

SUMMARY
In rat liver epithelial (WB) cells, the protein kinase C inhibitor H7 blocked gap junctional intercellular communication (GJIC) and reduced virus infectivity. Octanol, 18-beta-glycyrrhetinic acid, and staurosporine, agents that reduce GJIC, had no effect upon virus infectivity. Previous studies demonstrated that herpes simplex virus- type 2 (HSV-2) infection was accompanied by attenuated GJIC. Of agents tested, only H7 reduced plaque forming unit (pfu) ability in a dose-dependent manner with 100% plaque reduction at 40 μM without evidence of cytotoxicity. Dye transfer indicated that H7 decreased GJIC, although Western blotting revealed that it did not alter phosphorylation of the gap junction protein, connexin 43 (Cx43). Using indirect immunofluorescence, Cx43 was found to localize in membrane plaques in uninfected cells and H-7 did not alter this distribution. However, Cx43 was lost from the membrane at 24 hrs in both H-7 treated and untreated cells infected with HSV-2. Viral infection increased serine phosphorylation, particularly in the nuclear region, and this effect was reduced following H-7 treatment. Thus, H-7 attenuated both GJIC and infectivity of HSV-2 in WB cells but the anti-viral effects were due to reduced nuclear protein phosphorylation rather than alterations in phosphorylation or localization of Cx43.
Keywords: Gap junctions, Connexin 43, protein kinase C, H7, HSV-2
1) INTRODUCTION
It has long been known that certain RNA and tumor viruses decrease gap junctional communication among infected cells (Atkinson et al., 1981; Crow et al., 1990; Danave et al., 1994; Ennaji et al., 1995; Faccini et al., 1996, Jou et al., 1995). Recent studies have shown electrophysiologically that down-regulation of gap junctions by herpes simplex virus-type 2 (HSV-2) begins before viral DNA replication, and long before morphological signs of infection (Fischer et al., 2001). In addition, Musee et al. (2002) demonstrated that antiviral agents affect electrical coupling in HSV-2 infected cells in different ways, depending on the mechanism of action of the agent. However, it was not clear if GJIC down-regulation was initiated by the infected cells or by their surrounding uninfected neighbors. If the latter, drugs that reduce GJIC might protect neighboring cells from infection.
How HSV-2 induces gap junctional down-regulation is unknown although both viral- or cell-mediated mechanisms are possible. Gap junction channels are dynamic macromolecular complexes capable of opening and closing in response to a multitude of stimuli including pH, divalent cations, signaling molecules, and phosphorylation (De Mello, 1988, Saez et al., 2003). Kinases and phosphorylation of connexins are involved in the regulation of intercellular communication at all levels ranging from the expression of connexin genes to the degradation of gap junction channels (Cruciani and Mikalsen, 2002; Lampe and Lau, 2000). Previous work suggests that viruses may alter tyrosine protein kinase activity (Fletcher et al., 1987) which phosphorylates tyrosine residues in the gap junctional protein connexin-43 (Cx43) ( Brissette et al., 1991, Crow et al., 1990; Filson et al., 1990; Goldstein et al., 1991, Warn-Cramer and Lau, 2004).
In this study, various pharmacological agents were used to disrupt GJIC immediately following infection to determine if the loss of GJIC was a protective mechanism initiated by infected cells or their neighbors. Octanol has been previously shown to block GJIC in a variety of cell types (Chanson et al., 1989) while 18β-glycyrrhetinic acid (18β-GA) inhibits GJIC by inducing the dephosphorylation of Cx43 (Guan et al., 1996). Staurosporine, a protein kinase C inhibitor, reduces cell-to-cell coupling by reducing phosphorylation of Cx43 (Moreno et al, 1994; Saez et al, 1997). 1-(5-isoquinolinesulfonyl)-2-methylpiperazine (H-7), another protein kinase C inhibitor (Hidaka et al., 1984), has been shown to reduce GJIC without altering Cx43 phosphorylation (Duthe et al., 2000; Herve et al., 2004). In this study we used a non-tumorigenic WB-F344 rat liver epithelial cell model system to study the mechanism responsible for the electrical uncoupling of HSV-2 infected cells because the WB cells express high levels of Cx43, form numerous gap junctions, and have a high percentage of communicating cells (Esinduy et al., 1995).
2) MATERIALS AND METHODS
2.1) Cell culture and chemicals
Fischer WB-F344 rat liver epithelial cells were used in this study because they serve as an excellent model for gap junction studies. The cells were a generous gift from Dr. James Trosko (Michigan State University) and were propagated as the continuous cell line to be used as the permissive cell type for these experiments. The cells were grown in Dulbecco’s modified Eagle’s medium (D-medium, Gibco BRL) and supplemented with 10% Newborn Calf Serum (Hyclone) with penicillin/streptomycin at 37°C in 5% CO2.
2.2) Plaque Reduction Assay
To determine if agents that inhibit gap junctional communication possess antiviral effects, WB-F344 cells were grown to confluence on Corning 25 cm2 tissue culture flasks and infected with diluted HSV-2 virus to yield 30–300 plaques per monolayer. Uninfected controls were mock-infected with serum free D-medium. After one hour incubation with virus, the infected controls received D-medium while treated cells received the test compounds diluted in D-medium. Chemicals tested for possible antiviral effects were octanol (Sigma, 0.5–10 mM), 18-beta-glycyrrhetinic acid (Sigma, 3–25 μM), staurosporine (1–200 nM), and H7 (Sigma, 1–100 μM). Compounds were dissolved in DMSO, if necessary, and the DMSO concentration in the medium during exposure was 0.1% (v/v) or lower for all concentrations of test substances. After 24–36 hours, the plates were stained with a 1 % crystal violet in 35 % methanol and plaque numbers were recorded. Significant differences were determined using an unpaired Student’s t-test.
