

Summary
Site-3 toxins are small polypeptide venoms from scorpions, sea anemones, and spiders that bind with a high specificity to the extracellular surface of voltage-gated Na channels. After binding to a site near the S4 segment in domain IV the toxin causes disruption of the normal fast inactivation transition resulting in a marked prolongation of the action potentials of excitable tissues including those of cardiac and skeletal muscle and nerve. In this review we discuss the specific binding interactions between residues of the toxin and those of the Na channel, and the specific modification of Na channel kinetic behavior leading to a change in fast inactivation focusing on interactions deduced primarily from the study of sea anemone toxins and the cardiac Na channel (NaV1.5). We also illustrate the usefulness of site-3 toxins in the study of altered Na channel behavior by drug-modification.
Introduction
An extremely interesting set of toxins, named site-3 toxins (Catterall, 1980), bind to the extracellular surface of voltage-gated Na channels and produce profound slowing of current decay. Site-3 toxins have been particularly useful in helping understand the structural basis of Na channel gating, and we will review them with particular attention to their effects on the cardiac isoform (NaV1.5). Members include the 60–70 residue α-scorpion toxins (from Leiurus quinquestriatus, Centruroides sculpturatus, Tityus serulatus, and Androctonus australis and others for which a database is available (Srinivasan et al., 2002)) as well as several peptide toxins isolated from sea anenome, including Anthopleurin A (Ap-A) and Anthopleurin B (Ap-B), which were isolated from Anemonia xanthogrammica (49 aa), and the Anemonia sulcata toxins (ATXI [46aa], ATXII [47aa], and ATXIII [27aa]) (Norton, 1991) as well as a several toxins isolated more recently from spiders, including several from funnel spiders, e.g. Hadronyche versuta (Little et al., 1998a; Little et al., 1998b) and Hadronyche versuta, (Fletcher et al., 1997), as well as from Plectreurys tristis (Corzo et al., 2003) and Phyllodiscus semoni (Xiao et al., 2005) (for a review see Nicholson & Graudins, 2002). In addition to an excellent summary published in 1991 (Norton, 1991), two recent reviews are also noteworthy (Blumenthal & Seibert, 2003; Honma & Shiomi, 2006). Also, a recent brief review gives additional details of the various identified spider toxins (Nicholson & Graudins, 2002).
Early Investigations
The anemone toxins were first identified in 1976: (1) by noting skeletal muscle cramps and paralysis after injection of venom in crabs (Beress et al., 1976); (2) after superfusion onto isolated cardiac muscle strips with resultant increase in inotrophy associated with action potential prolongation (Shibata et al., 1976); (3) by noting extracellular but not intracellular application of toxin caused inhibition of INa decay in frog node of Ranvier (Bergman et al., 1976b). Initially, the idea that Na channels were represented by multiple isoforms was not appreciated, although it was noted that several of the site-3 toxins exhibited specificity for cardiac preparations. These included ATXII from A. sulcata (Beress & Beress, 1975) and Ap-A, isolated from A. xanthogrammica (Norton et al., 1976). In addition, several others have more recently been identified, including Jinghaotoxin I (Xiao et al., 2005) while others have been described that specifically target insect/crustacean channels (Salgado & Kem, 1992).
Early data not only reported very different apparent affinities between preparations but even within a single preparation, which depended on the technique used for the assay. For example, ED50’s for ATXII based on binding/flux in rat brain synaptosomes were estimated to be 150–240 nM, (Vincent et al., 1980) while voltage clamp affinities in myelinated nerve were estimated to be 30 to 100 fold lower, i.e. ED50’s at 5 μM(Schmidtmayer et al., 1982) and 20 μM.(Bergman et al., 1976a). It was then recognized that the action of the toxin was specific for Na channels, and that these toxins have a low affinity for the inactivated state of the channel because of voltage-dependent binding (Wang & Strichartz, 1985; Strichartz & Wang, 1986; Warashina et al., 1988). In binding and flux studies site-3 toxins were used in combination with toxins that promoted activation (usually veratridine) where binding and/or flux was augmented (Lawrence & Catterall, 1981; Frelin et al., 1984). Such data were interpreted to indicate a synergistic effect of the two agents (Corbett & Krueger, 1989), or more often a high affinity of the open state (or an open-like state) for toxin, a conclusion also suggested by early patch clamp studies (Schreibmayer et al., 1987b), but questioned by others in which other promoters of the Na channel open state were unable to mimic the effect of scorpion toxin (Rando et al., 1986).
Determinants of the Specific Interactions between Site-3 Toxins and Na Channels
Here we focus on the major toxins from A. xanthogrammica and their kinetic actions principally on cardiac Na channels. Extensive structure-function data are available for Ap-B, one toxin from the venom of A. xanthogrammica, which when assayed electrophysiologically exhibits a high affinity for multiple Na channel isoforms with a 50 fold preference for rat cardiac channels over neuronal isoforms (0.1 nM vs. 5 nM), although when assayed by flux the affinities were similar and appeared to be high affinity (9 nM vs. 22 nM) (Gallagher & Blumenthal, 1992; Khera et al., 1995). Ap-A, the major toxin from A. xanthogrammica, in which only seven of the 49 amino acids are different from Ap-B, was the first noted to have cardiac effects and to exhibit a 50-fold preference for cardiac over neuronal channels (2.5 nM vs. 120 nM). Ap-A toxin also exhibited isoform preference by flux assay of approximately 30-fold (14 nM to 400 nM) (Khera et al., 1995). Other studies also reported data to support the idea that these toxins display a 20 to 40-fold greater affinity for cardiac over neuronal channels (Vincent et al., 1980; Lawrence & Catterall, 1981; Nagy, 1988; Gallagher & Blumenthal, 1994). It should be noted that Ap-B is generally described as a high affinity variant, but it can also discriminate between isoforms. Cationic residues on these anemone toxins have been identified as underlying the affinity differences (Khera et al., 1995), with Arg12 being the most important, although in combination with Lys-49 it accounts for most of the cardiac specificity of Ap-A over Ap-B for cardiac channels. Although these toxins have a quite high affinity, i.e.ED50’s ~1 nM, their on-rates are extremely slow. Ap-B toxin, for example, which has an ED50of 0.1 nM estimated by electrophysiology for rat NaV1.5, has an on-rate of only 1.4 x 106 s−1M−1, 2–3 orders of magnitude less than predicted for diffusion of small molecules (Khera et al., 1995). Very slow rates of modification and unmodification require that affinities be measured after exposing preparations to toxin for long periods of time or be estimated from kinetic analyses of washin and washout experiments at toxin concentrations many times the EC50 in order to promote channel modification over a time frame achievable in a typical voltage clamp experiment. This method is illustrated in Figure 1.
Figure 1.
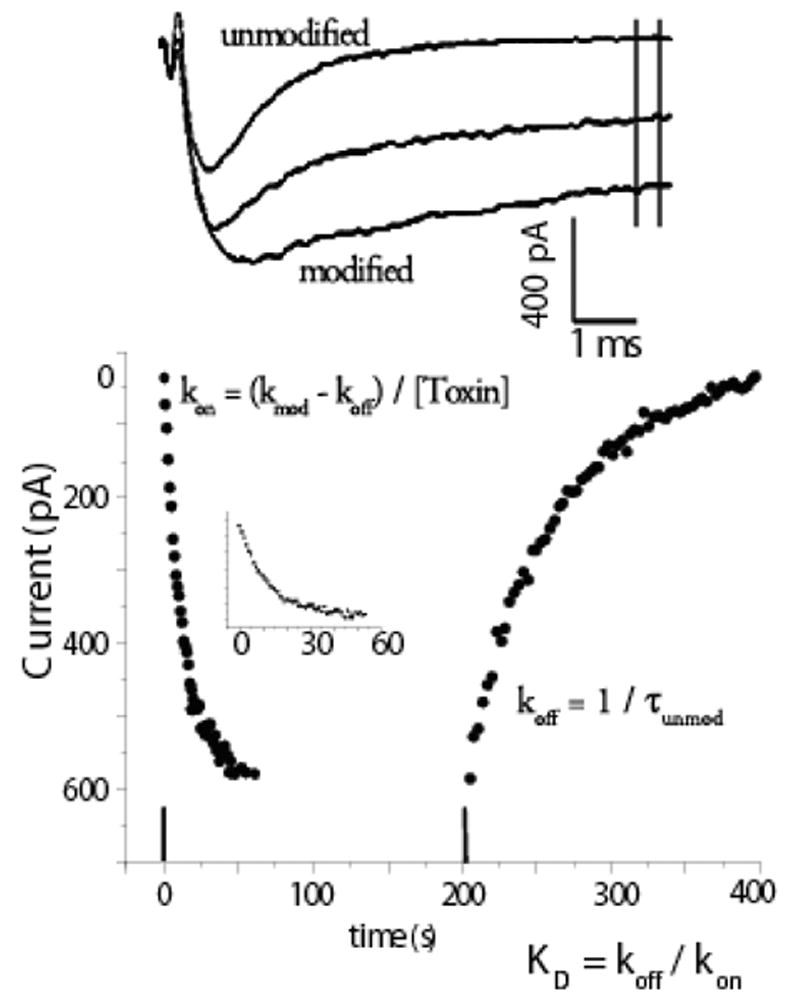
Calculation of affinity from kinetics. The time course of modification and unmodification of INa by site 3 toxins occurs so slowly that rate constants can be calculated from currents measured from multiple depolarizations. The upper panel shows superimposed example time course of INa recorded in a RT4B cell, which expresses cardiac INa (Zeng et al., 1996) at 0, 7.5, and 25 minutes after 40 nM of an Ap-B mutant toxin (R12S-K49Q) was removed from the bath. Currents were in response to trains of step depolarizations to −10 mV. The mean of the currents between the vertical lines was calculated and these currents are plotted in the panel below. Inset shows washin data with x-axis expanded. Data were fit with single exponentials, Ae−t/τ + b or A(1- e−t/τ) + b as appropriate, and rate constants were calculated as indicated in figure. Modified from Khera et al., 1995 with permission.
