

Abstract
Complementary interacting molecules on myelin and axons are required for long-term axon-myelin stability. Their disruption results in axon degeneration, contributing to the pathogenesis of demyelinating diseases. Myelin-associated glycoprotein (MAG), a minor constituent of central and peripheral nervous system myelin, is a member of the Siglec family of sialic acid-binding lectins, and binds to gangliosides GD1a and GT1b, prominent molecules on the axon surface. Mice lacking the ganglioside biosynthetic gene Galgt1 fail to express complex gangliosides, including GD1a and GT1b. In the current studies, CNS and PNS histopathology and behavior of Mag-null, Galgt1-null and double-null mice were compared on the same mouse strain background. When backcrossed to >99% C57BL/6 strain purity, Mag-null mice demonstrated marked CNS, as well as PNS, axon degeneration, in contrast to prior findings using mice of mixed strain background. On the same background, Mag- and Galgt1-null mice exhibited quantitatively and qualitatively similar CNS and PNS axon degeneration, and nearly identical decreases in axon diameter and neurofilament spacing. Double-null mice had qualitatively similar changes. Consistent with these findings, Mag-, Galgt1-null mice had similar motor behavioral deficits, with double-null mice only modestly more impaired. Despite their motor deficits, Mag- and Galgt1-null mice demonstrated significant hyperactivity, with spontaneous locomotor activity significantly above that of wild type mice. These data demonstrate that MAG and complex gangliosides contribute to axon stability in both the CNS and PNS. Similar, neuropathological and behavioral deficits in Galgt1-, Mag-, and double-null mice support the hypothesis that MAG binding to gangliosides contributes to long-term axon-myelin stability.
Keywords: MAG, axon degeneration, axon diameter, neurofilament spacing, motor deficit, hyperactivity
Introduction
Axon degeneration secondary to dysmyelination is responsible for long-term pathogenesis in demyelinating disorders, such as multiple sclerosis and Charcot-Marie-Tooth disease (Bjartmar et al., 1999; Bjartmar et al., 2003; Coleman and Perry, 2002; Wujek et al., 2002; Bjartmar et al., 2000; Bruck and Stadelmann, 2003; Krajewski et al., 2000; Frei et al., 1999). The relationship between myelination and axon survival is supported by studies of mice lacking certain myelin proteins, in which progressive axonal degeneration is a major phenotype (Bjartmar et al., 1999; Frei et al., 1999). Recognition molecules on the innermost myelin sheath bind to complementary ligands on the apposing axon surface to initiate signaling cascades that control axon physiology (Edgar and Garbern, 2004).
Knowledge of the molecules underlying axon-myelin interactions may reveal insights into axonal degeneration in demyelinating diseases. Among these molecules is myelin-associated glycoprotein (MAG), a minor constituent found on periaxonal myelin in the CNS and PNS (Trapp et al., 1989; Trapp, 1990). Although Mag-null mice myelinate axons, older mice exhibit perturbations in axon integrity (Fruttiger et al., 1995; Weiss et al., 2001). In addition to mild deficits in myelination, Mag-null mice exhibit late-onset progressive PNS axonal atrophy and increased Wallerian degeneration (Bjartmar et al., 1999). MAG also modulates the axonal cytoskeleton. Myelinated axons of Mag-null mice have smaller diameters and fail to exhibit myelin-regulated neurofilament phosphorylation and increased neurofilament spacing, as do wild type mice (Yin et al., 1998). Additionally, MAG, on residual myelin, inhibits axon outgrowth from adult nerve cells after injury (Mukhopadhyay et al., 1994; McKerracher et al., 1994; Filbin, 2003; Yiu and He, 2003; Sandvig et al., 2004). Thus, MAG is required for long-term axon stability, controls axon cytoarchitecture, and regulates axon outgrowth. These effects may be mediated by multiple complementary MAG ligands on the axon, including the Nogo receptor, NgR (Liu et al., 2002; Domeniconi et al., 2002), and gangliosides, the major glycans on nerve cells and axons (Schnaar, 2000) that are the focus of the current studies.