2.3) Cell communication assay
To determine if agents that block gap junctions were preventing communication with adjacent cells, Gap Junctional Intercellular Communication (GJIC) was assessed using the scrape loading/dye transfer (SL/DT) technique as described previously (El-Fouly et al., 1987; Opsahl and Rivedal, 2000). Briefly, WB cells were grown on 35 mm plates in D-medium until cells nearly reached confluence. Agents that block gap junctions, such as octanol (1.0 mM added 15 min prior to SL/DT) or virus (10 pfu/cell added 12 hr prior to SD/DT), were added before the communication assay was performed. The high multiplicity of infection (m.o.i = 10) was assessed on WB cells by dividing the HSV-2 infectious dose (titer assayed on WB cells) by the number of cells present in the plate.
The medium was removed and the cultures were rinsed three times with PBS. Three parallel scrapes were made in the center of each plate in the presence of 1 ml of a 1% solution of Lucifer Yellow (which loads into scrape damaged cells and crosses gap junctions) and Texas Red Dextran (which loads into cells but is too large to pass through the gap junctions, both dyes from Molecular Probes, Eugene, OR) in D-medium. Cells were viewed immediately using an Olympus IX71 inverted microscope fitted with fluorescence optics (Olympus America, Melville, NY). Images were recorded either with a Nikon CoolPix 995 digital camera or with an IFC-300 cooled CCD camera (Dage-MTI, Michigan City, IN).
2.4) Cytotoxicity assay
The MTT assay was utilized to assess the cytotoxicity of all agents on WB cells. Only the cytotoxicity data for H7 is shown because none of the other agents demonstrated antiviral effects at sub-toxic doses. WB cells grown to confluence on Corning twelve-well culture plates were treated with H7. Two wells per plate were treated with iodoacetic acid (1 mM) to serve as a metabolically inhibited control. After the 24 hours, each well was washed three times with serum free D-medium, MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide, 5.0 mg/ml, Sigma Chemical) was added to each well, and plates were incubated for an additional five hours at 37°C. Following this incubation 1.0 ml of DMSO was added to each well to solubilize formazan, and absorbance was measured at 550 nm.
2.5) Western blot
The ability of H7 to alter phosphorylation of Cx43 was assessed using Western blot. Membrane-enriched fractions of control and H7-treated WB cells grown in 25 cm2 flasks were prepared using Fraction 2 of the ProteoExtract Subcellular Proteome Extraction Kit (EMD Biosciences, Damstadt, Germany) containing protease inhibitor cocktail. Protein concentration was determined by the Bio-Rad DC protein assay (Bio-Rad, Hercules, CA, USA) and the cell extracts were immunoprecipitated using a Protein G Immunoprecipitation kit (Sigma, St. Louis, MO). For Western blot, protein was separated on a 12% SDS–polyacrylamide gel using a mini-Protein III electrophoresis apparatus (Bio-Rad, Hercules, CA) run at 100 V. The separated proteins were transferred to PVDF membranes using a mini-Transblot apparatus (Bio-Rad, Hercules, CA) at 30 V overnight. The membranes were incubated for at least 2 h with an anti-Connexin 43 antibody (1:1000, Sigma, Catalog No. C-6219). The bound antibody was detected using an anti-rabbit secondary antibody (1:1000) conjugated with alkaline phosphatase and visualized with the alkaline phosphatase color development reagents, BCIP (5-bromo-4-chloro-3-indoylphosphate p-toluidine salt) and NBT (p-nitro blue tetrazolium chloride).
2.6) Immunofluorescent labeling
Immunofluorescent labeling of WB cells was used to determine the localization of Cx43 gap junction protein and phosphoserine following viral infection in the absence and presence of H7. Cells grown in 12-well plates were incubated with Herpes Simplex Virus Type-2 (HSV-2) at 1 pfu/cell (m.o.i. assayed on WB cells) for 1 hr. Virus was washed off and cells were incubated for 24 hours in D-medium in the presence or absence of H7. Following incubation, the cells were washed twice with PBS, fixed in methanol for 20 min at −20°C, and permeabilized with PBS containing 0.1% (v/v) Tween 20 (PBT). To block unspecific binding of antibodies, cells were incubated for at least 30 min with PBT containing 5% Fetal Calf Serum (FCS-PBT). Cells were incubated for at least two hours in primary antibody (rabbit polyclonal or mouse monoclonal, Zymed/Invitrogen, Carlsbad, CA) at a 1:100 dilution in blocking buffer. Excess antibody was removed by washing wells three times with PBT for 5 min. Secondary antibody tagged with fluoroscein isothiocyanate (FITC) or Alexa Fluor (Molecular Probes, Eugene, OR) in a 1:500 dilution with blocking buffer was added to the cells for at least 60 min at room temperature.
3) RESULTS
3.1) Cell communication assay
Using the scrape loading technique, green fluorescing Lucifer yellow can pass into coupled cells (Fig. 1A) but octanol prevented this spread (Fig 1B). While only the original scrape-loaded cells showed the fluorescing Texas Red (Fig. 2B, D, F), cells treated with H7 (10 μM) demonstrated a reduction in GJIC (Fig. 2C). Compared to control untreated cells, by 12 hrs post infection, GJIC was reduced in HSV-2 infected WB cells (Fig. 2E).
Figure 1.
