

Abstract
Helper-free herpes simplex virus type-1 (HSV-1) amplicon vectors elicit robust immune responses to encoded proteins, including human immunodeficiency virus type-1 (HIV-1) antigens. To improve this vaccine delivery system, seven amplicon vectors were constructed, each encoding HIV-1 Gag under the control of a different promoter. Gag expression levels were analyzed in murine and human cell lines, as well as in biopsied tissue samples from injected mice; these data were then compared with Gag-specific T cell responses in BALB/c mice. The magnitude of the amplicon-induced immune response was found to correlate strongly with the level of Gag production both in vitro and in vivo. Interestingly, the best correlation of the strength of the amplicon-induced immune response was with antigen expression in cultured DC rather than expression at the tissue site of injection or in cultured cell lines. These findings may have implications for the generation of improved HSV-1 amplicon vectors for HIV-1 vaccine delivery.
Keywords: HSV-1 amplicon vectors, transcriptional control elements, HIV vaccines, Gag, dendritic cells (DC)
1. INTRODUCTION
Helper-free herpes simplex virus type-1 (HSV-1) amplicons represent a replication-defective gene transfer vector system with several biological features that make them an attractive choice for vaccine delivery applications [1, 2]. Among the appealing characteristics of these vectors are their large insert capacity, favorable safety profile and broad cellular/tissue tropism - which extends to antigen presenting cells, such as dendritic cells (DC) [3]. As a result, helper-free HSV-1 amplicons encoding mammalian or microbial proteins can elicit strong, antigen-specific immune responses, and may have utility in a range of applications, including cancer treatment and prophylactic vaccination against pathogens such as human immunodeficiency virus type-1 (HIV-1) [3–10].
Considerable attention has been paid to the rational improvement of amplicon vectors for gene transfer applications in the central nervous system (CNS) [11–15], but systematic attempts to enhance the performance of amplicon-based vaccine vectors have been more limited. We therefore sought to carefully characterize the effect of various transcriptional control elements on amplicon-driven expression of an HIV-1 antigen (Gag), and also on the strength of the host immune response to that antigen, following vector delivery to BALB/c mice. As part of this analysis, we also examined whether the magnitude of the in vivo immune response was correlated with the level of antigen expression, either at the local tissue site of vector injection in vivo, or in cultured cells that were representative of key potential in vivo target cells (i.e., fibroblasts, dendritic cells, keratinocytes and epithelial cells).
To date, most studies involving HSV-1 amplicon-vectored vaccines have relied on the use of the HSV-1 immediate early 4/5 promoter (HSV IE4/5) [3–5, 7]. However, it is unclear whether this is the ideal transcriptional control element for use in vaccine studies - especially since the activity of this promoter is strongly influenced by the HSV-1 tegument protein, VP16 [16–18]. We therefore generated a series of seven amplicon constructs, each of which contained the HIV-1 gag gene, under the transcriptional control of a different promoter element. The constructs included four promoters that were derived from the human cytomegalovirus (HCMV) immediate-early promoter (one wild-type promoter and three hybrid promoters, incorporating additional regulatory elements), as well as two retroviral promoters and the HSV IE4/5 promoter.
Amplicon vectors harboring the various promoters were used to transduce a panel of murine and human cell lines, and protein expression levels were assessed by immunoblot analysis and p24 Gag ELISA. The vectors were also injected into BALB/c mice, via an intradermal route, and Gag expression levels were measured at the local site of injection by p24 Gag ELISA. Finally, Gag-specific T cell immune responses were also analyzed. These studies revealed that constructs that contained two of the hybrid CMV promoters elicited the strongest immune responses, and significantly outperformed all of the other constructs. Interestingly, the magnitude of the in vivo immune response elicited by the various constructs was found to correlate significantly with the level of Gag expression both in vitro (in cultured cell lines and primary murine dendritic cells) and in vivo (in biopsied tissue from the local site of amplicon injection). These findings have implications for the rational improvement of HSV-1 amplicon-vectored vaccines.
2. MATERIALS AND METHODS
2.1 Transcriptional promoter elements
Seven helper virus-free HSV-1 amplicon vectors encoding HIV-1 Gag were developed, each with a different promoter (Figure 1). These promoters included: two retroviral promoters (the Rous sarcoma virus [RSV] and the myeloproliferative sarcoma virus [MPSV] long terminal repeat [LTR]) as well as the major human cytomegalovirus immediate-early promoter [CMV], and several hybrid promoters derived from this element; these included a composite CMV/chicken ß-actin promoter [CAG] [19], a modified CMV promoter containing regulatory sequences from the human T-cell leukemia virus type-1 LTR [CMV-R] [20], and a CMV promoter linked to the CMV Intron A sequence [CMV-I] [21, 22].
Figure 1. Schematic diagram of the promoter elements used in the various HSV-1 amplicon constructs.
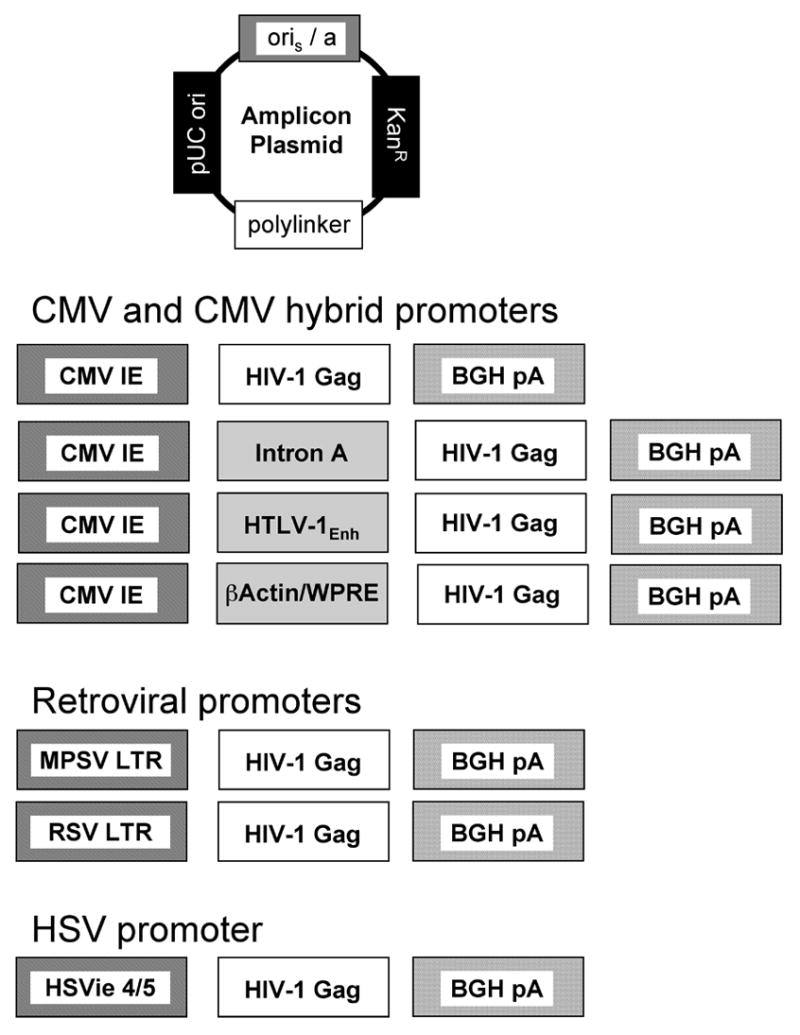
Seven helper virus-free HSV-1 amplicon vectors encoding HIV-1 Gag were developed, each with a different promoter. Promoters that were studied included: the major human cytomegalovirus immediate-early promoter [CMV], and several hybrid promoters derived from this element - including a CMV promoter linked to the CMV Intron A sequence [CMV-I], a modified CMV promoter containing regulatory sequences from the human T-cell leukemia virus type-1 LTR [CMV-R] and a composite CMV/chicken ß-actin promoter [CAG], as well as two retroviral promoters (the Rous sarcoma virus [RSV] and the myeloproliferative sarcoma virus [MPSV] long terminal repeat [LTR]). The Herpes Simplex Virus Type 1 immediate early 4/5 promoter [HSVie] was also used. Each of the indicated Gag expression cassettes was then inserted into the polylinker region of the parental amplicon plasmid.
2.2 Construction of HSV-1 amplicon vectors with different promoter elements driving HIV-1 gag expression
The HSV-1 amplicon contains the HSV-1 packaging signal and origin of DNA replication [2], inserted into the backbone of pVax (Invitrogen, Carlsbad, CA). The indicated gene cassettes (Figure 1) were inserted into the plasmid polylinker. The basic CMV promoter/enhancer was used from a modified pVax-based amplicon plasmid and the gag insert cloned in with HindIII and XbaI. The CMV promoter/enhancer and the associated Intron A sequence were PCR amplified from plasmid VRC5309 (provided by the NIH VRC) using forward (5′-ACCATATGCACGCGTGTGTGAAATACCGCA-3′) and reverse oligonucleotide primers (5′-TGGCGATATCTCTAGAGCGGCCGCGATATC-3′) [20, 23] (VRC5309 was generously provided by Dr. Gary Nabel of the NIH Vaccine Research Center, VRC). The 1.6 kb PCR product was digested with MluI and XbaI and cloned into the amplicon backbone to create CMV-I. The CMV promoter/enhancer and the HTLV-1 R region together with the gag insert were excised from the plasmid VRC4401 (also provided by the NIH VRC; [24]) using HindIII and XbaI. The resulting DNA fragment was cloned into the promoterless amplicon vector backbone to create CMV-R.