Even though site-3 toxins from different species are not similar in their sequence, they share, along with other peptide toxins such as charydotoxin and μ-conotoxin, multiple disulfide bonds (either 3 or 4) that form a tight core. Structural information is available for several of the site-3 toxins, including a 1.2 Å crystal structure for the α-scorpion toxin from Androctonus australis (Housset et al., 1994). NMR coordinates for Ap-A and Ap-B, although determined and reported earlier, were released in 1996 (Torda & Norton, 1989; Scanlon & Norton, 1994). For more information see Smith and Blumenthal in this issue.
Almost certainly loop flexibility near Arg-12 contributes to the very low on-rate. Substitution of the glycines at either position 10 or 15 in Ap-B markedly decrease affinity (Seibert et al., 2003). Since only a small subset of loop confirmations can achieve high affinity channel interactions, most toxin/channel interactions will be unfruitful. It appears that most of the contacts in the loop with the channel are likely to be hydrophobic since replacement of Asn-16 or Ser-19 with the smaller alanine has little effect on affinity, while introduction of charge at either site dramatically reduces toxin’s ability to modify channel activity (Seibert et al., 2004). In addition, consistent with this idea Leu-18 is critical for toxin action, both for its size and shape as well as its hydrophobicity since it cannot even be replaced by a valine, which reduces toxin affinity 100-fold (Dias-Kadambi et al., 1996b).
Early experiments, primarily in the laboratory of Catterall, used antibodies and photo affinity labeling to identify parts of the channel that interacted with site-3 toxins (Tejedor & Catterall, 1988; Thomsen & Catterall, 1989). These studies were primarily carried out with neuronal channels and α-scorpion toxins, and they identified extracellular regions in domain I and domain IV as controlling binding affinity. Later mutagenesis experiments failed to identify any specific residue in the extracellular loops of domain I, although an anionic residue in the S3-S4 loop of domain IV was identified (Rogers et al., 1996) that affected toxin affinity. At about this time other experiments took advantage of the isoform preference of Ap-A for cardiac Na channels over skeletal muscle Na channels, and showed that the isoform preference for this toxin was associated with domain IV (Benzinger et al., 1997). Recently, additional contacts in the domain IV S3–S4 linker have been suggested based on experiments with site-3 toxins isolated from Anthopleura fuscoviridis and Bunodosoma caissarum tested against neuronal Na channels (Oliveira et al., 2004).
A strong test of whether the residue identified by channel mutagenesis interacted directly with the toxin, in contrast to mutagenesis causing a general conformational change in the channel, is mutant cycle analysis. With this technique one explores the interactions of wild-type toxin with wild-type channels, and combinations of toxin mutated at the putative interaction residue with the channel mutated at the putative interacting residue (Horovitz & Fersht, 1990). If the two residues of interest do not interact then the change in affinity measured when either the toxin or channel is mutated is similar so that the ratio of the KD’s is approximately unity. If, on the other hand, the residues interact, then the combination of mutated toxin and mutated channel is not much worse than either the wild-type and mutant combinations since the binding site is disrupted by either change. In this case the ratio of the KD’s is significantly different from unity. This analysis established a specific interaction between the anionic residue in the domain IV S3-S4 linker (either an aspartate or glutamate depending on channel isoform) and Lys37, a well conserved, positively charged amino acid on the toxin (Benzinger et al., 1998). In addition to this specific electrostatic interaction between toxin and channel, and interaction(s) involving the hydrophobic residues in the flexible loop of the toxin with, as yet, unidentified channel binding determinants, there is a tryptophan near the important Lys-37 (Trp-33) that also makes a structural contribution to toxin action (Dias-Kadambi et al., 1996a).
Complementing the slow on-rates, off-rates are also exceedingly slow and state dependent, and determine, for the most part, differences in affinity between Na channel isoforms. For Ap-B and cardiac Na channels koff, for example, from the closed state is only 0.0001 s-1, which means even after an hour an appreciable amount of toxin remains bound to the channel (Khera et al., 1995). On the other hand, depolarization greatly augments off-rate and toxin can be washed out in 8–10 minutes (Hanck & Sheets, 1995). Similar disparities in off-rate between negative and positive potentials is shared among all site-3 toxins. Even when overall affinity is in the 100 to 1000 nM range, the action of the toxin is not reversed even after many minutes following removal of toxin from the bath when the cell is held at negative holding potentials (Wang & Strichartz, 1985). In myelinated nerve when the membrane was depolarized the off-rate was so fast that it could be appreciated in the time course of the current (Wang & Strichartz, 1985; Strichartz & Wang, 1986). However, all forms of these toxins share a high affinity for the closed state of the channel.
Together available data indicate that site-3 toxins bind with highest affinity when the channel is closed. It is also clear that they bind to the domain IV S3–S4 linker best when the S4 voltage sensor is deployed towards the inside of the cell (closed state), and bind with the lowest affinity when S4 voltage sensors are deployed towards the outside (inactivated state). This suggests that the S3–S4 linker is located at the extracellular surface independent of the state of the channel. This seems at odds with the suggestion from the crystal structure of bacterial potassium channels in which the S4 is buried in the bilayer when the channel is closed (Lee et al., 2005). The original structure is now recognized to be non-native, but there is considerable evidence that for bacterial channels the voltage sensor is buried in the bilayer when the channel is closed. It might be the case that the toxin partitions into the bilayer to reach its binding site, although this seems unlikely since these toxins are highly hydrophilic, and they do not bind to phospholipids. This is in contrast to site 4 toxins, which are known to bind to a distinct site, and to affect activation (Smith et al., 2005).
Modification of Na Channel Ionic Current by Site-3 Toxins
The mechanism of action of site 3 toxins has been most extensively studied in cardiac Na channels with anemone toxins, the channels with the highest affinities for toxin. As a consequently, low concentrations of the toxins could be used to modify nearly all Na channels, and the channels remained modified for long intervals even after toxin is removed from the bath solution. Consistent with the observation of long lasting sodium currents in whole cell voltage clamp recordings that suggested these toxins inhibited fast inactivation, single channel studies of ATX II toxin-modified rabbit ventricular Na channels found that mean channel open-time durations were monotonically prolonged as test potentials became more positive with toxin-modified open-times prolonged by a factor of 3.5 at −20 mV compared to control and by 4.5 times at −10 mV (Schreibmayer et al., 1987a; El-Sherif et al., 1992).
Fast inactivation is known, however, to occur for Na channels both from the open state and from closed, non-conductive state(s) (i.e. closed-state inactivation) (Bean, 1981; Lawrence et al., 1991). Ap-A does not slow or inhibit closed-state inactivation of cardiac Na channels. In canine cardiac Purkinje cells, closed-state inactivation remained intact after exposure to Ap-A toxin at conditioning potentials from −80 mV to −120 mV, membrane potentials that are negative to Na current threshold (see Figure 2) (Hanck & Sheets, 1995), even though at positive potentials Ap-A modified Na channels behave as though they are driven into an open state from which inactivation becomes much less likely, at least for short durations of time (Hanck & Sheets, 1995). Figure 3 illustrates the difference in the amount of inactivation developed during conditioning potentials between −20 mV and +60 mV for durations up to 24 ms. In contrast to unmodified Na channels, as the conditioning potential became more positive, the fraction of inactivated Na channel became less in Ap-A toxin. This relationship is best illustrated in Figure 3C which shows an inverse relationship between the fraction of Na channels undergoing inactivation at conditioning potentials from −50 mV to 100 mV in control solution compared to those in Ap-A toxin.