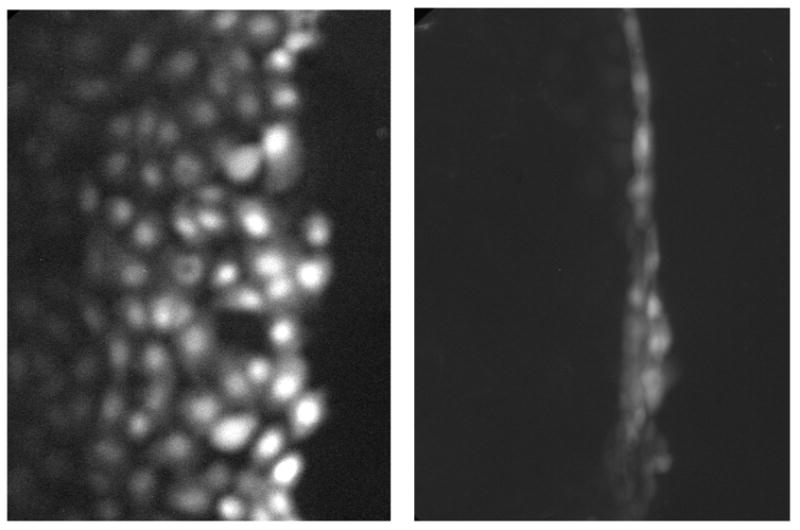
Octanol (1 mM) disrupted GJIC in WB-F344 cells. Lucifer yellow dye moved perpendicular to the vertical scrape line from initially loaded cells to contiguous cells in control (left) but not in octanol-treated (right) cells.
Figure 2.
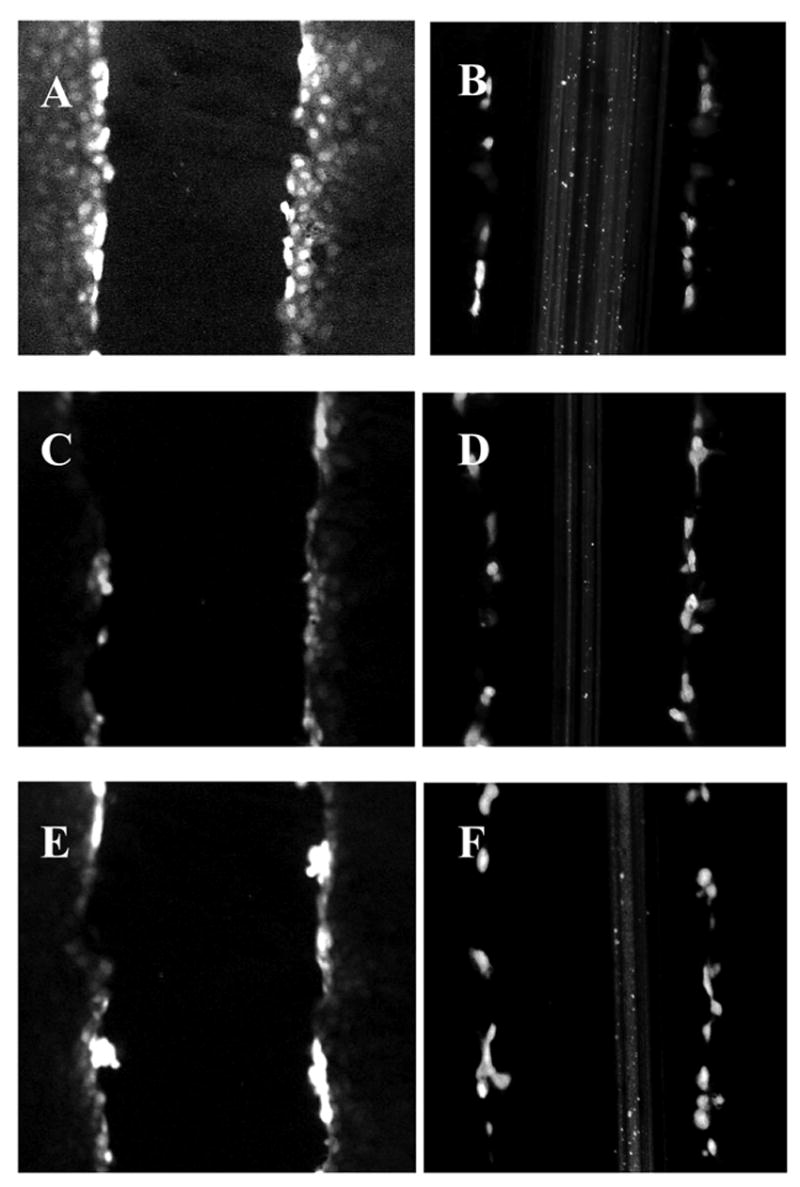
H7 and HSV-2 infection blocked GJIC in WB cells. Dye transfer in WB cells using Lucifer yellow (A, C, E) or Texas Red Dextran (B, D, F) was used to demonstrate functional gap junctions since Lucifer yellow passes into adjacent cells but Texas red is not transported to adjacent cells due to its high molecular weight. In control cells, Lucifer yellow penetrated gap junctions (A) but both H7 (24 hr treatment, C) and HSV-2 infection (12 hrs post-infection, E) prevented spread of Lucifer yellow.