The CAG promoter was contained in a plasmid kindly provided by Dr. Joshi Jacob [19]. To construct the CAG-containing amplicon, the HIV-1 gag gene was first PCR amplified using forward (5′-ACTGGCGGCCGCCTCGAGCACCATGGGCGCCCGCGCCAGCGTG-3′) and reverse oligonucleotide primers (5′-GACTTCTAGAAGATCTTTATTGTGACGAGGGGTCGCTG-3′). This PCR amplimer was then digested with NotI and XbaI, and inserted into the amplicon backbone cut with the same enzymes. The CAG promoter element was inserted into the amplicon backbone upstream of the gag gene, using NdeI and NotI.
The RSV promoter was contained in a commercially obtained plasmid (pREP4; Invitrogen, Carlsbad, CA), which was digested with BamHI and HindIII to incorporate the gag insert that was digested using the same enzymes. The RSV-gag insert was PCR amplified using 5′ GGCGTTTAAACACTCTCAGTACAATCTGCTCTGATGCCGC 3′ forward primer and 5′ GCCTCGAGGAGCTCGGTACCTATCTTT 3′ reverse primer; the resulting PCR fragment was cut with PmeI and XhoI and then ligated into the amplicon backbone that was digested with HincII and XhoI; this was then cloned into the amplicon backbone to create RSV. The MPSV [25, 26] promoter was PCR amplified using 5′GTTGACCCCGGGGCAAGGCATGGAAAATA 3′ forward primer and 5′GCAAGCTTGGATCCATTGAGAACACGGG3′ reverse primer. The PCR product was digested with HincII and HindIII and cloned into the amplicon vector backbone. All of the plasmids include a human codon-optimized insert encoding HIV-1 Gag, that was derived from the clade B HXB2 strain [24]. The gag insert was excised from plasmid VRC4401 (provided by the NIH VRC; [24]) with HindIII and XbaI and cloned into the amplicon plasmid cut with the same enzymes.
2.3 Helper virus-free HSV amplicon packaging
Packaging of viral particles was performed as described [27]]. Briefly, on the day prior to transfection, 2 x 107 BHK cells were seeded in a T-150 flask and incubated overnight at 37°C in 5% CO2. On the day of transfection, 1.8 ml of OptiMEM (Invitrogen), 25 μg of pBAC-V2 DNA [28], 7 μg of pBSKS (vhs) [27], and 7 μg of amplicon vector DNA were combined in a sterile polypropylene tube. Subsequently, 70 μl of Lipofectamine Plus reagent (Invitrogen) was added over a period of 30 s to the DNA mix, and the mixture was incubated at 22°C for 20 min. In a separate tube, 100 μl of Lipofectamine (Invitrogen) was mixed with 1.8 ml of OptiMEM, and this mixture was also incubated at 22°C for 20 min. After the incubation, the contents of the two tubes were combined over a period of 30 s and incubated for an additional 20 min at 22°C. During this second incubation, the medium in the seeded T-150 flask was removed and replaced with 14 ml of OptiMEM. The transfection mix was added to the flask and allowed to incubate at 37°C for 5 h. The transfection mix was diluted with an equal volume of DMEM plus 20% FBS, 2% penicillin-streptomycin, and 2 mM hexamethylene bisacetamide (HMBA) and then incubated overnight at 34°C. The following day the medium was removed and replaced with DMEM plus 10% FBS, 1% penicillin-streptomycin, and 2 mM HMBA. The packaging flask was incubated an additional 3 days before the virus was harvested and stored at −80°C until purification. Viral preparations were subsequently thawed, sonicated, clarified by centrifugation, and concentrated by ultracentrifugation through a 30% sucrose cushion. Viral pellets were resuspended in 100 μl of phosphate-buffered saline (PBS) and stored at −80°C until use. Vector titers were determined as described by using expression and transduction titering methods on NIH 3T3 cells [29]. Vector was diluted in sterile Dulbecco’s PBS (D-PBS; Invitrogen) to the appropriate concentration prior to injection. In addition, a separate irrelevant amplicon stock was prepared as a control (this vector encoded the gene for Escherichia coli ß-galactosidase [LacZ]).
2.4 Cell culture
All cells were incubated at 37°C/5% CO2 under humidified conditions. 3T3 cells (a murine fibroblast cell line) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen) supplemented with 10% bovine calf serum (Hyclone, Logan, UT) and 0.1% penicillin, streptomycin, and L-glutamine (PSG; obtained from GIBCO-BRL). XB2 cells (a murine keratinocyte cell line) were obtained from ATCC and cultured in DMEM with 4mM L-glutamine supplemented with 20% fetal bovine serum (FBS) and 0.1% PSG. These cells were propagated using a feeder layer of irradiated mouse fibroblasts. EMT6 cells (a mouse mammary epithelial cell line) were maintained in MAT/P medium (U.S. patent 4,816,401; cells and medium were kindly provided by Dr. Edith Lord) supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, and 2% FBS. Primary murine bone marrow derived dendritic cells were isolated as described, with minor modifications [30]. Briefly, femurs and tibiae of mice were removed and purified from the surrounding muscle tissue. Intact bones were left in 70% ethanol for 2–5 min for disinfection and washed with PBS. Then both ends of the bone were cut with scissors and the marrow flushed with PBS using a 1ml syringe with a 26G needle. Clusters within the marrow suspension were disintegrated by vigorous pipetting. The single cell suspension was spun down once (1200x, 7min) and the bone marrow cell population was subsequently purified by positive selection with anti-CD14 MACS beads (Miltenyi Biotec, Auburn, CA). These monocytes were then cultured in RPMI 1640 medium supplemented with 50 ng/ml mouse recombinant GM-CSF (R&D Systems, Minneapolis, MN) and 25 ng/ml IL-4 (R&D Systems). Media was replenished every 2 days. After 7 days of culture, DCs were transduced with amplicon particles.
2.5. Amplicon transduction
One day before exposure to amplicon particles, cells were seeded into 24-well plates and allowed to adhere overnight at 37°C/5% CO2. Cells were then either transduced with the Gag-encoding amplicon constructs or with lacZ-encoding amplicon particles (as a negative control) at a multiplicity of infection (MOI) of 1, in a volume of 0.5 ml at 37°C/5% CO2. After 1 hour, 0.5 ml of the corresponding culture medium was added to the cells; 24 hours later, cultures were harvested for analysis. In some experiments, cells were transfected with 1μg of VRC4401 plasmid DNA as a positive control (this plasmid encodes HIV-1 Gag, under the transcriptional control of the CMV-R promoter element, and was generously provided by the NIH VRC). For these control experiments, cells were transfected in 6-well plates using Lipofectamine2000 (Invitrogen), and then harvested 24 hours later.
2.6 In vitro protein expression analysis
After infection or transfection, cells were lysed with 500 μl of RIPA buffer (0.5M Tris-HCl, pH 7.4, 1.5M NaCl, 2.5% deoxycholic acid, 10% NP-40, 10mM EDTA) (Millipore/Upstate, Charlottesville, VA) supplemented with a 1:500 dilution of broad specificity protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Lysates were clarified by centrifugation at 13,000 rpm for 10 min at 4°C and protein concentration was measured by using Bradford reagent according to the manufacturer’s protocol (BioRad, Hercules, CA). 20 μg of lysate from each sample was then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted to nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ). The membranes were then probed with a 1:1000 dilution of a primary monoclonal antibody to HIV-1 p24 (AG3.0) obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH from Dr. Jonathan Allan [31]. A 1:1000 dilution of horseradish peroxidase-conjugated sheep anti-mouse IgG was used as the secondary antibody (Amersham). The blots were developed with enhanced chemiluminescence (ECL) reagent (Pierce Biochemical Co., Rockford, IL) and imaged using the ChemiDocTMXRS system (BioRad).
2.7 HIV-1 p24 ELISA
To measure Gag levels in cell and tissue lysates, we used an HIV-1 p24 enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (ZeptoMetrix, Buffalo, New York). Briefly, test samples were incubated overnight at 37oC in ELISA plates coated with a monoclonal antibody specific for p24 Gag. The captured antigen was then reacted with a human anti-HIV-1 p24 antibody conjugated to biotin; detection of bound antibody was achieved using streptavidin-peroxidase. To quantify the levels of HIV-1 p24, a standard curve was created using wells coated with known amounts of p24 antigen in serial 2-fold dilutions starting at 250pg/ml. In each test reaction, 1 μg of cell or tissue extract was analyzed, in a reaction volume of 400 μl. Thus, all assays for p24 protein production were normalized in terms of total input cell/tissue protein. All results (Figures 2 thru 4) are expressed as ng of HIV-1 p24 protein per ml of reaction volume, where 1 ml of the reaction contained 2.5 μg of cell or tissue extract.
Figure 2. Gag expression following transduction of a panel of cell lines and primary cells with HSV-1 amplicon constructs.
