tRNAs and various ncRNAs from yeast and mouse demonstrate that RCC is a powerful, efficient, reproducible and convenient method for isolating multiple species of RNA molecules from a single sample of total RNA. To establish this method as a generally applicable method for RNA research, it will be necessary to optimize and improve some aspects of this method. As shown in this study, RNA molecules were isolated with various purities and yields. These parameters depend on several factors, including probe design, temperature control and salt concentration. For isolating stable RNA molecules like tRNAs, probe design is the most important factor to achieve maximum purity and yield. Chain length, GC content and sequence specificity must also be considered for each DNA probe. For 44 E. coli tRNAs, we designed 30 mer DNA probes complementary to the anticodon or 3′ region of each tRNA, depending on the sequence specificity. To optimize design of probes for each target, northern or dot blots are convenient for checking the hybridization efficiency of each probe (28,29). RCC might also be used to compare the efficiency and performance of several different probes for a single target RNA.
According to our theoretical model for RCC, the total yield of target RNA was closely correlated to the equilibrium constant (K ) of ligand–target interaction. For efficient isolation of RNA molecules, it might be better to choose nucleic-acid-related compounds as probes (30), such as the locked nucleic acid (LNA) or the peptide nucleic acid (PNA), which have higher hybridization efficiencies than a DNA probe. Since a small fraction of biotinylated DNA probes detached from the streptavidin sepharose during the reciprocal circulating step, further investigation of resins and methods for immobilization of probes is needed. DNA probes having an amino-linker covalently bound to sepharose resin activated with N-hydroxysuccinimidyl ester may be a suitable alternative choice for immobilization. Furthermore, the temperature for hybridization and the buffer composition for each purification step is largely dependent on the particular probe. Since RCC basically has to use a single condition for 8 different probes in one operation, it is important to find conditions which are suitable for all the probes being used. Theoretical predictions of target RNA yield derived from a simplified model of the RCC method were closely correlated with the experimental results. The required number of cycles and yields of individual RNA species can be predicted by this model if the value of K is known. The theoretical model is useful to optimize the conditions and operation of the RCC instrument.
Preparation of total RNA is another critical factor for isolation of RNA species. Highly concentrated total RNA provides the best starting material, especially for isolation of minor RNA species. However, highly concentrated total RNA is often very viscous due to polysaccharides and other cellular components which are co-extracted from the cell. Prior to RCC, an initial separation of total RNA by anion exchange chromatography is required to reduce the viscosity of the concentrated total RNA solution.
RCC could easily be scaled up for high throughput applications. The number of channels in the RCC prototype instrument we assembled could be varied from one to eight, but a 96- or 384-channel liquid handling instrument could easily be used for RCC. In such cases, the RCC instrument could be used not only as a high-throughput purification machine, but also as an analytical device for various applications in RNA research. In addition, since the sample volume can be changed, minor RNA species can be purified from large volumes of an initial RNA sample, and ncRNAs such as miRNAs or piRNAs are good targets to be isolated by this method. Most of the miRNAs are regulatively expressed in specific physiological conditions, such as pathological cells, tissues and organs. It is impossible to isolate each miRNA from limited quantity of specimens. To profile miRNA expression pattern routinely, high sensitive detection systems such as microarray analysis or RT-PCR are now available. However, qualitative aspects of miRNAs are also important, because it is known that miRNAs have several dicing variants with different length and variable termini, which might modulate their activity and change its target specificity. To investigate such qualitative information, it will be of importance to isolate each miRNA once at least (not routinely) from relatively large amount of specimen. Mass spectrometry analysis of the purified RNAs can precisely quantify processing variants and/or identify the chemical structure of RNA modifications if present.
The RCC method can be used in a variety of applications. For example, when antibodies are immobilized on the tip-columns, RCC could be used for automated multiple-immunoprecipitations for interactome analysis. RCC has great potential to be used in a wide variety of applications requiring isolation of multiple components. Continued refinement of the operating conditions and testing of additional applications will validate the potential of RCC.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the Suzuki lab members, especially Takeo Suzuki and Yuriko Sakaguchi for many fruitful discussions and for technical support. We would also like to thank the Japanese companies Sakaguchi-giken and Nichiryo for assistance in assembling the RCC instrument. This work was supported by grants-in-aid for scientific research on priority areas from the Ministry of Education, Science, Sports, and Culture of Japan (to T.S.) and by a grant from the New Energy and Industrial Technology Development Organization (NEDO) (to T.S.). Funding to pay the Open Access publication charge was provided by Japan Ministry of Education, Science, Sports and Culture.
Conflict of interest statement. None declared.