3.2) Antiviral activity
Although octanol blocks GJIC in WB cells (Fig. 1B), treatment of HSV-2 infected WB cells with 1 mM octanol did not inhibit the production of virus in these cells (Fig. 3). 18β-glycyrrhetinic acid (18β-GA; 25 μM), which inhibits GJIC and induces the dephosphorylation of Cx43-P2 to Cx43-NP (Guan et al., 1996), did not confer HSV-2-resistance on WB cells (Fig. 3). Staurosporine, a protein kinase C inhibitor, did not demonstrate antiviral effects at non-toxic doses (Fig. 3). However, plaque formation was significantly reduced by 10 μM H7 with one hundred percent inhibition at 40 μM H7 (Fig. 4A). In addition, when H7 was added at various time points following infection, it continued to prevent plaque formation up to 12 hours post-infection (Fig. 4B). H7 began to exert cytotoxic effects at dosages greater than 60 μM (Fig 4C), above the dose for maximal antiviral activity.
Figure 3.
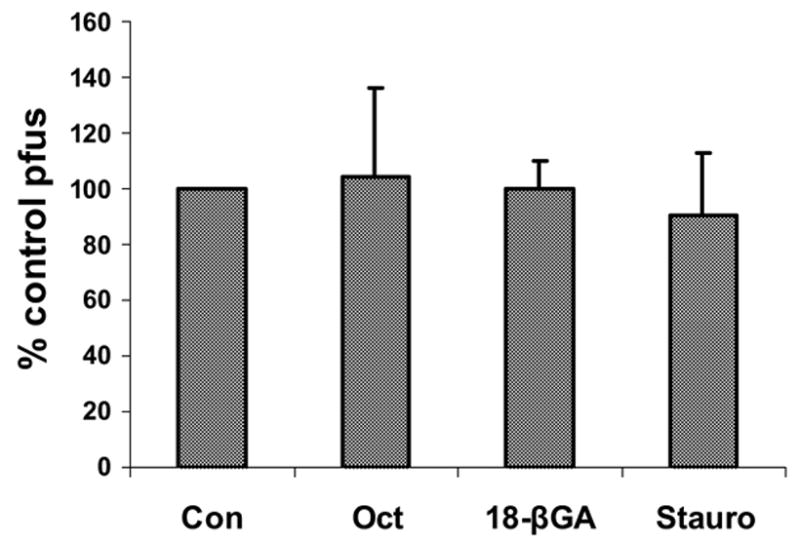
Agents that block gap junction communication, such as Octanol (Oct, 1 mM), 18β-glycyrrhetinic acid (18 βGA, 25 μM), and staurosporine (Stauro, 50 nM), did not prevent plaque forming unit ability of HSV-2 on WB cells at non-toxic doses.
Figure 4.
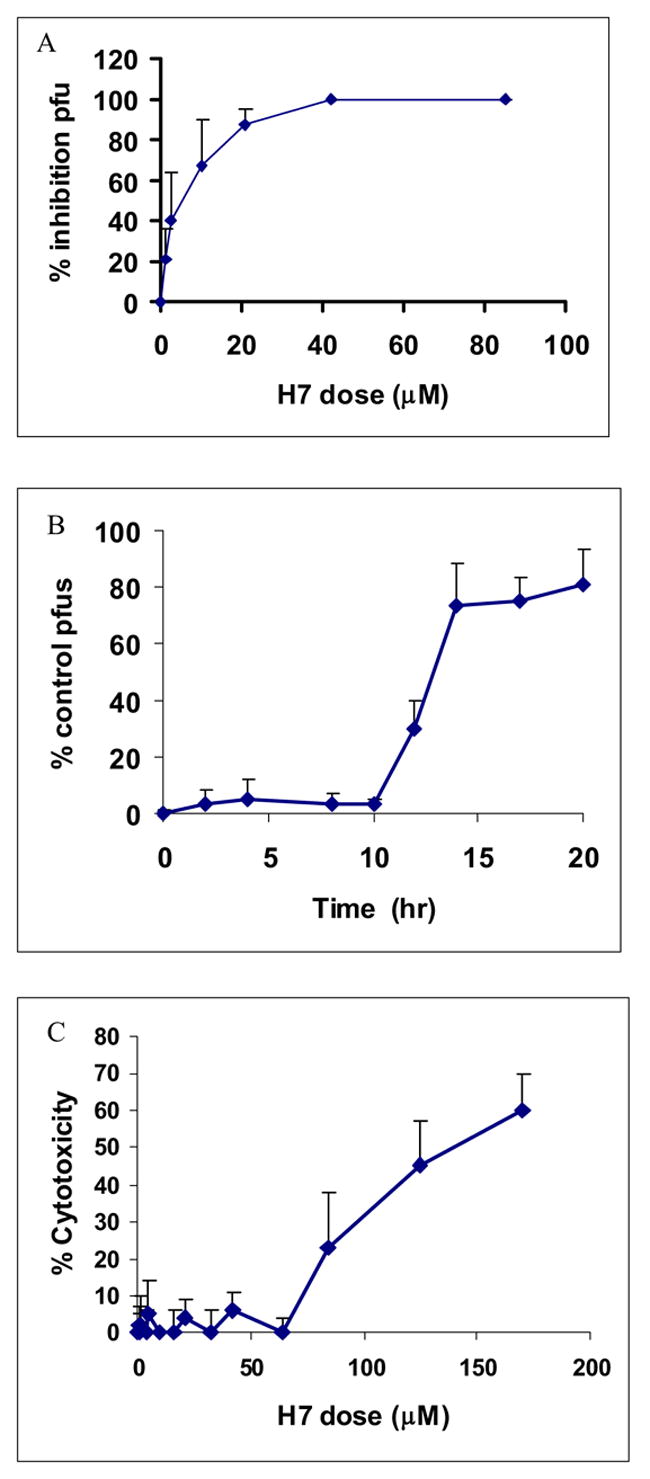
Plaque reduction following treatment of HSV-2 infected WB cells with varying doses of H7. A significant decrease in plaque forming unit (pfu) ability occurred at 10 μM H7 and increased to complete reduction at 40 μM (A). H7 (40 μM) continued to completely prevent pfu formation when added up to 12 hours post infection (B). Cytotoxic effects became evident only at doses greater than 60 μM (C).