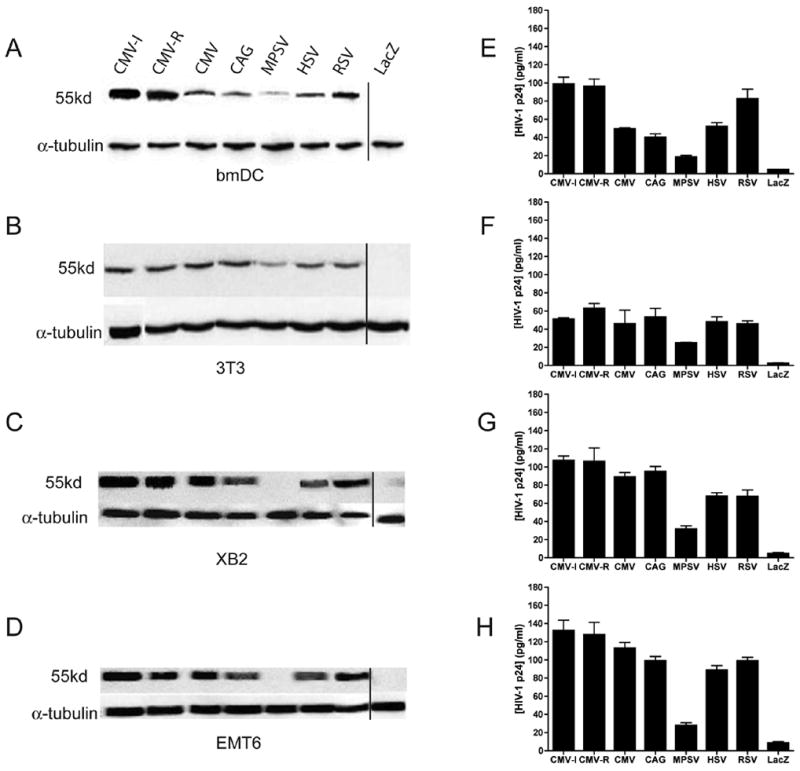
Murine bone marrow-derived dendritic cells (bmDC; panels A and E), NIH 3T3 cells (a murine fibroblast cell line; panels B and F), XB2 cells (a murine keratinocyte cell line; panels C and G) and EMT6 cells (a murine mammary epithelial cell line; panels D and H) were transduced with HSV-1 amplicon constructs encoding HIV-1 Gag from seven different promoters (as indicated in Figure 1) at an MOI of 1. Twenty-four hours later, cell lysates were prepared, and protein content within the lysates was determined by Bradford assay. A fixed amount of each lysate was then loaded onto a SDS/PAGE gel for subsequent immunoblot analysis with a Gag-specific monoclonal antibody (panels A–D), or used in a HIV-1 p24 Gag ELISA (panels E–H). For the immunoblot assays shown in panels A–D, the cell lysates were also probed with an anti-α-tubulin antibody (as a loading control). The p24 values in panels E–H represent mean values of triplicate measurements; bars denote the standard deviation of those values. The results shown (panels A–H) are representative of two independent experiments that yielded very similar data. Note that, due to the limited number of wells available on the SDS/PAGE gels, the lacZ control sample on all of the immunoblots was run on a different gel than the other samples; this is denoted by the vertical line on each of the four immunoblot panels. Results (panels E–H) are expressed as ng of HIV-1 p24 protein per ml of reaction volume, where 1 ml of the reaction contained 2.5 μg of cell extract.
Figure 4. Preformed Gag is present in HSV amplicon particles, but decays rapidly.
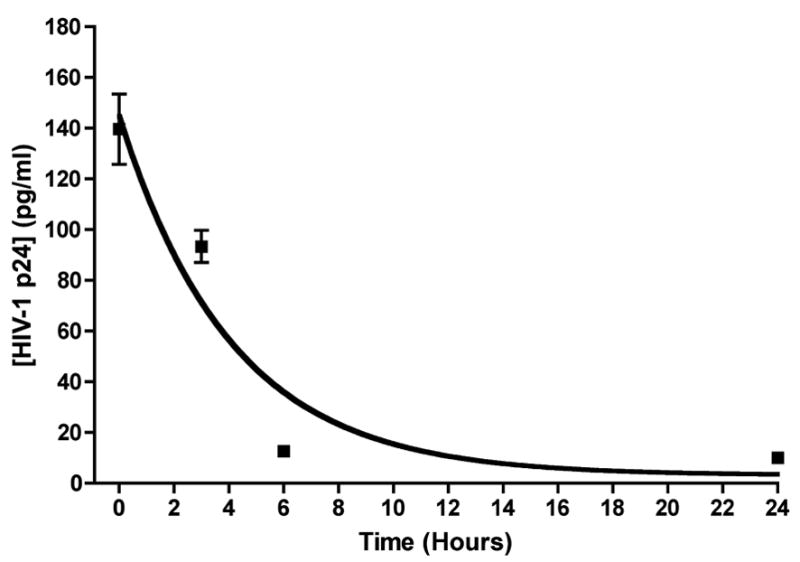
BALB/c mice (3/group) were inoculated in the tail base with 5 x 105 transducing units of UV-inactivated amplicon particles containing the (CMV-R) driven Gag insert (Figure 1). Groups of mice were then sacrificed at timed intervals following amplicon inoculation (5 min, as well as 3, 6 and 24 h), and the injection site was recovered using a tissue punch. A tissue lysate was prepared, and protein content in each lysate was determined by Bradford assay. A fixed amount of lysate was then used in a HIV-1 p24 ELISA; the results are expressed as ng of HIV-1 p24 protein per ml of reaction volume, where 1 ml of the reaction contained 2.5 μg of tissue extract. The data shown represent mean values of triplicate measurements, and bars denote the standard deviation of those values; the sensitivity cutoff of the Gag ELISA is 10 pg/ml, and samples in which the measured Gag level was below this were assigned a value of 10 pg/ml. A nonlinear curve for a single-phase exponential decay was fitted to the data using GraphPad Prism (and is shown in the Figure). The R2 value (goodness of fit) for this curve was 0.88, and the in vivo half-life of the preformed Gag antigen present within the HSV-1 amplicon inoculum was estimated at 2.84 hours (with 95% confidence intervals ranging from 1.73 to 7.94 hours).
2.8 Mice
Female BALB/c mice aged 5 to 6 weeks were obtained from Taconic Laboratories (Germantown, N.Y.) and maintained according to University of Rochester and NIH guidelines. Mice were injected with amplicon vectors using a 28G 0.5-in. 1-ml insulin syringe (Becton Dickinson [BD], Franklin Lakes, N.J.)
2.9 Tissue processing for in vivo protein expression analysis
BALB/c mice were injected with 5 x 105 amplicon particles via the intradermal (i.d.) tailbase route. The site of vector injection was marked with an indelible marker, and the tissue surrounding this site was harvested at defined time intervals following vector administration. The fur around the injection area was removed, and the entire injection site was recovered by taking a 6-mm skin specimen using a sterile punch (Acuderm, Fort Lauderdale, FL). These tissue samples were then snap frozen in liquid nitrogen and pulverized using a mortar and pestle, prior to resuspension in passive lysis buffer (Promega, Madison, WI). All samples were then homogenized using a polytron (Ultrathurrax, Ika-Werke, Germany) at 3000 rpm for 1 min. Lysates were subsequently cleared by high-speed centrifugation, and protein concentration was determined using Bradford reagent (BioRad). Samples were then analyzed by HIV-1 p24 ELISA as described above.
2.10 UV inactivation of amplicon particles
100 μl of a stock of HSV amplicon viral particles was transferred to an open 15mm Petri dish. The sample was then irradiated by placing it in a Stratalinker® 1800 UV Crosslinker (Stratagene, LaJolla, CA), at distance of 10 cm from the germicidal lamps; the sample was then irradiated for 10 minutes at 1,200 μW/cm2. To verify that this UV-irradiation protocol was sufficient to inactivate the amplicon particles, a pilot experiment was conducted, using HSV amplicon particles that encoded a CMV promoter-driven luciferase reporter gene (CMV:luc). After exposure to UV irradiation, the CMV:luc amplicon particles were added to HEK293 cells at an MOI of 1. Twenty four hours later, cell lysates were prepared, and clarified cell lysate (1μg total protein/μl) was assayed for luciferase activity using 20 μl firefly luciferase substrate (Promega). Light emission was measured in a white 96-well plate, using a luminometer (SpectraCount Version 3.0, Packard). This experiment confirmed that the UV treatment resulted in complete loss of vector infectivity (as defined by reduction in luciferase expression to baseline levels, that were indistinguishable to those present in untransduced control cells).
2.11 Tetramer staining
Gag-specific CD8+ T cells were quantitated by staining splenocytes with a H-2Kd AMQMLKETI tetramer obtained from the National Institutes of Health (NIH) Tetramer Facility (Emory University, Atlanta, GA); the AMQMLKETI peptide is known to be immunodominant in BALB/c mice, and is restricted by H2-Kd. Mice were immunized with 5 x 105 amplicon particles via the intradermal (i.d.) tailbase route. Ten days post immunization, mice were sacrificed and spleens removed. Splenocytes were harvested and cultured in RPMI (Invitrogen) supplemented with 10% FCS (Hyclone), 50 μM β-mercaptoethanol and PSG (Invitrogen). Cell suspensions were then stained at 4°C for 30 min with FITC-conjugated anti-CD8, PE-conjugated anti-CD3 (BD Biosciences, Franklin Lakes, NJ) and APC-conjugated Gag tetramer. Cells were washed in FACS buffer (PBS, 2% FCS). Samples were acquired using a FACSCalibur (BD Biosciences) and analyzed using CellQuest software by gating on CD3+ T cells and then displaying CD8 versus tetramer staining. The results were expressed as the percentage of tetramer-positive CD8+ T cells.
2.12 Intracellular cytokine staining (ICS)
Splenocytes from immunized mice were harvested and single cell suspensions were prepared as described above. 1 x 106 splenocytes were then stimulated with HIV-1 Gag peptide pools (15-mer peptides at 2μg/ml each, derived from strain HXB2, obtained from the NIH AIDS Reagent Program). Cultures were incubated for 6 hours and Golgi Plug (BD Biosciences) was added during the final 4 h of incubation. Cells were then permeabilized and fixed with Cytofix/Cytoperm (BD Biosciences). Samples were stained with a cocktail of conjugated antibodies consisting of anti-CD8 Pacific Blue, anti-CD3 Alexa 405 (Caltag Laboratories, Burlingame, CA), anti-CD4 APC, anti-IL-2 FITC and anti-IFN-γ PE (BD Biosciences). Stained cells were analyzed on a LSR II flow cytometer (BD) and the flow data was subsequently analyzed using FLOWJO software (TreeStar, Ashland, OR).