Figure 2.
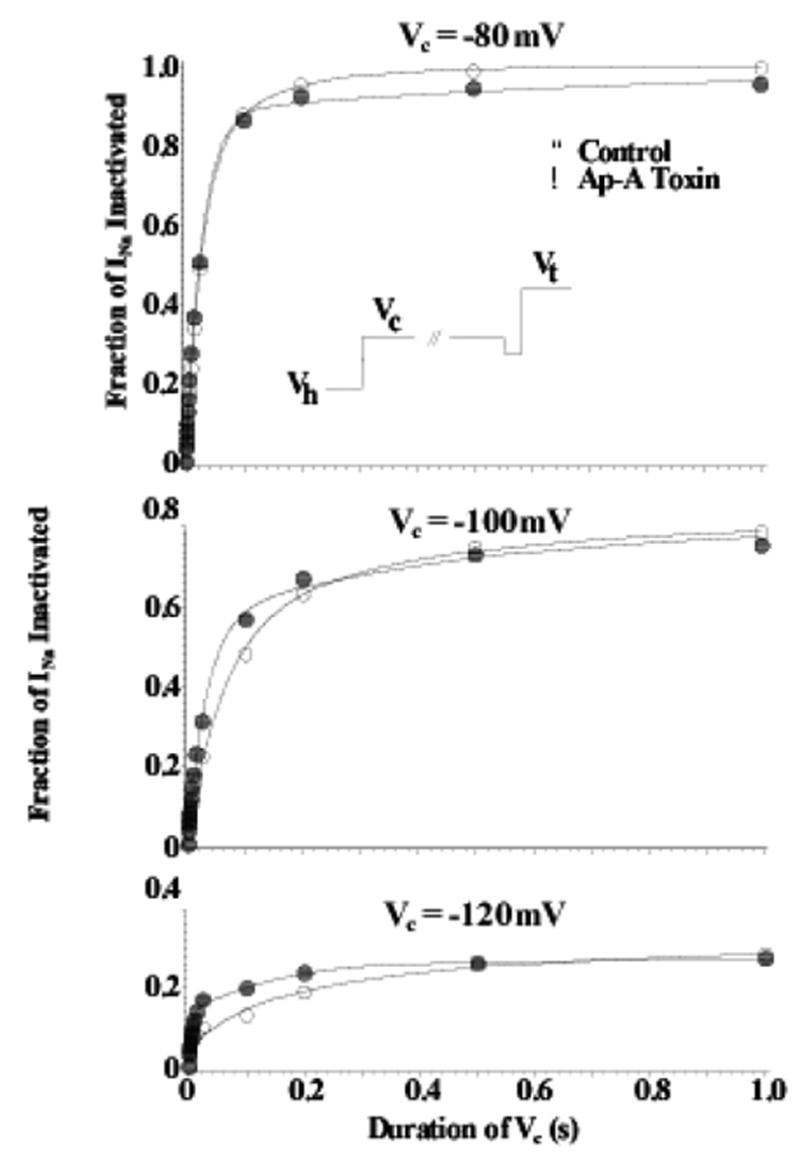
Closed-State Inactivation Remains Intact after Ap-A Toxin Modification of Na Channels. Shown are two-pulse development of inactivation relationships for Na channels in canine cardiac Purkinje cells at conditioning potentials (Vc) of −120 mV (bottom), −100 mV (middle), and −80 mV (top) in control (○) and after modification by Ap-A toxin (●). The voltage protocol is shown in the inset where the membrane potential was clamped-back to −110 or −120 mV for 2 ms before stepping to the test potential, Vt. Note that inactivation after Ap-A toxin occurs at least as fast as that in control at conditioning potentials negative to channel threshold. From Hanck and Sheets, 1995 with permission.
Figure 3.
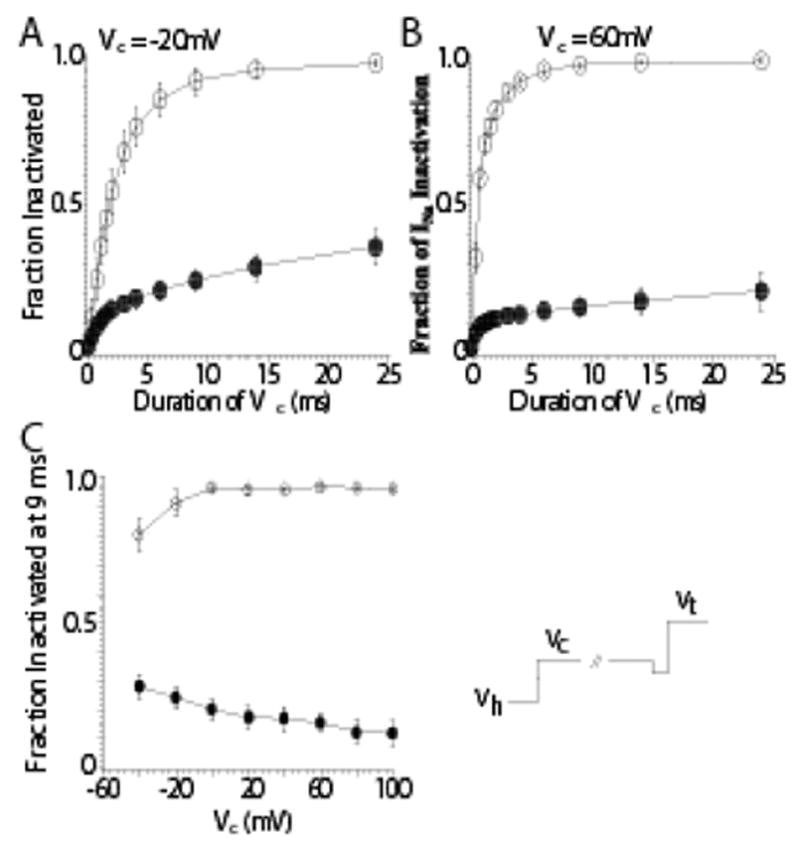
Inhibition of Inactivation from the Open-State after Anthopleurin-A Modification. Shown are two-pulse development of inactivation protocols at more positive conditioning potentials in control (○) and after modification by Ap-A toxin (●;). The voltage protocol (bottom right) is similar to that shown in the previous figure. Prior to stepping to the conditioning potential there was a brief voltage step to −40 mV for 0.2 ms. Peak INa measurements were normalized to the peak INa measured in the absence of a conditioning step. Panel A: At a conditioning potential of −20 mV INa had inactivated by 92% within 9 ms in control solution, but only by 24% after Ap-A toxin. Panel B: At a conditioning potential of 60 mV, 95% of the peak INa had inactivated at 9 ms in control while only 16% had inactivated after Ap-A toxin. Panel C: The mean fraction of INa inactivated by 9 ms as a function of potential in control (○) and after Ap-A toxin (●). Note that inactivation became more complete at positive potentials in control solution, while after Ap-A toxin there was less inactivation at positive potentials compared to negative conditioning potentials. From Hanck and Sheets, 1995 with permission.
Conversely, recovery from inactivation is augmented by site-3 toxins. This was established in single channel studies comparing the action of site-3 toxins in preparations expressing cardiac and neuronal channels (Benzinger et al., 1999). This action can be appreciated in Figure 4, where the shorter recovery time causes the development of inactivation to reach steady state more quickly at negative potentials. In summary the fast inactivation state is destablized by both slowing the forward rate constant into it and by becoming non-absorbing, i.e. channels can now recover. In contrast to these effects on fast inactivation, activation parameters remain unaffected. Deactivation, i.e. open to the closed, rested state, remained unchanged (Hanck & Sheets, 1995) and channel mean open times at negative potentials were like those of modified channels (El-Sherif et al., 1992). Consequently, site-3 toxins appear to target a single kinetic transition, the open-to-inactivated state transition, without an apparent change in channel activation, deactivation or closed-state inactivation.
Figure 4.