3.3) Western blot analysis of Cx43
Western blot analysis was performed to determine whether H7 had any effect on Cx43 content or phophorylation status (Fig. 5). WB cells treated with 40 μM H7 for 24 hrs did not demonstrate any difference in Cx43 content or distribution of the phosphorylated version of Cx43 (P2 band) versus the unphosphoryated state (NP band) when compared to control cells.
Figure 5.

Effects of H-7 on Cx43 levels in WB-F344 cells. After treatment with 40 μM H7 for 24 hours, membrane enriched fractions were prepared and analyzed by Western blotting. The non-phosphorylated (NP) and phosphorylated (P2) forms of Cx43 are indicated with lane 1, control and lane 2, H-7 treated.
3.4) Immunofluorescent labeling
In control uninfected cells, gap junctions were present as plaques in the plasma membrane and the phosphoserine levels were low (Fig. 6A and B). In infected cells, membrane plaques were lost and there was some redistribution of Cx43 to the cytoplasmic compartment (Fig. 6C). Phosphoserine labeling was enhanced in infected cells, particularly in the nuclear region (Fig. 6D). In H7 treated cells that were infected with HSV-2, there was a loss of Cx43 from the membrane but the phosphoserine labeling in the nucleus was reduced compared to infected cells (Fig. 6 E and F).
Figure 6.
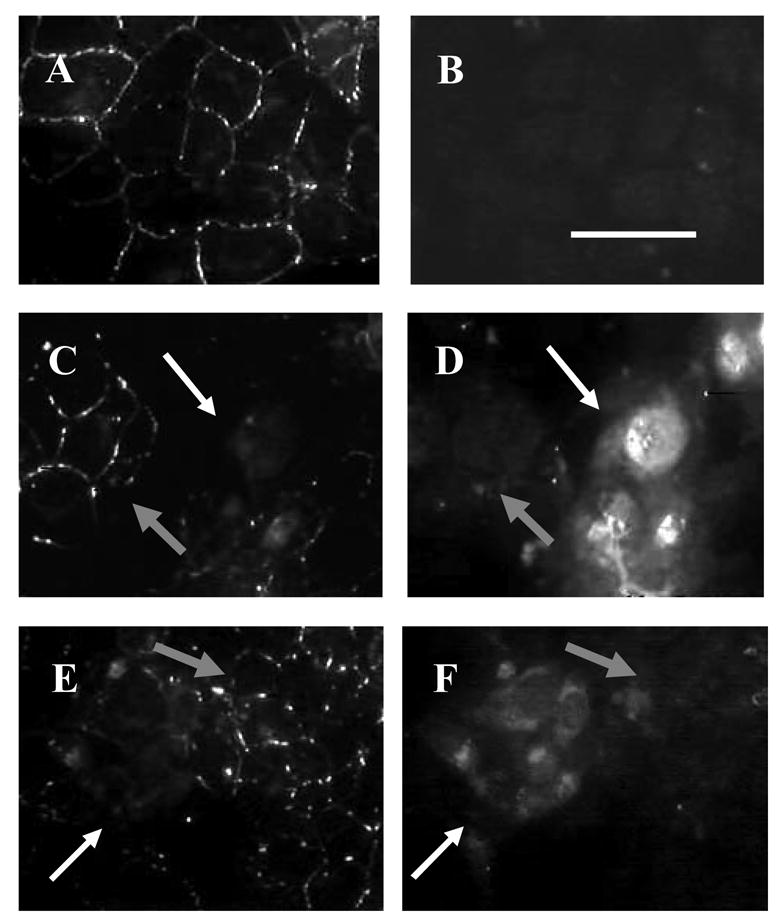
Fluorescent immunostaining of control, infected, and H-7 treated WB cells demonstrated decreased nuclear phosphorylation in infected cells treated with H-7. Cells were dual-labeled with Mouse Cx43 primary antibody with AlexaFluor 488 anti-mouse secondary antibody (A, C, E) and Rabbit phosphoserine primary antibody with AlexaFluor 546 anti-rabbit secondary antibody (B, D, F) in control (A, B), HSV-2 infected for 24 hours (C, D), and infected 20 μM H7 treated cells (E, F). White arrows in B–F identify infected areas while gray arrows identify uninfected areas in the same field. (40x, white bar = 50 μm)
4) DISCUSSION
In this study we have shown that the protein kinase inhibitor H7 demonstrated both disruption of gap junctional intercellular communication (GJIC) and antiviral activity in WB rat liver epithelial cells. However, this effect did not appear to be due to a change in the distribution of gap junctions in the membrane. The levels and distribution of phosphoserine were enhanced with viral infection, particularly in the nucleus, and H7 diminished this effect. No direct links between GJIC and the antiviral effects of H7 were identified.
Previous experiments by Fisher et al. (2000) and Musee et al. (2002) demonstrated that HSV-2 infection caused a significant decrease in electrical coupling in Vero cells by 3 hours post infection. In Musee et al.’s study, depending on their mechanism of action, various antiviral agents had differential effects on electrical coupling among infected cells. There was no direct correlation between prevention of electrical uncoupling that occurs with infection and antiviral activity.