MAG is a member of the Siglec family of carbohydrate binding proteins (Crocker, 2002). Siglecs bind to cell surface glycans that carry a terminal sialic acid residue. We previously identified potential MAG ligands as gangliosides GD1a and GT1b (Fig. 1). These two gangliosides, major sialic acid-containing glycans on nerve cells and axons, bind to MAG in vitro (Yang et al., 1996; Collins et al., 1997b; Collins et al., 1997a). Mice engineered to lack the MAG binding glycan determinant on GD1a and GT1b (Galgt1-null mice, see Fig. 1) exhibit progressive axon degeneration and other sequelae similar to Mag-null mice (Sheikh et al., 1999; Chiavegatto et al., 2000).
Fig. 1.
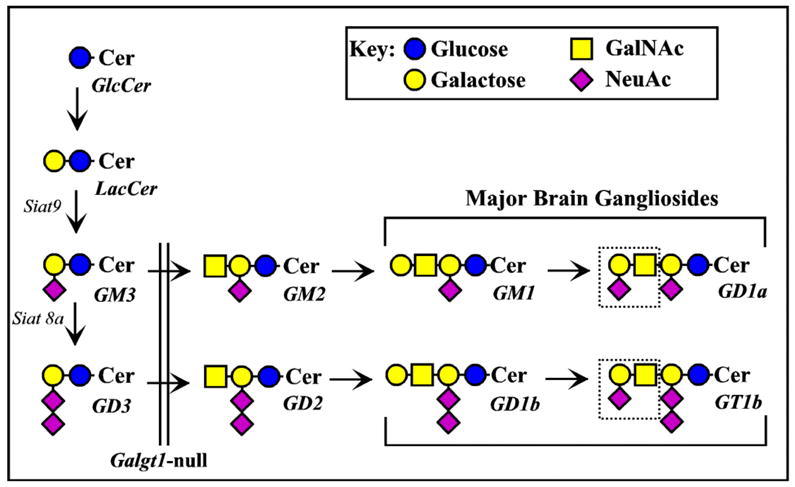
Biosynthesis of major brain gangliosides. The relationships between major brain gangliosides and their precursors are shown schematically, along with the ganglioside nomenclature of Svennerholm (Svennerholm, 1994). The block in ganglioside biosynthesis due to disruption of the Galgt1 gene (Liu et al., 1999) is indicated by a vertical double line (the genes responsible for synthesis of GM3 and GD3, Siat9 and Siat8a, respectively, are also indicated). MAG ligands GD1a and GT1b appear at the right, with the key MAG-binding determinant (NeuAc α2–3 Gal β1–3 GalNAc) indicated with a dotted line.
We now report results from a genetic approach that compares the functional relationship between MAG and gangliosides in vivo. We engineered Mag-null, Galgt1-null and double-null mice on the same mouse strain background (C57BL/6). Quantitative analysis of neuropathic and behavioral changes reveal remarkable similarities in the phenotypes of the three mouse strains.
Materials and Methods
Animals
Mag-null founder mice, kindly provided by Dr. Bruce Trapp, The Cleveland Clinic Foundation, Cleveland, OH, were constructed by disruption of exon 5 of the Mag gene as previously reported (Li et al., 1994). The strain provided (identical to that available from The Jackson Laboratory, Bar Harbor, ME) is a mixture of C57BL/6 and 129 inbred strains and the CD1 random bred strain. Mice with a disrupted Galgt1 gene, which lack the “NeuAc α2-3 Gal β1-3 GalNAc” terminus on gangliosides (Fig. 1), were constructed as described previously (Liu et al., 1999). Founder mice were kindly provided by Dr. Richard Proia, National Institutes of Health, Bethesda, MD. The strain provided was a mixture of C57BL/6 and 129 inbred strains.
To enhance comparisons between mutant strains, mutant mice were repeatedly back-crossed onto a C57BL/6 background. A PCR-based 80-loci full genome screen revealed 99.5% strain purity of the Mag-null mice and 94% strain purity of the Galgt1-null mice (personal communication, Peter Sobieszczuk, Ph.D., Director, Transgenics Core, Consortium for Functional Glycomics, The Scripps Research Institute, La Jolla, CA). The two strains were crossed to generate double-null offspring. Both back-crossed single-null strains were deposited with the Consortium for Functional Glycomics (www.functionalglycomics.org).