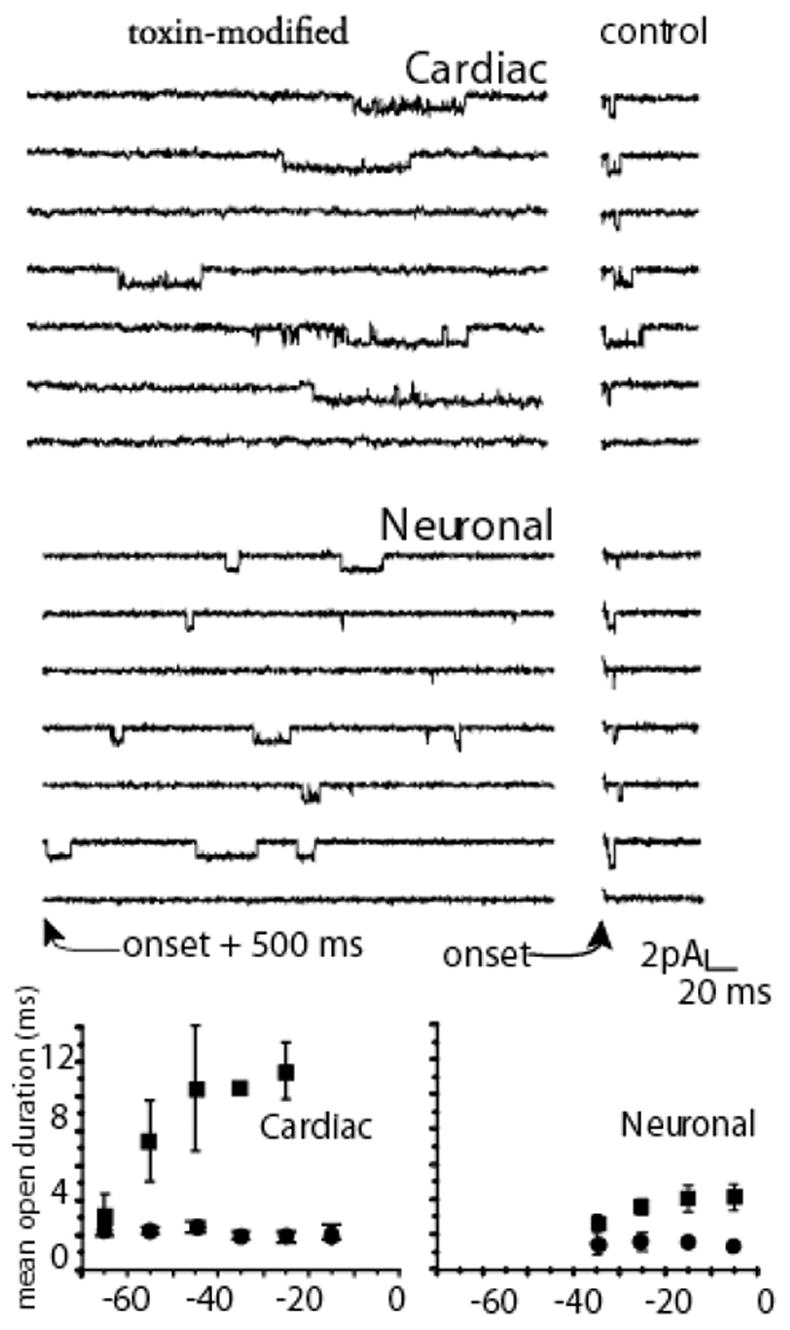
Mean Channel Lifetime. Upper panels show cell attached single channel recordings with 280 mM Na at 6 C. Right panels shows control (measured immediately after the onset of a step depolarization to −10 mV) in cardiac and neuronal channels. Left panels show records with 1 μM Ap-B in the pipette in which recordings are shown beginning 500 ms after the depolarization. Bottom panels show mean open durations measured from amplitude histograms for multiple patches measured at the potentials indicated. Data analysis techniques are completely described in Benzinger et al., 1999. Mean open time was prolonged by toxin in both isoforms, although more dramatically for cardiac than for neuronal channels.
Modification of Na Channel Gating Current by Site-3 Toxins
Further information about the action of site-3 toxins has been gained from gating current (Ig) measurements, the small electrical currents arising from the movement of the voltage sensors formed by the S4 segments in each of the four domains of the α-subunit (Bezanilla, 2000). One of the first studies (Neumcke et al., 1985) to directly investigate the molecular action of site-3 toxins on voltage-gated Na channels using this technique studied the effects of ATX-II toxin on gating currents in myelinated nerve. They found that ATX-II reduced the maximum gating charge (Qmax) and shortened the time constant of gating current (Ig) decay in response to step depolarizations. Later, a more detailed analysis of the effects of Ap-A toxin on gating currents found that Qmax was reduced by about 33% and resulted from the inhibition of the gating charge associated with the open-to-inactivated state transition (Sheets & Hanck, 1995). Because site-3 toxins only inhibit the open-to-inactivated state transition, subtraction of the gating currents of Ap-A modified Na channels from those in control recorded during step depolarizations represented the gating charge resulting from channels undergoing inactivation from the open state, a transition that was found to have a voltage-dependence (electronic charge) of 0.75 e−. Assuming symmetrical energy barriers the total gating charge associated with both forward and backward open-to-inactivated state transitions was estimated to 1.5 e−. An unexpected finding from studies with site-3 toxins (Sheets & Hanck, 1995) resulted from the observation that only two-thirds of the gating charge of voltage-gated Na channels moved before the Na channel opened. This is in sharp contrast to voltage-gated K channels, which are formed from four subunits in which all gating charge moves prior to channel opening (Bezanilla, 2000).
Further gating current experiments, in which the three outermost arginine residues in the S4-DIV were neutralized (one by one) to cysteine residues demonstrated that the reduction in gating charge by site-3 toxins resulted specifically from inhibition of the S4 segment in domain IV (Sheets et al., 1999). The substitution experiments were based upon the concept that if the movement of the basic residues in DIV-S4 were to account for most or all of the reduction in gating charge by Ap-A toxin, then site-3 toxin modification of mutant Na channels that had individual basic residues previously neutralized should demonstrate a smaller reduction in Qmax than the nearly 33% found for wild-type Na channels. The studies confirmed that expectation, and additionally they found that the three outermost arginine residues did not contribute an equal amount to the gating charge in the S4-DIV but that the outermost arginine was responsible for nearly two-thirds of all the gating charge contributed by the S4-DIV (Sheets et al., 1999) and predicted total channel charge to be 5–6 e−.
Although the above studies involved site-3 toxin interactions with the cardiac Na channel, the findings appear to be applicable to the general class of voltage-gated Na channels. Detailed studies of the effects of site-3 toxins on the gating currents of the human skeletal Na channel demonstrated that Ap-B toxin also reduced Qmax by about 33% in that isoform with only minor changes in the half-points and slope factors of the Q-V relationship (Sheets & Hanck, 1999). These findings were nearly identical to those for the cardiac channel. This was somewhat surprising because the channel kinetics of skeletal muscle Na channels are more rapid than those of cardiac Na channels (Chahine et al., 1996; Wang et al., 1996), and it had been suggested that the maximal number of electronic charges (e−) associated with skeletal muscle channel may be greater than that for cardiac channel (Hirschberg et al., 1995). Recently, a report on the effects of the site-3 α-scorpion toxin, Ts3, using site-specific fluorescence labeling of individual S4 segments in skeletal muscle Na channels has confirmed that site-3 toxins inhibit the movement of the S4-DIV by stabilizing the voltage sensor in its deactivated position accompanied by a slight shift of the voltage-dependent movement of S4-DI to slightly more positive potentials (Campos et al., 2006). Thus the action of site-3 toxins indicates their primary action is to inhibit the outward movement of the S4 segment in domain IV. All experiments with site-3 toxins support the idea that the S4 of domain IV has a unique role in the coupling of Na channel activation with inactivation (Chahine et al., 1994; Yang & Horn, 1995; Chen et al., 1996; Yang et al., 1996; Kontis et al., 1997; Kuhn & Greeff, 1999). The issue of the number of charges associated with channel gating, i.e. whether Na channels gate with > 10 e− as do K channels or whether they are overall less voltage dependent remains to be settled.