2.13 Statistical data analysis
Data are presented as means with standard deviations. Statistical analysis was performed with GraphPad PRISM software version 4.0 (GraphPad Software, Inc). Comparison of cellular immune responses between groups was performed by one-way ANOVA (nonparametric). In all cases, P values of less than 0.05 were considered as significant. Immune response data (tetramer staining or intracellular cytokine staining results) were compared in a pairwise fashion with protein expression data, to determine whether there was a correlation between immune responses and protein expression (either in vivo or in vitro). For this analysis, we assumed that protein expression and immune response data would be sampled from populations that follow a Gaussian distribution (at least approximately), and that covariation between immune response values and protein expression levels would be linear. We therefore calculated a linear correlation coefficient (Pearson r value) and a corresponding P value (two tailed) based on these assumptions. In some cases, we also performed a linear regression analysis using the same core assumptions.
3. RESULTS
3.1 Effect of different transcriptional control elements on in vitro Gag expression from HSV-1 amplicon vectors
The strength of the immune response elicited by virally vectored HIV-1 vaccines is likely determined by multiple factors, including the interplay between the properties of the encoded antigen expression cassette and the inherent characteristics of the vector itself (such as host cell tropism and immunoregulatory effects associated with receptor binding and host cell entry). As a result, the optimization of virally vectored vaccine platforms is likely to depend on individualized tailoring of the antigen expression cassette to the specific vector platform and intended application. As a first step towards this, the present study was designed to analyze the effect of different transcriptional control elements on immune responses to a virally-vectored HIV-1 antigen (Gag), and to determine whether levels of antigen expression in various cultured cells and cell lines, or in intact animals, might predict in vivo immunogenicity.
To optimize the helper-free HSV-1 amplicon platform for HIV-1 vaccine delivery, we inserted a series of seven different promoter elements upstream of the HIV-1 gag gene (Figure 1). Promoters chosen for this study included the ubiquitously active human cytomegalovirus immediate early (CMV) promoter, as well as three hybrid regulatory elements derived from this promoter. These hybrid promoters contained combinations of the CMV promoter/enhancer together with (i) its associated Intron A element (CMV-I) [21, 22, 32–34], (ii) the regulatory R region from the long terminal repeat (LTR) of the human T-cell leukemia virus type 1 (CMV-R) [20], and (iii) the chicken β-actin promoter and the woodchuck hepatitis virus regulatory element (CAG) [19]. Each of these composite CMV-based promoters has been reported to mediate improved levels of gene expression and/or enhanced immune responses to encoded antigens, when compared to the basic CMV promoter [35, 36]. The other transcriptional regulatory elements that were incorporated into our amplicon panel included two retroviral LTRs and the HSV-1 immediate early 4/5 promoter, which was used in our previous studies on the immunogenicity of HSV amplicon-vectored HIV-1 antigens [4]. The retroviral LTRs used were (i) the broadly active Rous sarcoma virus (RSV) LTR [22] and (ii) the mouse myeloproliferative sarcoma virus (MPSV) LTR, which has been reported to be highly active in hematopoietic cell types [25, 26].
The various Gag expression cassettes were inserted into our parental amplicon plasmid (Figure 1) and amplicon stocks were derived using published methods [4]. The resulting amplicon particles are expected to contain multiple head-to-tail concatemers of each amplicon plasmid, as described [1, 2].
In vitro studies were then conducted to analyze relative Gag expression levels in a number of cell lines and primary bone marrow-derived dendritic cells, following transduction with each of the amplicon constructs at a defined multiplicity of infection (MOI = 1). Cells were harvested at 24 hours following vector transduction, and Gag expression was assessed by immunoblot analysis and p24 Gag ELISA of cell lysates. As shown in Figure 2 (panels A–D), the immunoblot analysis included an evaluation of the expression of an irrelevant cellular “housekeeping” protein (α-tubulin); this was used both as a loading control to confirm that approximately equivalent levels of cell lysate were analyzed and as a normalization control for densitometric analysis of the immunoblots (not shown). The p24 Gag ELISA data (panels E–F) were also normalized; in this case, normalization was achieved by performing the assay on a constant, fixed amount of cell lysate (as defined in terms of total protein content, measured in a Bradford assay).
When Gag expression levels were analyzed in the three established cell lines - 3T3 cells (fibroblasts), EMT6 cells (mammary epithelial cells) and XB2 cells (keratinocytes) - all of the constructs were found to elicit very similar levels of Gag expression (except for the MPSV-based vector) (Figure 2; panels B–D, F–H). In contrast, analysis of protein expression in primary bmDC revealed a greater divergence amongst the various vectors (Figure 2; panels A and E). In this cell type, the expression of Gag from the CMV-I and CMV-R amplicons appeared to be approximately 2-fold higher than expression from the amplicon that contained the HSV 4/5 promoter, which has been the promoter of choice for our previous studies [4]. Importantly, densitometric analysis of the immunoblots (panels A–D) yielded quantitative Gag expression values that were consistent with the results from the Gag ELISA (panels E–H; data not shown).
3.2 In vivo protein expression analysis
To investigate if our panel of amplicon constructs elicited different levels of protein expression in vivo, BALB/c mice were injected with 5 x 105 amplicon particles via the intradermal (i.d.) tailbase route; the tissue site of injection was marked and then recovered 24 hours later, using a skin punch. Lysates of these tissue specimens were subjected to Bradford assay to determine their protein content, and a fixed amount of the sample (normalized in terms of total protein) was then analyzed in a HIV-1 p24 Gag ELISA. Gag expression levels were found to be very similar for all of the constructs, except for the MPSV vector, which generated levels of Gag antigen that were substantially inferior to those produced by any of the other constructs (Figure 3).
Figure 3. Gag expression following injection of HSV-1 amplicon constructs into mice.
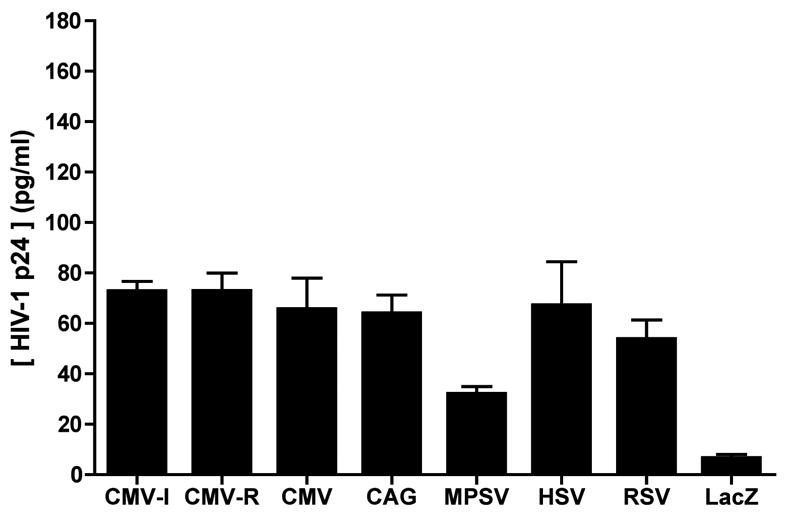
HSV-1 amplicon constructs encoding HIV-1 Gag from seven different promoters (Figure 1) were inoculated into the tail base of BALB/c mice (4 mice/group), at a dose of 5 x 105 transducing units. The injection site was marked, and 24 h later, the injection site was recovered with a tissue punch. A tissue lysate was prepared, and subjected to Bradford assay in order to determine the protein concentration. A fixed amount of lysate was then used in a HIV-1 p24 ELISA; the results are expressed as ng of HIV-1 p24 protein per ml of reaction volume, where 1 ml of the reaction contained 2.5 μg of tissue extract. The data shown represent mean values of triplicate measurements; bars denote the standard deviation of those values. The results shown are representative of two independent experiments that yielded very similar data.
An important consideration when using HIV-1 p55 Gag as a model antigen is the fact that, in the absence of any other viral proteins or viral RNA, the unprocessed Gag precursor has the ability to form virus-like particles when expressed in mammalian cells (reviewed in [37]). Virus-like particles of this kind might have the potential to copurify with amplicon particles. As a result, one might expect a small amount of preformed Gag protein to be present within our amplicon preparations. We therefore performed an experiment to examine both the amount of preformed Gag antigen present in our amplicon stocks, and the in vivo half-life of that preformed material at the tissue site of injection. To do this, a stock of CMV-I amplicon particles was UV-inactivated (to prevent new protein synthesis in vector-transduced cells) and this material was then inoculated into BALB/c mice (3/group) via the tail-base route. The site of injection was marked, and groups of animals were sacrificed at timed intervals following amplicon injection (5 min, as well as 3, 6 and 24 h); the tissue site of injection was then recovered with a skin punch. Lysates were prepared from the tissue specimens, and protein content determined by Bradford assay; a fixed amount of each lysate was then analyzed by p24 Gag ELISA. Results are shown in Figure 4. We assumed that Gag levels would decay with single-phase exponential kinetics over time, and tested whether such a decay curve could be fitted to the experimental data. As shown in Figure 4, a nonlinear curve for a single-phase exponential decay fit the data well, yielding a R2 (goodness of fit) value of 0.88. Based on this, we calculated the in vivo half-life of the preformed Gag antigen at the tissue site of injection to be 2.84 hours (with 95% confidence intervals ranging from 1.73 to 7.94 hours). It should be noted that this half-life measurement reflects both intrinsic protein stability/turnover and the effects of protein redistribution in vivo.