In this study, we used WB rat liver epithelial cells rather than Vero cells due to the high expression of Cx43 in the WB cells (Spray et al., 1991) and tested the antiviral activities of four GJIC blockers, octanol, 18-β glycyrrhetinic acid, staurosporine, and H7. Octanol and other long chain alcohols rapidly close gap junction channels in a non-specific manner. It has been suggested that such aliphatic compounds dissolve in membrane lipids and induce localized changes in membrane fluidity that eventually result in the compression and closing of intercellular channels (Chanson et al., 1989; Cotrina et al., 1998; Guan et al., 1997). Non-specific blocking of gap junction channels with octanol had no observable effect on the ability of HSV-2 to mount an infection in WB cells.
Numerous studies show that phosphorylation plays a key role in connexin expression and function, as well as overall gap junction permeability (Cruciani and Mikalsen, 2002; Lampe and Lau, 2000; Lowenstein, 1985; Musil and Goodenough, 1991). For example, phosphorylation of Cx43 on Ser368 by protein kinase C (PKC) decreases gap junctional communication by reducing single channel conductance (Richards et al., 2004). In previous studies, staurosporine was used to block gap junctional communication through inhibition of protein kinase C (PKC) specific phosphorylation of Cx43 (Moreno et al, 1994; Saez et al, 1997). In addition, dephosphorylation of Cx43 by 18β-glycyrrhetinic acid (18β-GA) induced the disassembly of gap junction plaques in WB cells which was correlated to decreased levels of Cx43-P2 and increased levels of Cx43-NP (Davidson et al., 1986; Davidson & Baumgarten, 1988; Guan et al., 1996). In this study, neither staurosporine nor 18β-GA had an observable effect on the ability of HSV-2 to infect WB cells.
H7 has been previously shown to reduce GJIC in cardiac cells without a change in Cx43 phosphorylation (Duthe et al., 2000; Herve et al., 2004). These results are consistent with the present study in that H7 showed a reduction in GJIC without altering Cx43 phosphorylation, membrane content, or distribution of Cx43. However, since none of the other three down-regulators of GJIC reduced infectivity, GJIC down-regulation by H7 does not explain its observed antiviral effects.
Previous work has shown that protein kinase inhibitors reduce herpes virus infectivity. Yura et al. (1993, 1997) found that genistein, a tyrosine kinase inhibitor, reduced plaque forming ability in Vero cells and prevented phosphorylation of viral polypeptides, particularly ICP 6, 19, and 26 which were necessary for HSV-1 replication. Calphostin C, a protein kinase C inhibitor, reduced HSV-1 infectivity and virus yield in astrocytes without causing an alteration in viral protein synthesis (Castagnino et al., 1995). Constantinescu et al. (1991) found that protein kinase C inhibitors reduced infectivity of enveloped, but not non-enveloped viruses, presumably exerting their effects at the level of viral entry. The antiviral xanthate D609 prevented HSV-1 replication by inhibiting the viral US3 protein kinase, protein kinase C, and viral protein phosphorylation (Walro and Rosenthal, 1997). Conversely, pharmacological cyclin-dependent kinase inhibitors exert their antiviral effects on herpes viruses by blocking cellular rather than viral proteins (Schang et al., 2002). Clearly the phosphorylation of both viral and cellular proteins is important for virus replication and protein kinase inhibitors exert their anti-viral effects by preventing phosphorylation of cellular and/or viral proteins.
Even though H7 had no effect on Cx43 distribution, phosphoserine labeling was markedly elevated in infected cells, particularly in the nuclear region, and H7 reduced phosphoserine labeling in HSV-2 infected cells. Recent work (Muranyi et al. 2002; Park and Baines, 2006) demonstrated that herpesvirus infection led to recruitment of PKC to the nuclear membrane and subsequent phosphorylation of lamin B. Lamin B disassembly is associated with specific serine phosphorylation sites (Stuurman, 1997). This modification of the nuclear lamina occurred 10–12 hours post infection and served to promote budding of nucleocapsids at the inner nuclear membrane. In this study, H7 continued to exert antiviral activity when administered up to 12 hours post infection, supporting a mechanism where H7 exerts its antiviral activity through prevention of viral egress through the nuclear membrane. However, staurosporine, a potent PKC inhibitor, did not demonstrate the same antiviral effects as H7, suggesting that H7 attenuates nuclear phosphorylation through either a PKC- independent mechanism or a PKC-dependent mechanism that is unaffected by staurosporine.
This study suggests that the loss of GJIC early in viral infection does not confer protection to the infected cell or its neighbors since agents that attenuate GJIC did not reduce HSV-2 infectivity. Although H7 reduced both GJIC and viral infection, its antiviral mechanism appears to be independent of its gap junction inhibitory effects. Further studies will focus on the role of viral proteins on the down regulation of GJIC in infected cells.