Although the movement of the S4 in domain IV is slower than other three S4 segments, after it has translocated Na channels inactivate quite quickly, i.e. within a few hundred μs (Sheets & Hanck, 1995). Attainment of the open state in wild-type Na channels would be expected to occur after movement of the three S4 segments in domains I, II and III but before full movement of the S4-DIV. Such a configuration for the four S4 segments in normal Na channels would be transient, and it would represent the configuration of the open channel that typically lasts no more than the few ms (Aldrich et al., 1983; Kunze et al., 1985; Grant & Starmer, 1987). However, after modification of the Na channel by site-3 toxins the Na channel would be able to occupy the open state for a prolonged time period thereby permitting other kinetic transitions to become apparent. During short, strong depolarizations toxin-modified Na channels remain principally in the open state, or burst, i.e. transitioning between the open and ultimate closed state (Sheets & Hanck, 1995). During long, sustained strong depolarizations toxin-modified Na channels have been shown to inactivate into a non-absorbing inactivated state in contrast to the absorbing inactivated state in control Na channels (Benzinger et al., 1999). As a consequence, the toxin-modified Na channels display persistent INa resulting from channels cycling between the toxin-induced destabilized inactivated state, the open state, and closed states. Not surprisingly, the magnitude of the persistent INa varies between different Na channel isoforms (Figure 4) with a smaller magnitude arising from cardiac Na channels while a larger magnitude occurs in neuronal Na channels (Benzinger et al., 1999)
The action of site-3 toxins to inhibit the outward movement of the S4 of domain IV would also be expected to manifest effects in the recovery from inactivation. As discussed above the recovery of ionic current is faster after toxin modification. It has long been appreciated that some of the gating charge that moved outward during depolarization returns slowly after hyperpolarization. The slow time course of return of charge, which reprises recovery of the ionic current, has been termed charge immobilization (Armstrong & Bezanilla, 1977; Bezanilla & Armstrong, 1977), and it has been shown to originate from the slow return of charge of the S4’s in domains III and IV but not those from domains I and II (Cha et al., 1999). When fast inactivation has been removed either by internal perfusion with proteolytic enzymes or by specific mutagenesis of the putative inactivation particle formed by the intracellular linker between domains III and IV (for example, by the substitution of a cysteine for the phenylalanine in the IFM motif with modification by intracellular MTSET) charge immobilization is either completely eliminated (Armstrong & Bezanilla, 1977) or reduced (Sheets et al., 2000). One might expect if site-3 toxins inhibit the outward movement of gating charge arising from domain IV, and that if a fraction of gating charge normally returns slowly reflecting the recovery of inactivated channels, then toxin modified channels should affect charge immobilization. In order to investigate whether charge immobilization could still occur in site-3 toxin modified Na channels where movement of the S4-DIV was prevented, we recorded gating currents from canine cardiac Purkinje cells before and after modification by Ap-A toxin. As discussed above, Na channels modified by Ap-A toxin are still able to undergo closed-state inactivation, therefore the membrane potential was first stepped to a conditioning potential of −70 mV for 500 ms to inactivate toxin-modified Na channels via closed-state inactivation prior to stepping the membrane potential to a second conditioning potential of 40 mV for 20 ms. The membrane potential was then repolarized to −190 mV, a potential where the fast and slow components of the time course of the recovery of gating charge can be readily differentiated in unmodified Na channels. Figure 5 shows the voltage-clamp protocol and the gating current integrals during repolarization for Na channels before and after modification by Ap-A toxin. Also shown are the gating current integrals during a step depolarization to 40 mV directly from a holding potential of −150 mV. As expected, control Na channels that have been inactivated demonstrate both a fast and slow component in the time course of the gating charge during repolarization. Interestingly, the toxin-modified Na channels also demonstrate two components in the gating charge during repolarization consistent with charge immobilization. However, the total magnitude of the gating charge during repolarization of the toxin-modified Na channel was reduced because the toxin inhibited only the movement of the S4-DIV leaving the gating charge from the slow return of charge of S4-DIII to contribute to charge immobilization. These data also demonstrate that the movement of the S4-DIV during a depolarizing step is not required for the putative inactivation particle to bind to its receptor causing channel inactivation and immobilization of the S4-DIII, and they explain the molecular basis of the augmentation of the rate of channel recovery from inactivation of channels bound to site-3 toxins.
Figure 5.
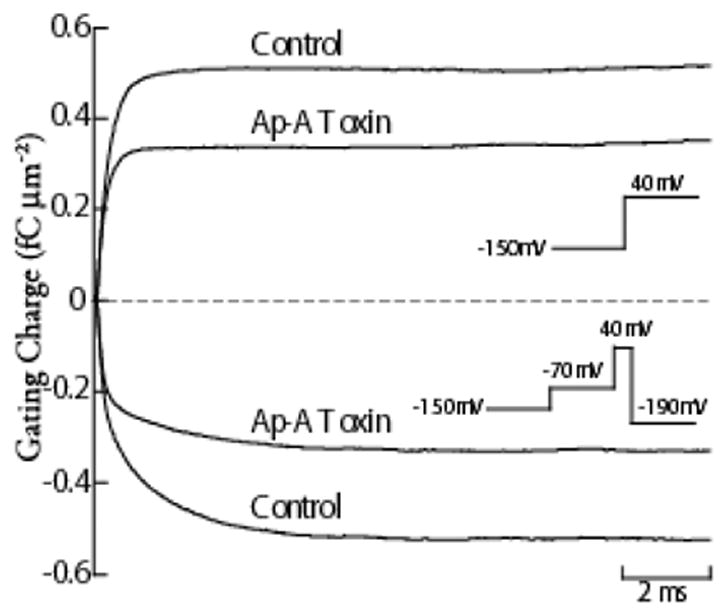
Charge Immobilization in Ap-A Modified Na Channels. Gating current integrals in response to a step depolarization to 40 mV from a holding potential of −150 mV are shown for a canine cardiac Purkinje cell in control and after exposure to Ap-A toxin (top two traces), and gating current integrals obtained during repolarization to -190 mV in the same cell (bottom two traces). The voltage protocols are shown as insets, and are adjacent to their corresponding gating charge traces. To inactivate both control and Ap-A toxin modified Na channels, the membrane potential was first stepped to −70 mV for 500 ms to allow for closed-state inactivation to occur before stepping to 40 mV for 20 ms. Note that the time course of the gating charge integral during repolarization has two readily apparent time constants for both control Na channels and for Ap-A toxin modified Na channels. In control, the magnitudes of the gating charge in both the fast and slow time constants are nearly equal while the magnitude of gating charge contained in the slow component of toxin-modified Na channels is only about one-half that in the fast component. The total magnitude of the gating charge during repolarization to −190 mV in control was 0.51 fC μm−2, and it equaled the maximum gating charge obtained during the step depolarization to 40 mV. After Na channel modification by 340 nM Ap-A toxin (a concentration that modifies at least 90% of cardiac Na channels) the magnitude of the gating charge during repolarization was reduced by nearly 35% to 0.33 fC μm−2, and it also equaled the gating charge at 40 mV.
Because the domain IV voltage sensor does not need to move in order for the channel to open, it raises an interesting question about the relationship of voltage sensor movement to the pore. In the available crystal structures of voltage-gated K channels the activation gate appears to be an intracellular structure formed at the bundle crossing of the four M2 (or S6) α helices (Jiang et al., 2002; for review see MacKinnon, 2003; Long et al., 2005). Upon depolarization the movement of all four S4 segments leads to channel opening by causing the intracellular ends of the S6 segments to bend away from the pore at hinge points formed by Glycine residues within the α helices of the S6 segments (MacKinnon, 2003). In contrast, the voltage-gated Na channel opens after movement of the first three S4 segments and before little, if any, movement of the S4-DIV. Although the three dimensional structure of the Na channel remains unknown, the S6 in domain IV differs from the other three S6 segments because it contains a serine at what would be by homology modeling its putative hinge point (Lipkind & Fozzard, 2005) while the other S6 segments have typical glycine residues (Goldin, 1995). In the voltage-gated K channel, the movement of the S4 segment tugs on the S4–S5 linker allowing its S6 segment to bend at the glycine hinge point thereby causing the channel to open (Long et al., 2005). It is tempting to suggest that the unique role of the S4-DIV in coupling activation to fast inactivation may depend upon the fact that the S6 in domain IV has a serine at in its putative hinge. The domain IV S6 may be already in an “open permissive” configuration such that movement of the domain IV voltage sensor produces a movement that promotes rapid binding of the putative inactivation particle (formed by the linker between domains III and IV) to its receptor rather than promoting channel opening.
Site-3 Toxins as Tools for Investigation into Drug-Modified Na Channels
Lastly, not only have site-3 toxins revealed important information about how voltage gated Na channels gate, but they have been used to help understand the action of other compounds, such as local anesthetic drugs, on the Na channel. It has long been proposed that local anesthetic drugs such as lidocaine block Na channel current by stabilization of the drug-bound channel in an inactivated state (Hille, 1977; Hondeghem & Katzung, 1977). If this were the case then it would be reasonable to conclude that the S4-DIV with its role in coupling channel activation to inactivation would be principally involved in the action of lidocaine block. Also supporting such an idea is the fact that the most important amino acid determining local anesthetic block is a Phe in domain IV, S6 (Ragsdale et al., 1994). With respect to gating charge lidocaine (Hanck et al., 2000) and other local anesthetic drugs (for example see Cahalan & Almers, 1979) cause a reduction in maximal gating charge (Qmax) by nearly 40%. To determine whether the reduction in Qmax by lidocaine was caused by stabilization of the S4-DIV, wild-type cardiac (Nav1.5) Na channels were expressed in HEK293 cells, and gating charge was measured in control, after exposure to lidocaine, and then after exposure to both lidocaine and Ap-A toxin. Consistent with previous studies lidocaine, by itself, decreased Qmax by approximately 37%, however in the presence of both lidocaine and Ap-A the decrease in Qmax was additive and was nearly 60% (Hanck et al., 2000) (Figure 6). These findings suggested that local anesthetic drugs and site-3 toxins stabilize different voltage sensors. Subsequently, it was shown that local anesthetic drugs appear to completely stabilize the S4 in domain III (Sheets & Hanck, 2003) with only a partial stabilization of the S4-DIV accompanied by a leftward shift in its gating.