REFERENCES
- 1.Eddy SR. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- 2.Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 3.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 4.Mattick JS, Makunin IV. Non-coding RNA. Hum. Mol. Genet. 2006;15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 5.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 8.Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- 9.Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- 10.Kim VN. Small RNAs just got bigger: Piwi-interacting RNAs (piRNAs) in mammalian testes. Genes Dev. 2006;20:1993–1997. doi: 10.1101/gad.1456106. [DOI] [PubMed] [Google Scholar]
- 11.Rozenski J, Crain PF, McCloskey JA. The RNA Modification Database: 1999 update. Nucleic Acids Res. 1999;27:196–197. doi: 10.1093/nar/27.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, Steward R, Chen X. Methylation as a crucial step in plant microRNA biogenesis. Science. 2005;307:932–935. doi: 10.1126/science.1107130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, Ebright YW, Yu B, Chen X. HEN1 recognizes 21-24 nt small RNA duplexes and deposits a methyl group onto the 2' OH of the 3. Nucleic Acids Res. 2006;34:667–675. doi: 10.1093/nar/gkj474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Yang Z, Yu B, Liu J, Chen X. Methylation protects miRNAs and siRNAs from a 3. Curr. Biol. 2005;15:1501–1507. doi: 10.1016/j.cub.2005.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD. A distinct small RNA pathway silences selfish genetic elements in the germline. Science. 2006;313:320–324. doi: 10.1126/science.1129333. [DOI] [PubMed] [Google Scholar]
- 16.Blow MJ, Grocock RJ, van Dongen S, Enright AJ, Dicks E, Futreal PA, Wooster R, Stratton MR. RNA editing of human microRNAs. Genome Biol. 2006;7:R27. doi: 10.1186/gb-2006-7-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luciano DJ, Mirsky H, Vendetti NJ, Maas S. RNA editing of a miRNA precursor. RNA. 2004;10:1174–1177. doi: 10.1261/rna.7350304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki T, Suzuki T, Wada T, Saigo K, Watanabe K. Taurine as a constituent of mitochondrial tRNAs: new insights into the functions of taurine and human mitochondrial diseases. EMBO J. 2002;21:6581–6589. doi: 10.1093/emboj/cdf656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaneko T, Suzuki T, Kapushoc ST, Rubio MA, Ghazvini J, Watanabe K, Simpson L, Suzuki T. Wobble modification differences and subcellular localization of tRNAs in Leishmania tarentolae: implication for tRNA sorting mechanism. EMBO J. 2003;22:657–667. doi: 10.1093/emboj/cdg066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakurai M, Ohtsuki T, Watanabe K. Modification at position 9 with 1-methyladenosine is crucial for structure and function of nematode mitochondrial tRNAs lacking the entire T-arm. Nucleic Acids Res. 2005;33:1653–1661. doi: 10.1093/nar/gki309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirino Y, Yasukawa T, Ohta S, Akira S, Ishihara K, Watanabe K, Suzuki T. Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15070–15075. doi: 10.1073/pnas.0405173101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsurui H, Kumazawa Y, Sanokawa R, Watanabe Y, Kuroda T, Wada A, Watanabe K, Shirai T. Batchwise purification of specific tRNAs by a solid-phase DNA probe. Anal. Biochem. 1994;221:166–172. doi: 10.1006/abio.1994.1393. [DOI] [PubMed] [Google Scholar]
- 24.Yokogawa T, Kumazawa Y, Miura K, Watanabe K. Purification and characterization of two serine isoacceptor tRNAs from bovine mitochondria by using a hybridization assay method. Nucleic Acids Res. 1989;17:2623–2638. doi: 10.1093/nar/17.7.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lease RA, Belfort M. Riboregulation by DsrA RNA: trans-actions for global economy. Mol. Microbiol. 2000;38:667–672. doi: 10.1046/j.1365-2958.2000.02162.x. [DOI] [PubMed] [Google Scholar]
- 26.Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 2001;11:941–950. doi: 10.1016/s0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 27.Majdalani N, Vanderpool CK, Gottesman S. Bacterial small RNA regulators. Crit. Rev. Biochem. Mol. Biol. 2005;40:93–113. doi: 10.1080/10409230590918702. [DOI] [PubMed] [Google Scholar]
- 28.Kumazawa Y, Yokogawa T, Tsurui H, Miura K, Watanabe K. Effect of the higher-order structure of tRNAs on the stability of hybrids with oligodeoxyribonucleotides: separation of tRNA by an efficient solution hybridization. Nucleic Acids Res. 1992;20:2223–2232. doi: 10.1093/nar/20.9.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mir KU, Southern EM. Determining the influence of structure on hybridization using oligonucleotide arrays. Nat. Biotechnol. 1999;17:788–792. doi: 10.1038/11732. [DOI] [PubMed] [Google Scholar]
- 30.Karkare S, Bhatnagar D. Promising nucleic acid analogs and mimics: characteristic features and applications of PNA, LNA, and morpholino. Appl. Microbiol. Biotechnol. 2006;71:575–586. doi: 10.1007/s00253-006-0434-2. [DOI] [PubMed] [Google Scholar]
- 31.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.