Mice were genotyped at the Galgt1 locus using PCR with tail-tissue DNA as template as described previously (Sun et al., 2004). The Mag locus was similarly genotyped using TGCCGCTGTTTTGGATAA and CGCCTCGGAAATAGTATTTG as the forward and reverse primers, resulting in products of 0.6 and 2 kb for wild type and disrupted alleles respectively.
At the time of tissue harvest for histopathology or behavioral studies, mice averaged approximately 6 months old (ages of each genotype are provided in the figure legends). Most mice used were males; no significant differences between males and females were observed.
Histopathology
Animals from each of Mag-null, Galgt1-null, double null and wild type C57BL/6 mice were perfused with 4% paraformaldehyde and 1.5% glutaraldehyde in Sorenson's buffer (Morris et al., 1980). Sciatic nerves and lumbosacral spinal cord were harvested from all animals. Tissues were trimmed and postfixed in osmium and embedded in Epon. Thick sections (1 μm) were stained with toluidine blue for light microscopy, and thin sections (80 nm) were examined by electron microscopy.
Axon degeneration
For evaluation of Wallerian degeneration in the PNS, the number of degenerating myelinated nerve fibers per total nerve transverse section was determined using 1-μm-thick Epon cross sections of sciatic nerve. For evaluation of nerve degeneration in the CNS, 1-μm-thick Epon transverse sections of the cervical spinal cord were used. Degenerating fibers were determined in the dorsal and ventral funiculus and the pathological features were confirmed on thin sections by electron microscopy.
Axon caliber
Sciatic nerves were analyzed at light and ultrastructural levels, respectively. Thick sections (1 μm) of sciatic nerve were sampled and nerve fiber counts were obtained at light level by standard stereological methods as described previously (Sheikh et al., 1999; Mayhew and Sharma, 1984; Mayhew, 1988). The mean axonal caliber of myelinated fibers in sciatic nerves was calculated from the diameter of a circle with an area equivalent to that of each axon.
Neurofilament Spacing
Electron micrographic images of myelinated axons of sciatic nerves at a final magnification of 100,000X were used to determine neurofilament spacing. Nearest-neighbor distance was used as a measure of neurofilament spacing in myelinated axons in sciatic nerve as described previously (Yin et al., 1998; Xu et al., 1996). The mean neurofilament nearest-neighbor distance for 15 or more axons from three animals in each group was compared.
Behavior
Mice were individually housed for one week prior to behavioral experiments and were maintained in a 12 h light-dark cycle in a temperature- and humidity-controlled room. The mice were allowed free access to food and water. Mice of each genotype (Mag-null, Galgt1-null, double-null and wild type C57BL/6) were used for behavioral testing. Spontaneous locomotor activity was tested for a 24-h period starting at 1700 h. All other behavioral tests were conducted between 1300–1800 h. Mice were allowed to acclimate in the testing room for 5 h prior to initiating experiments. All mice were tested on the same day for each behavioral parameter. Care and handling procedures were in accordance with institutional and government guidelines.
Motor coordination and balance
A rotarod apparatus (Economex, Columbus Instruments, Columbus, OH) was used in fixed speed mode. Each mouse was placed in a separate lane of the apparatus on a rotating cylinder (5 cm diameter) at 10 rpm (for up to 10 min) or 5 rpm (for up to 5 min). Their performance was recorded as the mean latency to fall from the cylinder in 3 consecutive trials with a resting period between each trial.
Hindlimb reflex extension
Each mouse was suspended by the tail for 10 s in three consecutive trials. The position of the hindlimbs was scored as follows: 0, one or both hindlimbs paralyzed; 1, loss of reflex and hindlimbs and paws held close to the body with clasping toes; 2, loss of reflex with flexion of hindlimbs; 3, hindlimbs extended to form <90° angle; 4, hindlimbs extended to form ≥90° angle.
Beam balance and platform
Each mouse was placed at the center and perpendicular to a rounded wooden beam (60-cm length, 2-cm diameter) suspended 60 cm above a foam pillow. The mean latency to fall (up to 120 s) was registered in three consecutive trials. Subsequently, each mouse was placed at the center of the same wooden beam set as a bridge, and facing one of two platforms (15 x 15 cm) set at either end of the bridge. The mean latency to reach either platform with four paws (up to 60 s) was registered in three consecutive trials. In each trial the mouse faced alternate platforms and a score of 60 s was given to the animal that failed to reach the platform or fell during the test.