Figure 6.
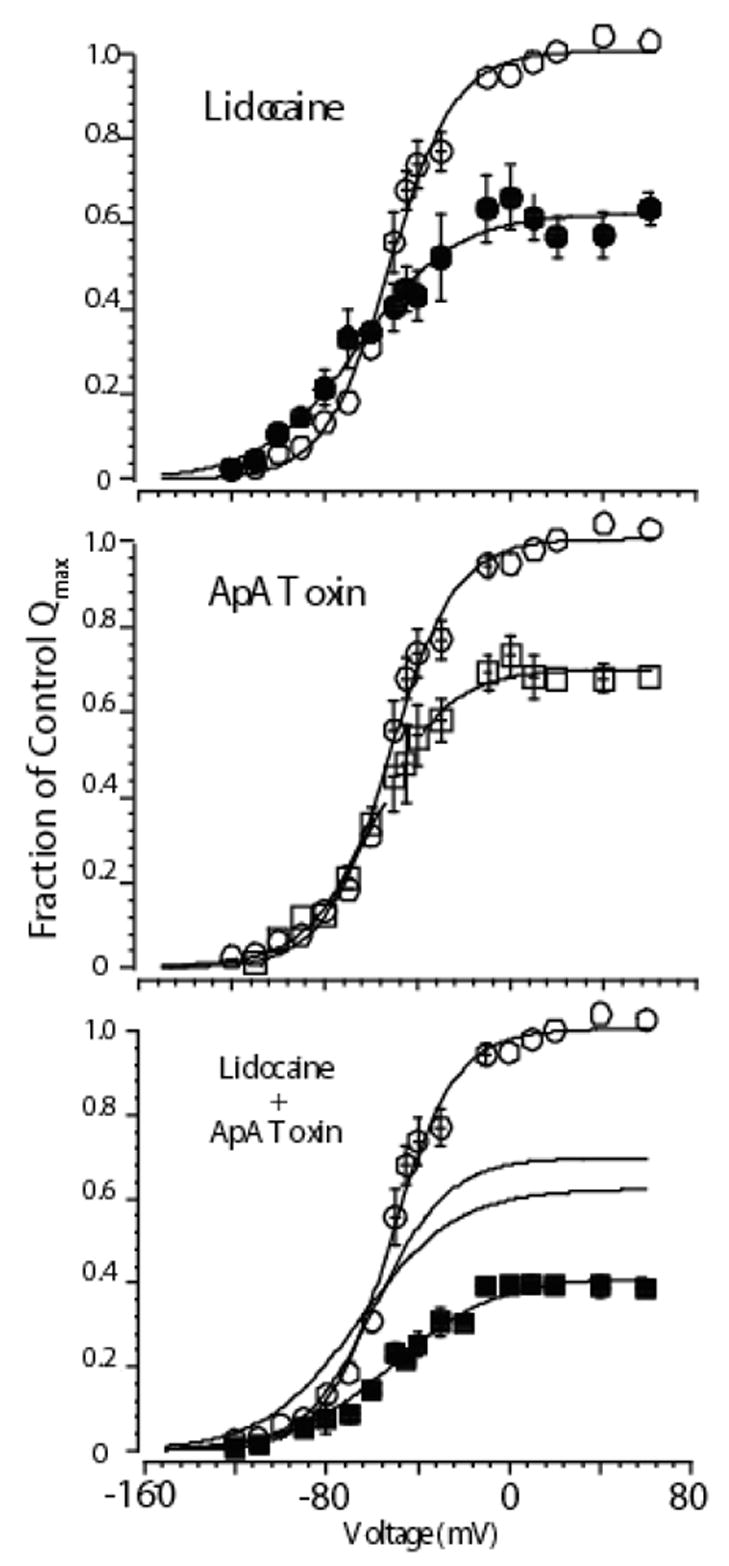
Mean charge voltage relationships for cells expressing cardiac Na channels exposed to high concentrations of either lidocaine alone (upper panel), Ap-A alone (middle panel), or both agents together (bottom panel). In each panel open circles show control data. Lidocaine modified data are shown as filled circles, Ap-A toxin modified data as open squares, and when both agents were present as filled squares. Solid lines are Boltzmann fits to the data. When both agents were present the effect on Qmax was additive, which can be appreciated from inspection of the fitted lines from each agent alone, which are also graphed in the lower panel. Interestingly, when both agents were present the Q-V relationship did not shift leftward as it did with lidocaine alone (see Hanck et al., 2000). Subsequent experiments determined that the gating charge responsible for the leftward shift in lidocaine is located in domain IV, the same gating charge that does not move when site-3 toxin is bound (Sheets & Hanck, 2003). Modified from Hanck et al., 2000.
Conclusion
Site-3 toxins, studied using whole cell voltage clamp, single channel recordings, and gating currents, have revealed a specific interaction with the voltage sensor of domain IV, and they have revealed the special role this voltage sensor plays in inactivation from the open state for voltage dependent Na channels. Unlike K channels, in which all four voltage sensors contribute to activation, for Na channels voltage sensors have specialized movements. Not only has investigation of toxin action revealed fundamental insights into molecular movements associated with kinetic states, but they have allowed the dissection of the mechanism of action of local anesthetics, which form an important class of therepeutic agents for treating pain and reducing arrhythmias. They provide, therefore, an important class of natural products that can be used as structure function probes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean B. Sodium channel inactivation in the crayfish giant axon: Must channels open before inactivating? Biophysical Journal. 1981;35:595–614. doi: 10.1016/S0006-3495(81)84815-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzinger GR, Drum CL, Chen LQ, Kallen RG, Hanck DA, Hanck D. Differences in the binding sites of two site-3 sodium channel toxins. Pflugers Arch. 1997;434:742–749. doi: 10.1007/s004240050460. [DOI] [PubMed] [Google Scholar]
- Benzinger GR, Kyle JW, Blumenthal KM, Hanck DA. A specific interaction between the cardiac sodium channel and site-3 toxin anthopleurin B. J Biol Chem. 1998;273:80–84. doi: 10.1074/jbc.273.1.80. [DOI] [PubMed] [Google Scholar]
- Benzinger GR, Tonkovich GS, Hanck DA. Augmentation of recovery from inactivation by site-3 Na channel toxins: A single-channel and whole-cell study of persistent currents. Journal of General Physiology. 1999;113:333–346. doi: 10.1085/jgp.113.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beress L, Beress R. Purification of three polypeptides with neuro- and cardiotoxic activity from the sea anemone Anemonia sulcata. Toxicon. 1975;13:359–367. doi: 10.1016/0041-0101(75)90196-8. [DOI] [PubMed] [Google Scholar]
- Beress R, Beress L, Wunderer G. Purification and characterisation of four polypeptides with neurotoxic activity from Condylactis aurantiaca. Hoppe Seylers Z Physiol Chem. 1976;357:409–414. doi: 10.1515/bchm2.1976.357.1.409. [DOI] [PubMed] [Google Scholar]
- Bergman C, Dubois JM, Rojas E, Rathmayer W. Decreased rate of sodium conductance inactivation in the node of Ranvier induced by a polypeptide toxin from sea anemone. Biochim Biophys Acta. 1976a;455:173–184. doi: 10.1016/0005-2736(76)90162-0. [DOI] [PubMed] [Google Scholar]
- Bergman C, Dubois JM, Rojas E, Rathmayer W. Decreased rate of sodium conductance inactivation in the node of Ranvier induced by a polypeptide toxin from sea anemone. Biochimica et Biophysica Acta. 1976b;455:173–184. doi: 10.1016/0005-2736(76)90162-0. [DOI] [PubMed] [Google Scholar]
- Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal KM, Seibert AL. Voltage-gated sodium channel toxins: poisons, probes, and future promise. Cell Biochem Biophys. 2003;38:215–238. doi: 10.1385/CBB:38:2:215. [DOI] [PubMed] [Google Scholar]
- Cahalan MD, Almers W. Interactions between quaternary lidocaine, the sodium channel gates, and tetrodotoxin. Biophys J. 1979;27:39–55. doi: 10.1016/S0006-3495(79)85201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos F, Chanda B, Beirao P, Bezanilla F. Electrophysiological and site-specific fluorescence measurements on alpha-scorpion toxin bound voltage-gated sodium channels. Biophyical Journal. 2006:382-Poster. [Google Scholar]
- Catterall W. Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Ann Rev Pharmacol Toxicol. 1980;20:15–43. doi: 10.1146/annurev.pa.20.040180.000311. [DOI] [PubMed] [Google Scholar]
- Cha A, Ruben PC, George AL, Jr, Fujimoto E, Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999;22:73–87. doi: 10.1016/s0896-6273(00)80680-7. [DOI] [PubMed] [Google Scholar]
- Chahine M, Deschene I, Chen LQ, Kallen RG. Electrophysiological characteristics of cloned skeletal and cardiac muscle sodium channels. American Journal of Physiology (Heart and Circulatory Physiology) 1996;271:H498–H506. doi: 10.1152/ajpheart.1996.271.2.H498. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Chen LQ, Santarelli V, Horn R, Kallen RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J Gen Physiol. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett AM, Krueger BK. Polypeptide neurotoxins modify gating and apparent single-channel conductance of veratridine-activated sodium channels in planar lipid bilayers. J Membr Biol. 1989;110:199–207. doi: 10.1007/BF01869150. [DOI] [PubMed] [Google Scholar]
- Corzo G, Gilles N, Satake H, Villegas E, Dai L, Nakajima T, Haupt J. Distinct primary structures of the major peptide toxins from the venom of the spider Macrothele gigas that bind to sites 3 and 4 in the sodium channel. FEBS Lett. 2003;547:43–50. doi: 10.1016/s0014-5793(03)00666-5. [DOI] [PubMed] [Google Scholar]
- Dias-Kadambi BL, Combs KA, Drum CL, Hanck DA, Blumenthal KM. The role of exposed tryptophan residues in the activity of the cardiotonic polypeptide anthopleurin B. J Biol Chem. 1996a;271:23828–23835. doi: 10.1074/jbc.271.39.23828. [DOI] [PubMed] [Google Scholar]
- Dias-Kadambi BL, Drum CL, Hanck DA, Blumenthal KM. Leucine 18, a hydrophobic residue essential for high affinity binding of anthopleurin B to the voltage-sensitive sodium channel. J Biol Chem. 1996b;271:9422–9428. doi: 10.1074/jbc.271.16.9422. [DOI] [PubMed] [Google Scholar]
- El-Sherif N, Fozzard HA, Hanck DA. Dose-dependent modulation of the cardiac sodium channel by sea anemone toxin ATXII. Circ Res. 1992;70:285–301. doi: 10.1161/01.res.70.2.285. [DOI] [PubMed] [Google Scholar]