Acknowledgments
This project was supported by a West Chester University College of Arts and Sciences undergraduate student research award to CAD and an NIH AREA grant (1 R15 AI055524-01) to Woodruff, Mbuy, Knabb. The authors would like to thank Blair DeFulvio and Radhika Bhatt for technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atkinson MM, Menko AS, Johnson RG, Sheppard JR, Sheridan JD. Rapid and reversible reduction of junctional permeability in cells infected with a temperature-sensitive mutant of avian sarcoma virus. J Cell Biol. 1981;91:573–578. doi: 10.1083/jcb.91.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissette JL, Kumar NK, Gilula NB, Dotto GP. The tumor promoter 12-o-tetradecanoylphorbol-13-acetate and the ras oncogene module expression and phosphorylation of gap junction proteins. Mol Cell Biol. 1991;11:5364–5371. doi: 10.1128/mcb.11.10.5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnino CG, Yakisich JS, Iacono RF, Berria MI, Idoyaga-Vargas V. Growth inhibition of herpes simplex virus-type 1 in calphostin C-treated astrocytes. Intervirology. 1995;38(6):332–338. doi: 10.1159/000150460. [DOI] [PubMed] [Google Scholar]
- Chanson M, Bruzzone R, Bosco D, Meda PJ. Effects of n-alcohols on junctional coupling and amylase secretion of pancreatic acinar cells. J Cell Physiol. 1989;139:147–156. doi: 10.1002/jcp.1041390121. [DOI] [PubMed] [Google Scholar]
- Constantinescu SN, Cernescu CD, Popescu LM. Effects of protein kinase C inhibitors on viral entry and infectivity. FEBS Lett. 1991;292(1–2):31–3. doi: 10.1016/0014-5793(91)80826-o. [DOI] [PubMed] [Google Scholar]
- Cotrina M, Lin JH, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, Kang J, Naus CCG, Nedergaard M. Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci USA. 1998;95:15735–15740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow DS, Beyer EC, Paul DL, Kobe SS, Lau AF. Phosphorylation of connexin43 gap junction protein in uninfected and Rous Sarcoma Virus-transformed mammalian fibroblasts. Mol Cell Biol. 1990;10:1754–1763. doi: 10.1128/mcb.10.4.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani V, Mikalsen SO. Connexins, gap junctional intercellular communication and kinases. Biol Cell. 2002;94:433–443. doi: 10.1016/s0248-4900(02)00014-x. [DOI] [PubMed] [Google Scholar]
- Danave RI, Tiffany-Castiglioni E, Zenger E, Barhoumi R, Burghardt RC, Collisson EW. Feline immunodeficiency virus decreases cell-cell communication and mitochondrial membrane potential. J Virol. 1994;68:6745–6750. doi: 10.1128/jvi.68.10.6745-6750.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM, Harley EH. Reversible inhibition of intercellular junctional communication by glycyrrhetinic acid. Biochem Biophys Res Commun. 1986;134(1):29–36. doi: 10.1016/0006-291x(86)90522-x. [DOI] [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM. Glycyrrhetinic acid derivatives: a novel class of inhibitors of gap-junctional intercellular communication. Structure-activity relationships. J Pharmacol Exp Ther. 1988;246(3):1104–7. [PubMed] [Google Scholar]
- De Mello WC. Increase in junctional conductance caused by isoproterenol in heart cell pairs is suppressed by cAMP-dependent protein-kinase inhibitor. Biochem Biophys Res Commun. 1988;154(2):509–14. doi: 10.1016/0006-291x(88)90169-6. [DOI] [PubMed] [Google Scholar]
- Duthe F, Dupont E, Verrecchia F, Plaisance I, Severs NJ, Sarrouilhe D, Herve JC. Dephosphorylation agents depress gap junctional communication between rat cardiac cells without modifying the connexin43 phosphorylation degree. Gen Physiol Biophys. 2000;19(4):441–449. [PubMed] [Google Scholar]
- el-Fouly MH, JE Trosko, CC Chang. Scrape-loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Exp Cell Res. 1987;168:422–430. doi: 10.1016/0014-4827(87)90014-0. [DOI] [PubMed] [Google Scholar]
- Ennaji MM, Schwartz JL, Mealing G, Belbaraka L, Parker C, Parentaux M, Jouishomme H, Arella M, Whitfield JF, Phipps J. Alterations in cell-cell communication in human papillomavirus type 16 (HPV16) transformed rat myoblasts. Cell Mol Biol. 1995;41:481–498. [PubMed] [Google Scholar]
- Esinduy CB, Chang CC, Trosko JE, Ruch RJ. In vitro growth inhibition of neoplastically transformed cells by non-transformed cells: requirement for gap junctional intercellular communication. Carcinogenesis. 1995;16(4):915–921. doi: 10.1093/carcin/16.4.915. [DOI] [PubMed] [Google Scholar]
- Faccini AM, Cairney M, Ashrafi BH, Finbow ME, Campo MS, Pitts JD. The bovine papillomavirus type E8 protein binds to ductin and causes loss of gap junctional intercellular communication in primary fibroblasts. J Virol. 1996;70:9041–9045. doi: 10.1128/jvi.70.12.9041-9045.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filson AJ, Azarnia R, Beyer EC, Loewenstein WR, Brugge JS. Tyrosine phosphorylation of a gap junction protein correlates with inhibition of cell-to-cell communication. Cell Growth Differ. 1990;1:661–668. [PubMed] [Google Scholar]
- Fischer NO, Mbuy GNK, Woodruff RI. HSV-2 disrupts gap junctional intercellular communication between mammalian cells in vitro. J Virol Methods. 2001;91:157–166. doi: 10.1016/s0166-0934(00)00260-3. [DOI] [PubMed] [Google Scholar]
- Fletcher WH, Shiu WW, Ishida TA, Haviland DL, Ware CF. Resistance to the cytolytic action of lymphotoxin and tumor necrosis factor coincides with the presence of gap junctions uniting target cells. J Immunol. 1987;139:956–962. [PubMed] [Google Scholar]