Our analysis of the tissue half-life of Gag yielded two important conclusions. First, it is clear that our amplicon stocks contained preformed Gag antigen, which might contribute to amplicon-induced immune responses. Second, this preformed Gag antigen decays rapidly in vivo and is likely to make only a minimal contribution to the measured Gag levels in biopsied tissue specimens at the 24 h time point (Figure 3). Specifically, preformed Gag can be expected to account for less than 1% of the total Gag levels measured at the 24 h time point (based on an in vivo half-life at the tissue site of injection of 3 h); even if the in vivo half-life were as much as 8 h (which corresponds to the far-end of the calculated 95% confidence interval), it would still contribute only about 10% of the total Gag levels present at 24 hours. This suggests that amplicon vectors provide a unique, biphasic mode of antigen delivery in vivo, with an initial bolus of preformed antigen that is available immediately upon vector delivery, followed by a subsequent wave of newly formed antigen that is generated following transduction of host cells and de novo protein synthesis.
3.3 In vivo immunogenicity
In light of the presence of preformed Gag in our amplicon stocks, we conducted an experiment to determine whether this might contribute to the immunogenicity of our gag-encoding amplicons. To do this, BALB/c mice were inoculated with untreated or UV-inactivated amplicon particles containing the (CMV-R) driven Gag insert, via the intradermal tail-base route, since this was shown to be an effective immunization route in our previous studies [4]. Immune responses were analyzed ten days later, by intracellular cytokine staining of splenocytes for IFN• (CD8+ T cells) following stimulation of cells using HIV-1 Gag peptide pools (Figure 5). A statistically significant reduction in T cell responses was detected in mice inoculated with UV-treated Gag-encoding amplicons (CMV-R, UV-treated), when compared to mice that were immunized with an untreated aliquot of the same amplicon stock (CMV-R; *, p < 0.01) (Figure 5). In fact, the data values for mice immunized with UV-treated Gag-encoding amplicons were not statistically different from those for mice immunized with a negative control amplicon (HSV-lacZ; p > 0.05). These results suggest that preformed Gag makes only a modest contribution to the amplicon-induced, Gag-specific, CD8+ T cell response in mice.
Figure 5. Preformed Gag makes only a modest contribution to the Gag-specific cellular immune response in mice.
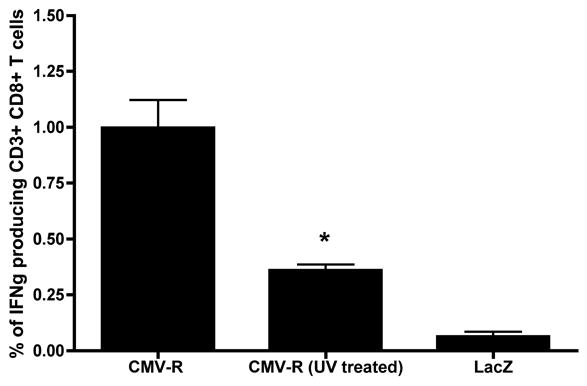
BALB/c mice (3/group) were inoculated in the tail base with 5 x 105 transducing units of untreated or UV-inactivated amplicon particles containing the (CMV-R) driven Gag insert (Figure 1). Animals were sacrificed 10 days later, and splenocytes were harvested. Gag-specific T cell responses were then assayed by intracellular cytokine staining for IFNγ (CD8+ T cells) following stimulation of cells using HIV-1 Gag peptide pools. The data represent mean values of triplicate measurements; bars denote the standard deviation of those values. Statistical analysis was performed by a nonparametric one-way ANOVA test (GraphPad Prism). A statistically significant reduction in the Gag-specific CD8+ T cell response was observed in mice inoculated with UV-treated Gag-encoding amplicons (CMV-R, UV-treated), versus mice immunized with untreated Gag-encoding amplicons (CMV-R) (*, p < 0.01). The data values for mice immunized with UV-treated Gag-encoding amplicons were not statistically different from those for mice immunized with a negative control amplicon (HSV-lacZ; p > 0.05).
We next analyzed the in vivo immunogenicity of our panel of amplicon constructs, by immunizing BALB/c mice via the tail-base route [4]. Mice were sacrificed at ten days post immunization, splenocytes harvested, and Gag-specific T cell responses were quantitated by performing tetramer analysis and intracellular cytokine staining. Tetramer staining was conducted using H-2Kd tetramers that were refolded with the immunodominant Gag-derived peptide, AMQMLKETI peptide [38], while intracellular cytokine staining was performed using cells that were briefly stimulated with HIV-1 Gag peptide pools, and then analyzed for IFNγ (CD8+ T cells) or IL-2 production (CD4+ T cells).
As shown in Figure 6, the CMV-I and CMV-R containing amplicons elicited the strongest in vivo immune responses, as assessed either by tetramer staining (Figure 6A) or analysis of Gag-specific cytokine production by CD8+ T cells (IFNγ) and CD4+ T cells (IL-2) (Figure 6B, 6C).
Figure 6. Gag-specific T cell responses following injection of HSV-1 amplicon constructs into mice.
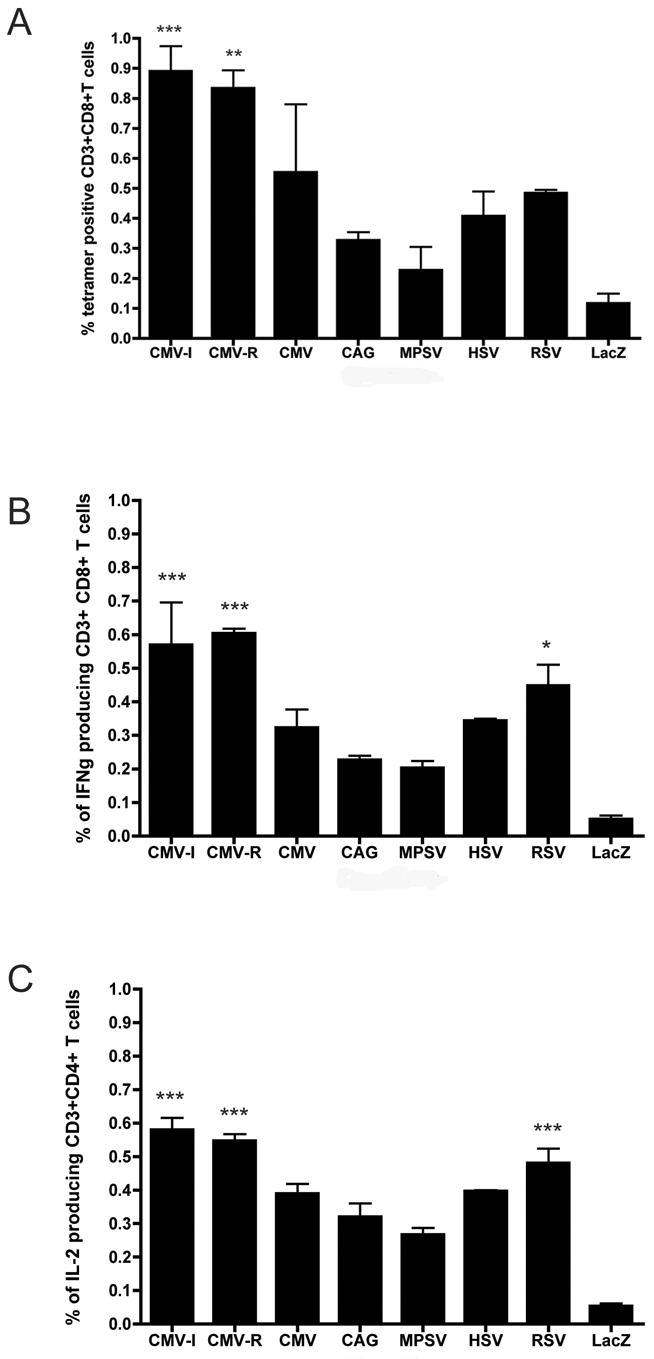
HSV-1 amplicon constructs encoding HIV-1 Gag from seven different promoters (Figure 1) were inoculated in the tail base of BALB/c mice (4 mice/group), at a dose of 5 x 105 transducing units. Animals were sacrificed 10 days later, and splenocytes were harvested. Gag-specific T cell responses were then assayed by (A) staining with a H-2Kd AMQMLKETI tetramer and by intracellular cytokine staining for (B) IFNγ (CD8+ T cells) and for (C) IL-2 (CD4+ T cells) following stimulation of cells using HIV-1 Gag peptide pools. The data represent mean values of triplicate measurements; bars denote the standard deviation of those values. Statistical analysis was performed by a nonparametric one-way ANOVA test (GraphPad Prism). Statistically significant T cell responses, compared to mice immunized with irrelevant amplicon particles (HSV-lacZ), are denoted by asterisks, as follows: *, p<0.05; **, p<0.01; ***, p<0.001. Results shown are representative of two independent experiments that yielded very similar data.
In addition to analyzing the magnitude of the Gag-specific T cell response following amplicon administration, we also performed statistical analyses to examine the relationship between vector-induced immune responses and the level of vector-mediated antigen expression either at the tissue site of injection in mice, or in various cultured cell lines that were representative of potential in vivo target cells (i.e., primary dendritic cells, as well as cell lines of fibroblast, epithelial and keratincyte lineage). A summary of this analysis is presented in Table 1. The data show that in vivo immune responses were significantly correlated with protein expression levels - whether they were measured in cultured cell lines, primary dendritic cells, or biopsied tissue collected from immunized animals (see correlation coefficients and associated P values in Table 1). Pairwise comparisons of vector-mediated Gag expression in cultured DC versus Gag-specific IFNγ production by CD8+ T cells or IL-2 production by CD4+ T cells are shown in Figure 7 (panels A and B, respectively). The graphs show the best-fit linear regression plot in each case; the R2 (goodness of fit) values for the plots shown are, respectively, 0.965 and 0.923 (Figure 7).