- Fletcher JI, Chapman BE, Mackay JP, Howden ME, King GF. The structure of versutoxin (delta-atracotoxin-Hv1) provides insights into the binding of site 3 neurotoxins to the voltage-gated sodium channel. Structure. 1997;5:1525–1535. doi: 10.1016/s0969-2126(97)00301-8. [DOI] [PubMed] [Google Scholar]
- Frelin C, Vigne P, Schweitz H, Lazdunski M. The interaction of sea anemone and scorpion neurotoxins with tetrodotoxin-resistant Na+ channels in rat myoblasts. A comparison with Na+ channels in other excitable and non-excitable cells. Mol Pharmacol. 1984;26:70–74. [PubMed] [Google Scholar]
- Gallagher MJ, Blumenthal KM. Cloning and expression of wild-type and mutant forms of the cardiotonic polypeptide anthopleurin B. J Biol Chem. 1992;267:13958–13963. [PubMed] [Google Scholar]
- Gallagher MJ, Blumenthal KM. Importance of the unique cationic residues arginine 12 and lysine 49 in the activity of the cardiotonic polypeptide anthopleurin B. J Biol Chem. 1994;269:254–259. [PubMed] [Google Scholar]
- Goldin AL. Voltage-Gated Sodium Channels. In: North RA, editor. Handbook of Receptors and Channels: Ligand and Voltage-Gated Ion Channels. CRC Press; Boca Raton: 1995. pp. 73–112. [Google Scholar]
- Grant AO, Starmer CF. Mechanisms of closure of cardiac sodium channels in rabbit ventricular myocytes: single channel analysis. Circulation Research. 1987;60:897–913. doi: 10.1161/01.res.60.6.897. [DOI] [PubMed] [Google Scholar]
- Hanck DA, Makielski JC, Sheets MF. Lidocaine alters activation gating of cardiac Na channels. Pflugers Arch. 2000;439:814–821. doi: 10.1007/s004249900217. [DOI] [PubMed] [Google Scholar]
- Hanck DA, Sheets MF. Modification of inactivation in cardiac sodium channels: Ionic current studies with Anthopleurin-A toxin. J Gen Physiol. 1995;106:601–616. doi: 10.1085/jgp.106.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. Journal of General Physiology. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg B, Rovner A, Lieberman M, Patlak J. Transfer of twelve charges is needed to open skeletal muscle Na+ channels. J Gen Physiol. 1995;106:1053–1068. doi: 10.1085/jgp.106.6.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG. Time- and voltage- dependent interaction of antiarrhythmic drugs with cardiac sodium channels. Biochimica et Biophysica Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- Honma T, Shiomi K. Peptide toxins in sea anemones: structural and functional aspects. Mar Biotechnol (NY) 2006;8:1–10. doi: 10.1007/s10126-005-5093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horovitz A, Fersht AR. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol. 1990;214:613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- Housset D, Habersetzer-Rochat C, Astier JP, Fontecilla-Camps JC. Crystal structure of toxin II from the scorpion Androctonus australis Hector refined at 1.3 A resolution. J Mol Biol. 1994;238:88–103. doi: 10.1006/jmbi.1994.1270. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- Khera PK, Benzinger GR, Lipkind G, Drum CL, Hanck DA, Blumenthal KM. Multiple cationic residues of anthopleurin B that determine high affinity and channel isoform discrimination. Biochemistry. 1995;34:8533–8541. doi: 10.1021/bi00027a003. [DOI] [PubMed] [Google Scholar]
- Kontis KJ, Rounaghi A, Goldin AL. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains [published erratum appears in J Gen Physiol 1997 Dec;110(6):763] J Gen Physiol. 1997;110:391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn FJ, Greeff NG. Movement of voltage sensor S4 in domain 4 is tightly coupled to sodium channel fast inactivation and gating charge immobilization. J Gen Physiol. 1999;114:167–183. doi: 10.1085/jgp.114.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze DL, Lacerda AE, Wilson DL, Brown AM. Cardiac Na currents and the inactivity, reopening, and waiting properties of single Na channels. Journal of General Physiology. 1985;86:697–719. doi: 10.1085/jgp.86.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JC, Catterall WA. Tetrodotoxin-insensitive sodium channels. Binding of polypeptide neurotoxins in primary cultures of rat muscle cells. J Biol Chem. 1981;256:6223–6229. [PubMed] [Google Scholar]
- Lawrence JH, Yue DT, Rose WC, Marban E. Sodium channel inactivation from resting states in guinea-pig ventricular myocytes. Journal of Physiology-London. 1991;443:629–650. doi: 10.1113/jphysiol.1991.sp018855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Lee A, Chen J, MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci U S A. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkind GM, Fozzard HA. Molecular modeling of local anesthetic drug binding by voltage-gated sodium channels. Mol Pharmacol. 2005;68:1611–1622. doi: 10.1124/mol.105.014803. [DOI] [PubMed] [Google Scholar]
- Little MJ, Wilson H, Zappia C, Cestele S, Tyler MI, Martin-Eauclaire MF, Gordon D, Nicholson GM. Delta-atracotoxins from Australian funnel-web spiders compete with scorpion alpha-toxin binding on both rat brain and insect sodium channels. FEBS Lett. 1998a;439:246–252. doi: 10.1016/s0014-5793(98)01378-7. [DOI] [PubMed] [Google Scholar]
- Little MJ, Zappia C, Gilles N, Connor M, Tyler MI, Martin-Eauclaire MF, Gordon D, Nicholson GM. delta-Atracotoxins from australian funnel-web spiders compete with scorpion alpha-toxin binding but differentially modulate alkaloid toxin activation of voltage-gated sodium channels. J Biol Chem. 1998b;273:27076–27083. doi: 10.1074/jbc.273.42.27076. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Potassium channels. FEBS Lett. 2003;555:62–65. doi: 10.1016/s0014-5793(03)01104-9. [DOI] [PubMed] [Google Scholar]
- Nagy K. Mechanism of inactivation of single sodium channels after modification by chloramine-T, sea anemone toxin and scorpion toxin. J Membr Biol. 1988;106:29–40. doi: 10.1007/BF01871764. [DOI] [PubMed] [Google Scholar]
- Neumcke B, Schwarz W, Stampfli R. Comparison of the effects of Anemonia toxin II on sodium and gating currents in frog myelinated nerve. Biochimica et Biophysica Acta. 1985;814:111–119. doi: 10.1016/0005-2736(85)90425-0. [DOI] [PubMed] [Google Scholar]
- Nicholson GM, Graudins A. Spiders of medical importance in the Asia-Pacific: atracotoxin, latrotoxin and related spider neurotoxins. Clin Exp Pharmacol Physiol. 2002;29:785–794. doi: 10.1046/j.1440-1681.2002.03741.x. [DOI] [PubMed] [Google Scholar]
- Norton RS. Structure and structure-function relationships of sea anemone proteins that interact with the sodium channel. Toxicon. 1991;29:1051–1084. doi: 10.1016/0041-0101(91)90205-6. [DOI] [PubMed] [Google Scholar]
- Norton TR, Shibata S, Kashiwagi M, Bentley J. Isolation and characterization of the cardiotonic polypeptide anthopleurin-A from the sea anemone Anthopleura xanthogrammica. J Pharm Sci. 1976;65:1368–1374. doi: 10.1002/jps.2600650927. [DOI] [PubMed] [Google Scholar]