- Goldstein DJ, Finbow ME, Andersson T, McLean P, Smith K, Bubb V, Schlegel R. Bovine papillomavirus E5 oncoprotein binds to the 16K component of vacuolar H(+)-ATPases. Nature. 1991;352:347–349. doi: 10.1038/352347a0. [DOI] [PubMed] [Google Scholar]
- Guan X, Wilson S, Schlender KK, Ruch RJ. Gap-junction disassembly and connexin 43 dephosphorylation induced by 18β-glycyrrhetinic acid. Mol Carcinog. 1996;16:157–164. doi: 10.1002/(SICI)1098-2744(199607)16:3<157::AID-MC6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Guan X, Cravatt BF, Ehring GR, Hall JE, Boger DL, Lerner RA, Gilula NB. The sleep-inducing lipid oleamide deconvolutes gap junction communication and calcium wave transmission in glial cells. J Cell Biol. 1997;139(7):1785–92. doi: 10.1083/jcb.139.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herve JC, Plaisance I, Loncarek J, Duthe F, Sarrouilhe D. Is the junctional uncoupling elicited in rat ventricular myocytes by some dephosphorylation treatments due to changes in the phosphorylation status of Cx43? Eur Biophys J. 2004;33(3):201–210. doi: 10.1007/s00249-003-0381-0. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- Jou YS, Layhe B, Matesic DF, Chang CC, de Feijter AW, Lockwood L, Welsch CW, Klaunig JE, Trosko JE. Inhibition of gap junctional intercellular communication and malignant transformation of rat liver epithelial cells by neu oncogene. Carcinogenesis. 1995;16:311–317. doi: 10.1093/carcin/16.2.311. [DOI] [PubMed] [Google Scholar]
- Lampe PD, Lau AF. Regulation of gap junctions by phosphorylation of connexins. Arch Biochem Biophys. 2000;384:205–215. doi: 10.1006/abbi.2000.2131. [DOI] [PubMed] [Google Scholar]
- Lowenstein WR. Regulation of cell-to-cell communication by phosphorylation. Biochem Soc Symp. 1985;50:43–58. [PubMed] [Google Scholar]
- Moreno AP, Saez JC, Fishman GI, Spray DC. Human connexin43 gap junction channels. Regulation of unitary conductances by phosphorylation. Circ Res. 1994;74(6):1050–1057. doi: 10.1161/01.res.74.6.1050. [DOI] [PubMed] [Google Scholar]
- Musee J, Mbuy GNK, Woodruff RI. Antiviral agents alter ability of HSV-2 to disrupt gap junctional intercellular communication between mammalian cells in vitro. Antiviral Res. 2002;56:143–151. doi: 10.1016/s0166-3542(02)00106-7. [DOI] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science. 2002;297(5582):778–9. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- Opsahl H, Rivedal E. Quantitative determination of gap junction intercellular communication by scrape loading and image analysis. Cell Adhes Commun. 2000;7:367–375. doi: 10.3109/15419060009109019. [DOI] [PubMed] [Google Scholar]
- Park R, Baines JD. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol. 2006;80(1):494–504. doi: 10.1128/JVI.80.1.494-504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards TS, Dunn CA, Carter WG, Usui ML, Olerud JE, Lampe PD. Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J Cell Biol. 2004;167(3):555–562. doi: 10.1083/jcb.200404142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez JC, Nairn AC, Czernik AJ, Fishman GI, Spray DC, Hertzberg EL. Phosphorylation of connexin43 and the regulation of neonatal rat, cardiac myocyte gap junctions. J Mol Cell Cardiol. 1997;29(8):2131–45. doi: 10.1006/jmcc.1997.0447. [DOI] [PubMed] [Google Scholar]
- Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83(4):1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Schang LM, Bantly A, Knockaert M, Shaheen F, Meijer L, Malim MH, Gray NS, Schaffer PA. Pharmacological Cyclin-Dependent Kinase Inhibitors Inhibit Replication of Wild-Type and Drug-Resistant Strains of Herpes Simplex Virus and Human Immunodeficiency Virus Type 1 by Targeting Cellular, Not Viral, Proteins. J Virol. 2002;76(15):7874–7882. doi: 10.1128/JVI.76.15.7874-7882.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spray DC, Chanson M, Moreno AP, Dermietzel R, Meda P. Distinctive gap junction channel types connect WB cells, a clonal cell line derived from rat liver. Am J Physiol. 1991;260(3 Pt 1):C513–527. doi: 10.1152/ajpcell.1991.260.3.C513. [DOI] [PubMed] [Google Scholar]
- Stuurman N. Identification of a conserved phosphorylation site modulating nuclear lamin polymerization. FEBS Lett. 1997;401(2–3):171–4. doi: 10.1016/s0014-5793(96)01464-0. [DOI] [PubMed] [Google Scholar]
- Thomas MA, Huang S, Cokoja A, Riccio O, Staub O, Suter S, Chanson M. Interaction of connexins with protein partners in the control of channel turnover and gating. Biol Cell. 2002;94:445–456. doi: 10.1016/s0248-4900(02)00015-1. [DOI] [PubMed] [Google Scholar]
- Walro DG, Rosenthal KS. The antiviral xanthate compound D609 inhibits herpes simplex virus type 1 replication and protein phosphorylation. Antiviral Res. 1997;36:63–72. doi: 10.1016/s0166-3542(97)00040-5. [DOI] [PubMed] [Google Scholar]
- Warn-Cramer BJ, Lau AF. Regulation of gap junctions by tyrosine protein kinases. Biochim Biophys Acta-Biomembranes. 2004;1662:81–96. doi: 10.1016/j.bbamem.2003.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yura Y, Yoshida H, Sato M. Inhibition of herpes simplex virus replication by genistein, an inhibitor of protein-tyrosine kinase. Arch Virol. 1993;132:451–461. doi: 10.1007/BF01309554. [DOI] [PubMed] [Google Scholar]
- Yura Y, Kusaka J, Tsujimoto H, Yoshioka Y, Yoshida H, Sato M. Effects of protein tyrosine kinase inhibitors on the replication of herpes simplex virus and the phosphorylation of viral proteins. Intervirology. 1997;40:7–14. doi: 10.1159/000150515. [DOI] [PubMed] [Google Scholar]