Forelimb grip strength
Each mouse was suspended by its forepaws on a rounded metal bar (diameter 3 mm) positioned 60 cm above a foam pillow. The forelimb grip strength was assessed by measuring the mean time (up to 30 s) the mice hung from the bar in three consecutive trials.
Tremor and Catalepsy
Each animal was scored for the presence or absence of whole body tremor (resting or during movement) in an observational plastic cage with acrylic cover in three consecutive observations of 10 s interspersed with an interval of 10 s. The scoring began 1 min after being placed in the test cage. To test catalepsy, each mouse was placed with its hindlimbs on a flat surface and its forelimbs on a horizontal metal bar suspended 5 cm above the surface. The immobility state (time that the animal spent motionless hanging from the bar) was recorded in three consecutive trials. If an animal fell or climbed the bar, it was placed immediately on the bar again; the test was terminated after a maximum of three such escapes.
Spontaneous locomotor activity
Locomotor activity was quantified by placing mice into individual automated Plexiglas boxes (29 x 50 cm), each with 12 2-cm high infrared beam detectors arranged in a 4 x 8 grid (San Diego Instruments; San Diego, CA). Computer-recorded beam breaks were accumulated every 20 min for the duration of a 24-h test period starting at 1700 h, with changes in beam status assessed 18 times/s. Animals were habituated to test cages for at least 5 h prior to data collection, and had access to food and water ad libitum throughout the experiments.
Results
Histopathology in Mag-null, Galgt1-null, and double-null mice
Axon degeneration
In both the CNS and the PNS, axon degeneration was the predominant pathological feature in mice lacking expression of MAG or complex gangliosides (Figs. 2–4). In comparison to wild type mice, in which essentially no degenerating fibers were seen, single- and double-null mice had a highly significant occurrence of degeneration of myelinated axons, including myelin figures and ovoids representing various stages of degeneration (Figs. 2 & 3). Quantification revealed highly significant axon degeneration in the PNS (sciatic nerve, Figs. 2 & 4) of all mutant strains compared to wild type mice. Galgt1-null and Mag-null mice displayed similar PNS axon degeneration, whereas double-null mice displayed significantly greater PNS axon degeneration than did either single-null strain. In the CNS (spinal dorsal and ventral columns, Figs. 3 & 4), all mutant strains displayed highly significant axon degeneration compared to wild type mice, with no significant differences among mutant strains. The discovery of axon degeneration in the CNS is in contrast to prior studies of Mag-null mice when studied on a mixed-strain background (see Discussion).
Fig. 2.
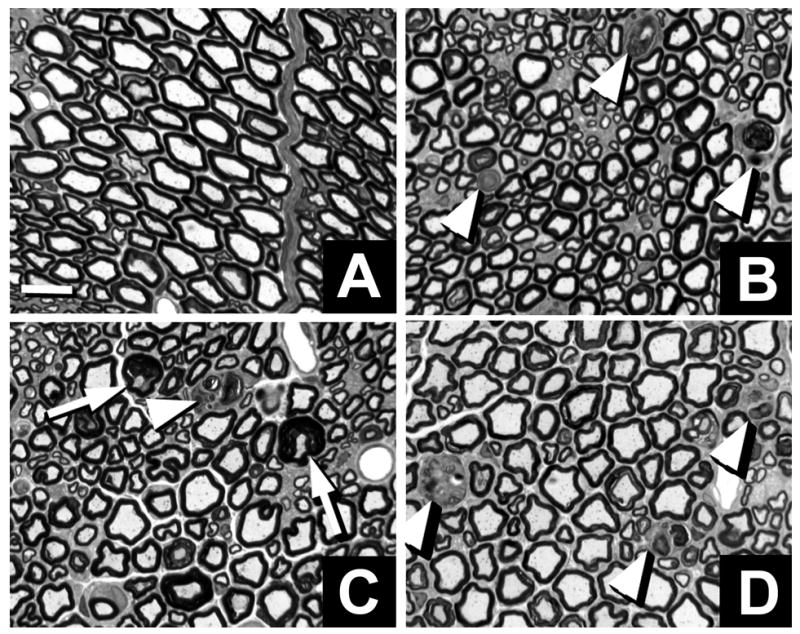
Neuropathology in sciatic nerve axons of Mag-, Galgt1- and double-null mice. Light microscopic toluidine blue-stained 1-μm Epon cross sections of sciatic nerve. Myelinated fibers undergoing axonal degeneration (arrowheads) and tomacula (arrows) in Galgt1-null (B), Mag-null (C) and double-null (D) nerves compared with wild type nerves (A). Bar = 10 μm.