TABLE 1. Pairwise comparison of immune response and protein expression data: summary of statistical data analysis.
The magnitude of the Gag-specific T cell response in vivo, as measured by tetramer staining or intracellular cytokine staining (ICS; data from Figure 6), was compared in a pairwise fashion with normalized data values for amplicon-directed Gag expression in cultured cells and tissue samples (data from Figure 2). The cells listed correspond to primary murine bone marrow-derived dendritic cells (DC) and to murine cell lines of fibroblast (3T3), keratinocyte (XB2) and epithelial (EMT6) lineage. A correlation coefficient (Pearson r value) was generated for each pairwise data comparison (using GraphPad PRISM), and is shown above. The corresponding P value is also shown (based on a two-tailed test). The data show that the magnitude of the in vivo T cell response was significantly correlated with the level of Gag antigen production both in vitro (in cultured cell lines and primary DC) and in vivo (in biopsied tissue samples).
| Immune response parameter | Protein (Gag) production parameter | Correlation coefficient (Pearson r) | P value (two-tailed) |
|---|---|---|---|
| CD8+ Tetramer+ cells | DC (in vitro) | 0.932 | 0.007 |
| 3T3 (in vitro) | 0.763 | 0.028 | |
| XB2 (in vitro) | 0.842 | 0.009 | |
| EMT6 (in vitro) | 0.882 | 0.004 | |
| Tissue site (in vivo) | 0.791 | 0.019 | |
| CD8+ IFNγ+ cells (ICS) | DC (in vitro) | 0.982 | <0.0001 |
| 3T3 (in vitro) | 0.810 | 0.015 | |
| XB2 (in vitro) | 0.797 | 0.018 | |
| EMT6 (in vitro) | 0.860 | 0.006 | |
| Tissue site (in vivo) | 0.797 | 0.018 | |
| CD4+ IL2+ cells (ICS) | DC (in vitro) | 0.961 | 0.0001 |
| 3T3 (in vitro) | 0.880 | 0.004 | |
| XB2 (in vitro) | 0.861 | 0.006 | |
| EMT6 (in vitro) | 0.911 | 0.002 | |
| Tissue site (in vivo) | 0.884 | 0.004 |
Figure 7. Linear regression analysis of the relationship between Gag expression in cultured dendritic cells and the magnitude of the T cell response.
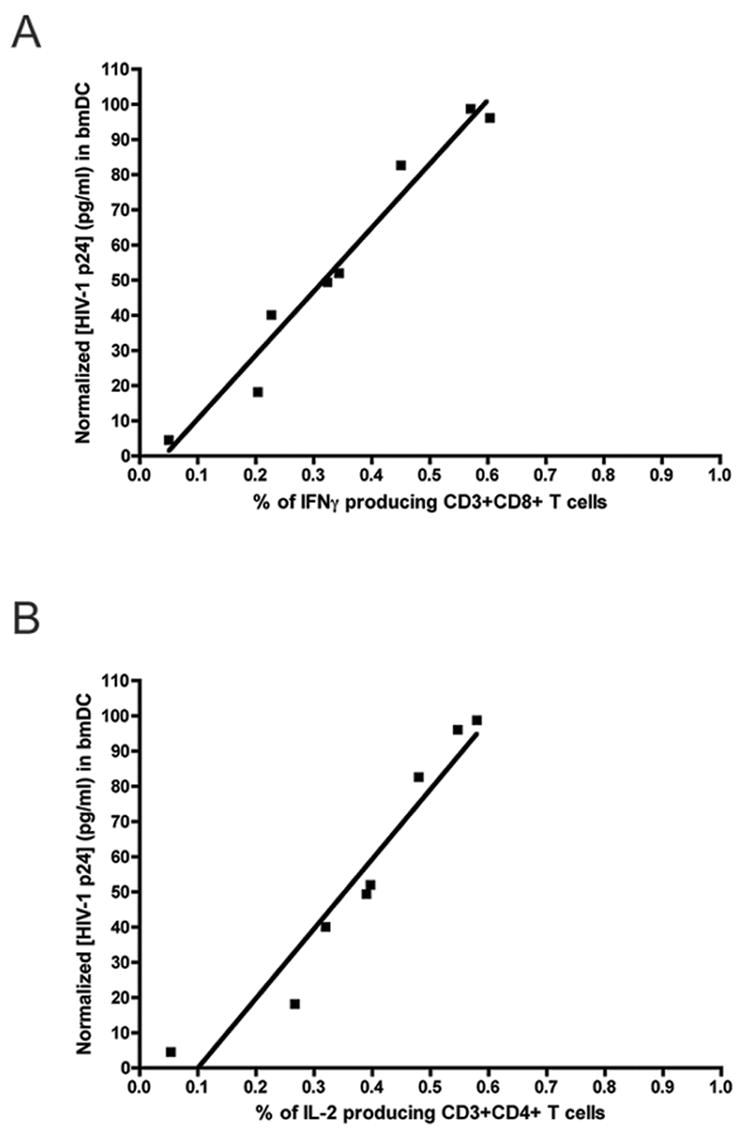
in vivo. Normalized data values for amplicon-directed Gag expression in cultured murine bone marrow dendritic cells (bmDC; data from Figure 2E) are plotted against the magnitude of the Gag-specific T cell response in vivo, as measured by intracellular cytokine staining for IFN-γ in CD8+ cells (data from Figure 6B) or IL-2 in CD4+ cells (data from Figure 6C). A correlation analysis was performed initially, which yielded a correlation coefficient (Pearson r value) of 0.982 (panel A; p <0.0001) or 0.961 (panel B; p=0.0001). The data were then fitted to a linear regression model, which yielded the plots shown; the R2 (goodness of fit) values for these plots were 0.965 (panel A) and 0.923 (panel B).
4. DISCUSSION
The experiments described here were conducted as part of an ongoing effort to optimize the HSV-1 amplicon vector platform for HIV-1 vaccine delivery, and to identify transcriptional control elements that might result in improved levels of antigen-specific T cell immune responses. A related objective was to carefully characterize protein expression both in vivo and in a panel of cultured cell lines and primary cells, and to determine whether protein expression data might correlate with or predict the relative strength of cellular immune responses in vivo. With these goals in mind, we selected HIV-1 Gag as a model antigen for our experiments, since this made it possible to directly quantitate antigen expression levels in vitro and in vivo, and to compare expression data to immunogenicity results.
Quantitative analysis of Gag expression in amplicon-transduced animals revealed an important and previously unappreciated aspect of the HSV-1 amplicon system. First, we detected substantial levels of performed Gag antigen in our amplicon stocks, and second, we showed that this preformed Gag antigen decayed very rapidly in vivo. Thus, HSV-1 amplicon vectors deliver a bolus of preformed Gag antigen that is immediately available for uptake and presentation at the time of vector delivery. This initial bolus of antigen decays within a matter of hours, and is followed by a second wave of newly synthesized Gag antigen, produced by vector-transduced target cells.
The unexpected biphasic nature of amplicon-vectored Gag antigen delivery may reflect, at least in part, the unusual nature of the Gag antigen and its propensity to form virus-like particles (VLPs) that may copurify with HSV-1 amplicon particles. It also suggests some novel approaches to enhance or modify the immunogenicity of amplicon vector (for example, by tethering antigen to HSV-1 tegument proteins, in order to drive the incorporation of preformed antigen into amplicon particles).
While HIV-1 Gag has a well characterized ability to form VLPs in human cells, this may not be the case in cells from other species (such as the baby hamster kidney cells used to generate our amplicon stocks). Indeed, it has been reported that the assembly of HIV-1 Gag, and the generation of Gag VLP, is blocked in murine cells [39, 40]. However, Nabel and colleagues (who provided the Gag construct used in our experiments) have shown that a plasmid which encoded both HIV-1 Gag and Env efficiently generated VLP in mouse cells [41]. These workers attributed this result to their to their use of a codon-optimized Gag construct and a strong transcriptional promoter [41], in contrast to the constructs used by Mariani et al. [39].
In the present case, while we cannot unequivocally assert that our amplicon stocks contain Gag VLPs (since we did not perform electron microscopy studies on our amplicon preparations), we think that this is very likely – in large part because of the high levels of Gag that copurify with our amplicon particles. Amplicons that encode a different exogenous gene product (luciferase), which lacks the ability to form VLP, contain much lower amounts of preformed protein (data not shown).
While preformed Gag antigen may potentially contribute to amplicon-induced immune responses in vivo, it is not likely to be a major contributor to the steady-state levels of Gag that were measured in our biopsied tissue specimens. This is because the estimated half-life of preformed Gag at the tissue site of injection was only on the order of 3 h (Figure 4). As a result, residual preformed protein would represent only a very small fraction of the Gag antigen present at the site of vector inoculation at 24 h following amplicon transduction. Analysis of steady-state Gag levels at the 24 h time point therefore allowed us to directly compare the efficiency of new Gag protein production by the various amplicon constructs (note that Gag levels peaked at 24 h following amplicon transduction, both in vitro and in vivo, and then declined rapidly; data not shown).