- Oliveira JS, Redaelli E, Zaharenko AJ, Cassulini RR, Konno K, Pimenta DC, Freitas JC, Clare JJ, Wanke E. Binding specificity of sea anemone toxins to Nav 1.1–1.6 sodium channels: unexpected contributions from differences in the IV/S3-S4 outer loop. J Biol Chem. 2004;279:33323–33335. doi: 10.1074/jbc.M404344200. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Rando TA, Wang GK, Strichartz GR. The interaction between the activator agents batrachotoxin and veratridine and the gating processes of neuronal sodium channels. Mol Pharmacol. 1986;29:467–477. [PubMed] [Google Scholar]
- Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3–S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- Salgado VL, Kem WR. Actions of three structurally distinct sea anemone toxins on crustacean and insect sodium channels. Toxicon. 1992;30:1365–1381. doi: 10.1016/0041-0101(92)90512-4. [DOI] [PubMed] [Google Scholar]
- Scanlon MJ, Norton RS. Multiple conformations of the sea anemone polypeptide anthopleurin-A in solution. Protein Sci. 1994;3:1121–1124. doi: 10.1002/pro.5560030717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtmayer J, Stoye-Herzog M, Ulbricht W. Rate of action of Anemonia sulcata toxin II on sodium channels in myelinated nerve fibres. Pflugers Arch. 1982;394:313–319. doi: 10.1007/BF00583695. [DOI] [PubMed] [Google Scholar]
- Schreibmayer W, Kazerani H, Tritthart HA. A mechanistic interpretation of the action of toxin II from {I Anemonia sulcata} on the cardiac sodium channel. Biochim Biophys Acta. 1987a;901:273–282. doi: 10.1016/0005-2736(87)90124-6. [DOI] [PubMed] [Google Scholar]
- Schreibmayer W, Kazerani H, Tritthart HA. A mechanistic interpretation of the action of toxin II from Anemonia sulcata on the cardiac sodium channel. Biochim Biophys Acta. 1987b;901:273–282. doi: 10.1016/0005-2736(87)90124-6. [DOI] [PubMed] [Google Scholar]
- Seibert AL, Liu J, Hanck DA, Blumenthal KM. Arg-14 loop of site 3 anemone toxins: effects of glycine replacement on toxin affinity. Biochemistry. 2003;42:14515–14521. doi: 10.1021/bi035291d. [DOI] [PubMed] [Google Scholar]
- Seibert AL, Liu J, Hanck DA, Blumenthal KM. Role of Asn-16 and Ser-19 in anthopleurin B binding. Implications for the electrostatic nature of Na(V) site 3. Biochemistry. 2004;43:7082–7089. doi: 10.1021/bi0496135. [DOI] [PubMed] [Google Scholar]
- Sheets MF, Hanck DA. Voltage-dependent open-state inactivation of cardiac sodium channels: Gating currents studies with Anthopleurin-A toxin. J Gen Physiol. 1995;106:617–640. doi: 10.1085/jgp.106.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF, Hanck DA. Gating of skeletal and cardiac muscle sodium channels in mammalian cells. J Physiol. 1999;514:425–436. doi: 10.1111/j.1469-7793.1999.425ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF, Hanck DA. Molecular action of lidocaine on the voltage sensors of sodium channels. J Gen Physiol. 2003;121:163–175. doi: 10.1085/jgp.20028651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF, Kyle JW, Hanck DA. The role of the putative inactivation lid in sodium channel gating current immobilization. J Gen Physiol. 2000;115:609–620. doi: 10.1085/jgp.115.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF, Kyle JW, Kallen RG, Hanck DA. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys J. 1999;77:747–757. doi: 10.1016/S0006-3495(99)76929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S, Norton TR, Izumi T, Matsuo T, Katsuki S. A polypeptide (AP-A) from sea anemone ({I Anthropleura Xanthogrammica}) with potent positive inotropic action. Journal of Pharmacology and Experimental Therapeutics. 1976;199:298–309. [PubMed] [Google Scholar]
- Smith JJ, Alphy S, Seibert AL, Blumenthal KM. Differential phospholipid binding by site 3 and site 4 toxins. Implications for structural variability between voltage-sensitive sodium channel domains. J Biol Chem. 2005;280:11127–11133. doi: 10.1074/jbc.M412552200. [DOI] [PubMed] [Google Scholar]
- Srinivasan KN, Gopalakrishnakone P, Tan PT, Chew KC, Cheng B, Kini RM, Koh JL, Seah SH, Brusic V. SCORPION, a molecular database of scorpion toxins. Toxicon. 2002;40:23–31. doi: 10.1016/s0041-0101(01)00182-9. [DOI] [PubMed] [Google Scholar]
- Strichartz GR, Wang GK. Rapid voltage-dependent dissociation of scorpion alpha-toxins coupled to Na channel inactivation in amphibian myelinated nerves. J Gen Physiol. 1986;88:413–435. doi: 10.1085/jgp.88.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor FJ, Catterall WA. Site of covalent attachment of alpha-scorpion toxin derivatives in domain I of the sodium channel alpha subunit. Proc Natl Acad Sci U S A. 1988;85:8742–8746. doi: 10.1073/pnas.85.22.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen WJ, Catterall WA. Localization of the receptor site for alpha-scorpion toxins by antibody mapping: implications for sodium channel topology. Proc Natl Acad Sci U S A. 1989;86:10161–10165. doi: 10.1073/pnas.86.24.10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torda AE, Norton RS. Proton nmr relaxation study of the dynamics of anthopleurin-A in solution. Biopolymers. 1989;28:703–716. doi: 10.1002/bip.360280303. [DOI] [PubMed] [Google Scholar]
- Vincent JP, Balerna M, Barhanin J, Fosset M, Lazdunski M. Binding of sea anemone toxin to receptor sites associated with gating system of sodium channel in synaptic nerve endings in vitro. Proc Natl Acad Sci U S A. 1980;77:1646–1650. doi: 10.1073/pnas.77.3.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DW, George AL, Jr, Bennett PB. Comparison of heterologously expressed human cardiac and skeletal muscle sodium channels. Biophysical Journal. 1996;70:238–245. doi: 10.1016/S0006-3495(96)79566-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Strichartz G. Kinetic analysis of the action of Leiurus scorpion alpha-toxin on ionic currents in myelinated nerve. J Gen Physiol. 1985;86:739–762. doi: 10.1085/jgp.86.5.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warashina A, Jiang ZY, Ogura T. Potential-dependent action of Anemonia sulcata toxins III and IV on sodium channels in crayfish giant axons. Pflugers Arch. 1988;411:88–93. doi: 10.1007/BF00581651. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Tang J, Hu W, Xie J, Maertens C, Tytgat J, Liang S. Jingzhaotoxin-I, a novel spider neurotoxin preferentially inhibiting cardiac sodium channel inactivation. J Biol Chem. 2005;280:12069–12076. doi: 10.1074/jbc.M411651200. [DOI] [PubMed] [Google Scholar]
- Yang N, Horn R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Yang NB, George AL, Jr, Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- Zeng DW, Kyle JW, Martin RL, Ambler KS, Hanck DA. Cardiac sodium channels expressed in a peripheral neurotumor-derived cell line, RT4-B8. American Journal of Physiology (Cell Physiology) 1996;270:C1522–C1531. doi: 10.1152/ajpcell.1996.270.5.C1522. [DOI] [PubMed] [Google Scholar]