Our experiments were focused principally on the comparative analysis of different promoter elements in the context of the HSV-1 amplicon vector system, since the optimization of transcriptional control sequences has been shown to substantially improve the immunogenicity of both DNA plasmid and adenovirally-vectored vaccines [20, 23, 32, 35, 36, 42–45]. In HIV-1 DNA vaccines, the use of the hybrid CMV-I transcriptional control element resulted in improved immunogenicity, compared to vectors that contained a simple CMV promoter without Intron A [23, 42, 43]. A similar effect was seen in protein expression studies using adenovirus vectors that contained a luciferase reporter gene under the transcriptional control of either the basic CMV promoter or a CMV promoter that had been coupled to the Intron A element [34]. In this case, addition of the Intron A element resulted in 2–20 fold higher levels of luciferase expression in HeLa, HepG2, ECV304, SKHep-1 and LN319 cells in vitro, and 10 fold higher expression in vivo [34].
More recent studies have shown that the hybrid CMV-R element can elicit even greater levels of antigen expression and immunogenicity, when compared directly to the CMV-I promoter element [20]. Barouch et al. reported 5–10 fold higher expression of a multivalent HIV-1 DNA vaccine that contained the CMV-R promoter versus the CMV-I promoter in 3T3 cells in vitro [20], and these authors further showed that the CMV-R containing plasmids elicited markedly higher levels of antigen-specific T cells in both mice and cynomolgus monkeys [20]. In the present study, amplicon constructs containing the CMV-I and CMV-R constructs elicited the strongest cellular immune responses in vivo, but the two promoters performed with essentially indistinguishable activity in all of our protein expression and immunogenicity assays. The lack of any obvious difference in the performance of the two promoter elements presumably reflects the fact that our experiments were conducted using helper-free HSV-1 amplicon particles, which deliver a DNA payload encapsidated in a herpesvirus coat, while previous comparisons of the two promoters were conducted using naked DNA plasmids [20]. It is possible that immunostimulatory and/or transcriptional activation events associated with cell binding and/or entry of HSV-1 particles may be responsible for these differing results. Also, the concatemeric nature of amplicon genomes could lead to intra-copy interactions and/or there could be differences in DNA transcription in vivo.
Comparative analysis of the in vivo immunogenicity and protein expression profiles for the various amplicon constructs revealed that the strength of the vector-induced immune response was correlated significantly with the level of (Gag) antigen production, both in vitro (in cultured cell lines and primary dendritic cells), and in vivo (in biopsied tissue specimens). One interpretation of these data is that the strength of the amplicon-induced immune response may reflect both direct and indirect priming of antigen-specific T cells.
The strong correlation between Gag production in cultured DC, and the magnitude of the Gag-specific in vivo immune response, suggests that direct amplicon-mediated transduction of DC in vivo might make an important contribution to the overall strength of the amplicon-induced immune response. In addition, cross-presentation is also likely to also contribute to amplicon-induced immune responses. This is suggested by the fact that the strength of the amplicon-induced immune responses was significantly correlated with the total level of protein production at the tissue site of amplicon delivery as well as with the level of protein production in cultured cell lines of epithelial (EMT6), fibroblast (3T3) and keratinocyte (XB2) lineages (all cell types that would represent potential targets for intradermally delivered amplicon particles).
One additional aspect of the correlation analysis that bears note is the fact that the magnitude of both the CD8+ and the CD4+ T cell response was correlated strongly with levels of Gag expression. Naive CD4+ cells reside only in the T cell areas of secondary lymphoid tissues such as lymph nodes and spleen, and must be stimulated by peptide:MHC II complexes on dendritic cells [46–48]. Since the class II antigen presentation pathway relies upon exogenously acquired antigen, this observation suggests that amplicon-transduced cells must migrate from the local site of amplicon injection and then either (i) secrete soluble Gag antigen within lymph nodes and spleen, or (ii) undergo apoptosis and become engulfed by local DC.
Overall, the findings reported here suggest that antigen expression may represent a valuable correlate of the strength of amplicon-induced immune responses in vivo, and that it may be possible to use in vitro studies to streamline the future optimization of amplicon-based vaccines. In this light, the unexpectedly strong correlation between Gag expression in cultured DC, and the strength of the amplicon-induced immune response, may merit further investigation. Finally, the present study provides important insight into the biological properties of helper-free HSV-1 amplicon-vectored HIV-1 vaccines, and provides a rational basis on which to design future improvements of this potent vector platform.
Acknowledgments
We would like to thank: Dr. Paul Johnson for helpful suggestions, Dr. Mike McDermott for statistical advice, the NIH Tetramer Facility (Emory University, Atlanta, GA) for the Gag-specific tetramer used in this study, Dr. Edith Lord (University of Rochester) for EMT6 cells, and Drs. Gary Nabel (NIH VRC), Paul Johnson (Harvard Medical School) and Joshi Jacob (Emory Vaccine Center) for gifts of plasmids/promoter elements; we also thank Ann Casey, Clark Burris and Louis Lotta for helper virus-free amplicon packaging. The monoclonal antibody to HIV-1 p24 (AG3.0) was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH from Dr. Jonathan Allan. This work was supported by NIH grants F31 AI054330 (to K.S.), and P01 AI056356 (to M.C.K. and other personnel).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Spaete RR, Frenkel N. The herpes simplex virus amplicon: a new eucaryotic defective-virus cloning-amplifying vector. Cell. 1982;30(1):295–304. doi: 10.1016/0092-8674(82)90035-6. [DOI] [PubMed] [Google Scholar]
- 2.Geller AI, Breakefield XO. A defective HSV-1 vector expresses Escherichia coli beta-galactosidase in cultured peripheral neurons. Science. 1988;241(4873):1667–9. doi: 10.1126/science.241.4873.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willis RA, Bowers WJ, Turner MJ, Fisher TL, Abdul-Alim CS, Howard DF, et al. Dendritic cells transduced with HSV-1 amplicons expressing prostate-specific antigen generate antitumor immunity in mice. Hum Gene Ther. 2001;12(15):1867–79. doi: 10.1089/104303401753153929. [DOI] [PubMed] [Google Scholar]
- 4.Hocknell PK, Wiley RD, Wang X, Evans TG, Bowers WJ, Hanke T, et al. Expression of human immunodeficiency virus type 1 gp120 from herpes simplex virus type 1-derived amplicons results in potent, specific, and durable cellular and humoral immune responses. J Virol. 2002;76(11):5565–80. doi: 10.1128/JVI.76.11.5565-5580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Wiley RD, Evans TG, Bowers WJ, Federoff HJ, Dewhurst S. Cellular immune responses to helper-free HSV-1 amplicon particles encoding HIV-1 gp120 are enhanced by DNA priming. Vaccine. 2003;21(19–20):2288–97. doi: 10.1016/s0264-410x(03)00099-9. [DOI] [PubMed] [Google Scholar]
- 6.Bowers WJ, Mastrangelo MA, Stanley HA, Casey AE, Milo LJ, Jr, Federoff HJ. HSV amplicon-mediated Abeta vaccination in Tg2576 mice: differential antigen-specific immune responses. Neurobiol Aging. 2005;26(4):393–407. doi: 10.1016/j.neurobiolaging.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Gorantla S, Santos K, Meyer V, Dewhurst S, Bowers WJ, Federoff HJ, et al. Human dendritic cells transduced with herpes simplex virus amplicons encoding human immunodeficiency virus type 1 (HIV-1) gp120 elicit adaptive immune responses from human cells engrafted into NOD/SCID mice and confer partial protection against HIV-1 challenge. J Virol. 2005;79(4):2124–32. doi: 10.1128/JVI.79.4.2124-2132.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zibert A, Thomassen A, Muller L, Nguyen L, Glouchkova L, Fraefel C, et al. Herpes simplex virus type-1 amplicon vectors for vaccine generation in acute lymphoblastic leukemia. Gene Ther. 2005;12(23):1707–17. doi: 10.1038/sj.gt.3302577. [DOI] [PubMed] [Google Scholar]
- 9.Herrlinger U, Jacobs A, Quinones A, Woiciechowsky C, Sena-Esteves M, Rainov NG, et al. Helper virus-free herpes simplex virus type 1 amplicon vectors for granulocyte-macrophage colony-stimulating factor-enhanced vaccination therapy for experimental glioma. Hum Gene Ther. 2000;11(10):1429–38. doi: 10.1089/10430340050057503. [DOI] [PubMed] [Google Scholar]
- 10.Tolba KA, Bowers WJ, Hilchey SP, Halterman MW, Howard DF, Giuliano RE, et al. Development of herpes simplex virus-1 amplicon-based immunotherapy for chronic lymphocytic leukemia. Blood. 2001;98(2):287–95. doi: 10.1182/blood.v98.2.287. [DOI] [PubMed] [Google Scholar]
- 11.Olschowka JA, Bowers WJ, Hurley SD, Mastrangelo MA, Federoff HJ. Helper-free HSV-1 amplicons elicit a markedly less robust innate immune response in the CNS. Mol Ther. 2003;7(2):218–27. doi: 10.1016/s1525-0016(02)00036-9. [DOI] [PubMed] [Google Scholar]
- 12.Tyler CM, Wuertzer CA, Bowers WJ, Federoff HJ. HSV Amplicons: Neuro Applications. Curr Gene Ther. 2006;6(3):337–50. doi: 10.2174/156652306777592045. [DOI] [PubMed] [Google Scholar]
- 13.Epstein AL. HSV-1-based amplicon vectors: design and applications. Gene Ther. 2005;12 :S154–8. doi: 10.1038/sj.gt.3302617. Suppl 1. [DOI] [PubMed] [Google Scholar]
- 14.Liu M, Tang J, Wang X, Yang T, Geller AI. Enhanced long-term expression from helper virus-free HSV-1 vectors packaged in the presence of deletions in genes that modulate the function of VP16, U L 46 and U L 47. J Neurosci Methods. 2005;145(1–2):1–9. doi: 10.1016/j.jneumeth.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 15.Breakefield XO, Geller AI. Gene transfer into the nervous system. Mol Neurobiol. 1987;1(4):339–71. doi: 10.1007/BF02935741. [DOI] [PubMed] [Google Scholar]
- 16.Hagmann M, Georgiev O, Schaffner W. The VP16 paradox: herpes simplex virus VP16 contains a long-range activation domain but within the natural multiprotein complex activates only from promoter-proximal positions. J Virol. 1997;71(8):5952–62. doi: 10.1128/jvi.71.8.5952-5962.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrera FJ, Triezenberg SJ. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004;78(18):9689–96. doi: 10.1128/JVI.78.18.9689-9696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnosti DN, Preston CM, Hagmann M, Schaffner W, Hope RG, Laughlan G, et al. Specific transcriptional activation in vitro by the herpes simplex virus protein VP16. Nucleic Acids Res. 1993;21(24):5570–6. doi: 10.1093/nar/21.24.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garg S, Oran AE, Hon H, Jacob J. The hybrid cytomegalovirus enhancer/chicken beta-actin promoter along with woodchuck hepatitis virus posttranscriptional regulatory element enhances the protective efficacy of DNA vaccines. J Immunol. 2004;173(1):550–8. doi: 10.4049/jimmunol.173.1.550. [DOI] [PubMed] [Google Scholar]
- 20.Barouch DH, Yang ZY, Kong WP, Korioth-Schmitz B, Sumida SM, Truitt DM, et al. A human T-cell leukemia virus type 1 regulatory element enhances the immunogenicity of human immunodeficiency virus type 1 DNA vaccines in mice and nonhuman primates. J Virol. 2005;79(14):8828–34. doi: 10.1128/JVI.79.14.8828-8834.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapman BS, Thayer RM, Vincent KA, Haigwood NL. Effect of intron A from human cytomegalovirus (Towne) immediate-early gene on heterologous expression in mammalian cells. Nucleic Acids Res. 1991;19(14):3979–86. doi: 10.1093/nar/19.14.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norman JA, Hobart P, Manthorpe M, Felgner P, Wheeler C. Development of improved vectors for DNA-based immunization and other gene therapy applications. Vaccine. 1997;15(8):801–3. doi: 10.1016/s0264-410x(96)00247-2. [DOI] [PubMed] [Google Scholar]
- 23.Chakrabarti BK, Ling X, Yang ZY, Montefiori DC, Panet A, Kong WP, et al. Expanded breadth of virus neutralization after immunization with a multiclade envelope HIV vaccine candidate. Vaccine. 2005;23(26):3434–45. doi: 10.1016/j.vaccine.2005.01.099. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Kong WP, Nabel GJ. Human immunodeficiency virus type 1-specific immunity after genetic immunization is enhanced by modification of Gag and Pol expression. J Virol. 2001;75(10):4947–51. doi: 10.1128/JVI.75.10.4947-4951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chinnasamy D, Chinnasamy N, Enriquez MJ, Otsu M, Morgan RA, Candotti F. Lentiviral-mediated gene transfer into human lymphocytes: role of HIV-1 accessory proteins. Blood. 2000;96(4):1309–16. [PubMed] [Google Scholar]
- 26.Halene S, Wang L, Cooper RM, Bockstoce DC, Robbins PB, Kohn DB. Improved expression in hematopoietic and lymphoid cells in mice after transplantation of bone marrow transduced with a modified retroviral vector. Blood. 1999;94(10):3349–57. [PMC free article] [PubMed] [Google Scholar]
- 27.Bowers WJ, Howard DF, Brooks AI, Halterman MW, Federoff HJ. Expression of vhs and VP16 during HSV-1 helper virus-free amplicon packaging enhances titers. Gene Ther. 2001;8(2):111–20. doi: 10.1038/sj.gt.3301340. [DOI] [PubMed] [Google Scholar]
- 28.Stavropoulos TA, Strathdee CA. An enhanced packaging system for helper-dependent herpes simplex virus vectors. J Virol. 1998;72(9):7137–43. doi: 10.1128/jvi.72.9.7137-7143.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowers WJ, Howard DF, Federoff HJ. Discordance between expression and genome transfer titering of HSV amplicon vectors: recommendation for standardized enumeration. Mol Ther. 2000;1(3):294–9. doi: 10.1006/mthe.2000.0039. [DOI] [PubMed] [Google Scholar]
- 30.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223(1):77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 31.Simm M, Shahabuddin M, Chao W, Allan JS, Volsky DJ. Aberrant Gag protein composition of a human immunodeficiency virus type 1 vif mutant produced in primary lymphocytes. J Virol. 1995;69(7):4582–6. doi: 10.1128/jvi.69.7.4582-4586.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia W, Bringmann P, McClary J, Jones PP, Manzana W, Zhu Y, et al. High levels of protein expression using different mammalian CMV promoters in several cell lines. Protein Expr Purif. 2006;45(1):115–24. doi: 10.1016/j.pep.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 33.Xu ZL, Mizuguchi H, Ishii-Watabe A, Uchida E, Mayumi T, Hayakawa T. Optimization of transcriptional regulatory elements for constructing plasmid vectors. Gene. 2001;272(1–2):149–56. doi: 10.1016/s0378-1119(01)00550-9. [DOI] [PubMed] [Google Scholar]
- 34.Xu ZL, Mizuguchi H, Ishii-Watabe A, Uchida E, Mayumi T, Hayakawa T. Strength evaluation of transcriptional regulatory elements for transgene expression by adenovirus vector. J Control Release. 2002;81(1–2):155–63. doi: 10.1016/s0168-3659(02)00059-7. [DOI] [PubMed] [Google Scholar]
- 35.Wang S, Farfan-Arribas DJ, Shen S, Chou TH, Hirsch A, He F, et al. Relative contributions of codon usage, promoter efficiency and leader sequence to the antigen expression and immunogenicity of HIV-1 Env DNA vaccine. Vaccine. 2006;24(21):4531–40. doi: 10.1016/j.vaccine.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 36.Egan MA, Megati S, Roopchand V, Garcia-Hand D, Luckay A, Chong SY, et al. Rational design of a plasmid DNA vaccine capable of eliciting cell-mediated immune responses to multiple HIV antigens in mice. Vaccine. 2006;24(21):4510–23. doi: 10.1016/j.vaccine.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 37.Deml L, Speth C, Dierich MP, Wolf H, Wagner R. Recombinant HIV-1 Pr55gag virus-like particles: potent stimulators of innate and acquired immune responses. Mol Immunol. 2005;42(2):259–77. doi: 10.1016/j.molimm.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 38.Mata M, Travers PJ, Liu Q, Frankel FR, Paterson Y. The MHC class I-restricted immune response to HIV-gag in BALB/c mice selects a single epitope that does not have a predictable MHC-binding motif and binds to Kd through interactions between a glutamine at P3 and pocket D. J Immunol. 1998;161(6):2985–93. [PubMed] [Google Scholar]
- 39.Mariani R, Rutter G, Harris ME, Hope TJ, Krausslich HG, Landau NR. A block to human immunodeficiency virus type 1 assembly in murine cells. J Virol. 2000;74(8):3859–70. doi: 10.1128/jvi.74.8.3859-3870.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong SB, Siliciano RF. Contribution of virus-like particles to the immunogenicity of human immunodeficiency virus type 1 Gag-derived vaccines in mice. J Virol. 2005;79(3):1701–12. doi: 10.1128/JVI.79.3.1701-1712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akahata W, Yang ZY, Nabel GJ. Comparative immunogenicity of human immunodeficiency virus particles and corresponding polypeptides in a DNA vaccine. J Virol. 2005;79(1):626–31. doi: 10.1128/JVI.79.1.626-631.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chakrabarti BK, Kong WP, Wu BY, Yang ZY, Friborg J, Ling X, et al. Modifications of the human immunodeficiency virus envelope glycoprotein enhance immunogenicity for genetic immunization. J Virol. 2002;76(11):5357–68. doi: 10.1128/JVI.76.11.5357-5368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mascola JR, Sambor A, Beaudry K, Santra S, Welcher B, Louder MK, et al. Neutralizing antibodies elicited by immunization of monkeys with DNA plasmids and recombinant adenoviral vectors expressing human immunodeficiency virus type 1 proteins. J Virol. 2005;79(2):771–9. doi: 10.1128/JVI.79.2.771-779.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith RL, Traul DL, Schaack J, Clayton GH, Staley KJ, Wilcox CL. Characterization of promoter function and cell-type-specific expression from viral vectors in the nervous system. J Virol. 2000;74(23):11254–61. doi: 10.1128/jvi.74.23.11254-11261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wright A, Semyonov A, Dawes G, Crameri A, Lyons R, Stemmer WP, et al. Diverse plasmid DNA vectors by directed molecular evolution of cytomegalovirus promoters. Hum Gene Ther. 2005;16(7):881–92. doi: 10.1089/hum.2005.16.881. [DOI] [PubMed] [Google Scholar]
- 46.Itano AA, Jenkins MK. Antigen presentation to naive CD4 T cells in the lymph node. Nat Immunol. 2003;4(8):733–9. doi: 10.1038/ni957. [DOI] [PubMed] [Google Scholar]
- 47.Jenkins MK, Khoruts A, Ingulli E, Mueller DL, McSorley SJ, Reinhardt RL, et al. In vivo activation of antigen-specific CD4 T cells. Annu Rev Immunol. 2001;19:23–45. doi: 10.1146/annurev.immunol.19.1.23. [DOI] [PubMed] [Google Scholar]
- 48.Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 2001;410(6824):101–5. doi: 10.1038/35065111. [DOI] [PubMed] [Google Scholar]