Fig. 4.
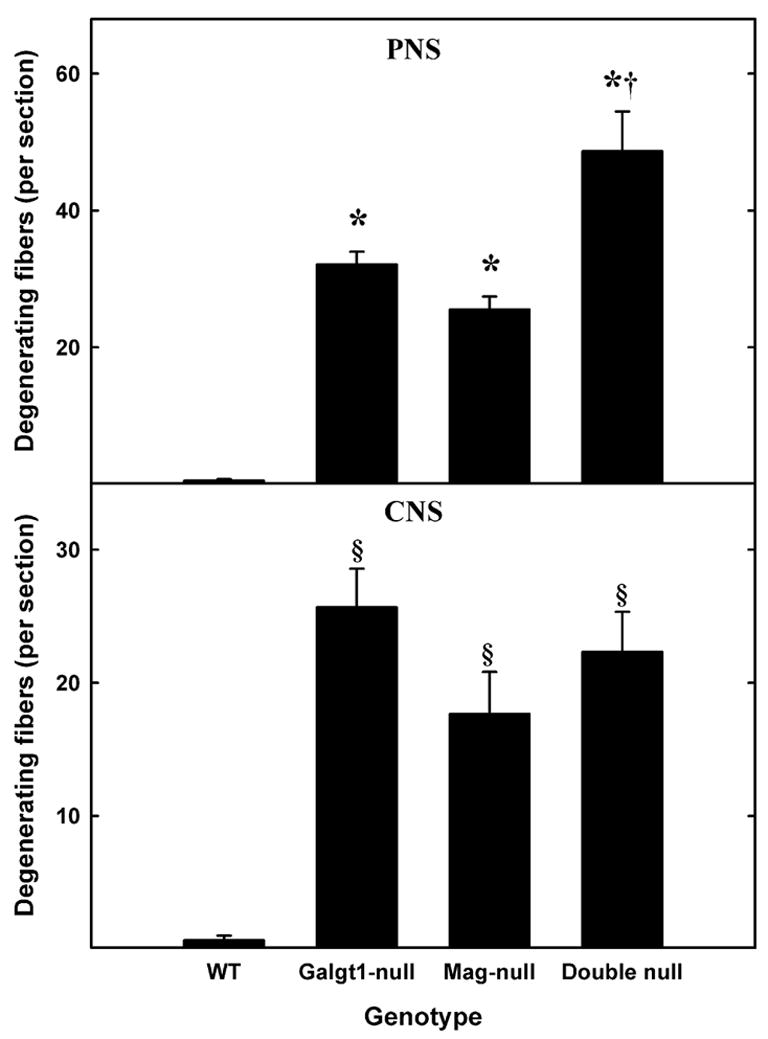
Increased axon degeneration in Mag-, Galgt1- and double-null mice. Sciatic nerve (PNS, top panel) and spinal dorsal column (CNS, bottom panel) cross sections were viewed for evidence of degenerating axons in the following mice (number, average age): Mag-null (n=6, 6.3 months), Galgt1-null (n=5, 7.4 months), double-null (n=6, 7.9 months), and wild type C57BL/6 (n=6, 6 months). Data are presented as means ± SE. One-way ANOVA for the number of degenerating axons revealed highly significant differences based on genotype (p < 0.01) for both PNS and CNS. Post-hoc analysis (Fisher’s PLSD) revealed significant increases (*, p < 0.0001) in the number of degenerating PNS axons in Galgt1-, Mag- and double-null mice compared to wild type mice, and significantly more degenerating axons in the double-null mice compared to the single-null mice (†, p < 0.01). Post-hoc analysis of the CNS data indicated highly significant increases in degenerating axons (§, p < 0.01) in all mutant strains compared to wild type, but no difference among the mutant strains.
Fig. 3.
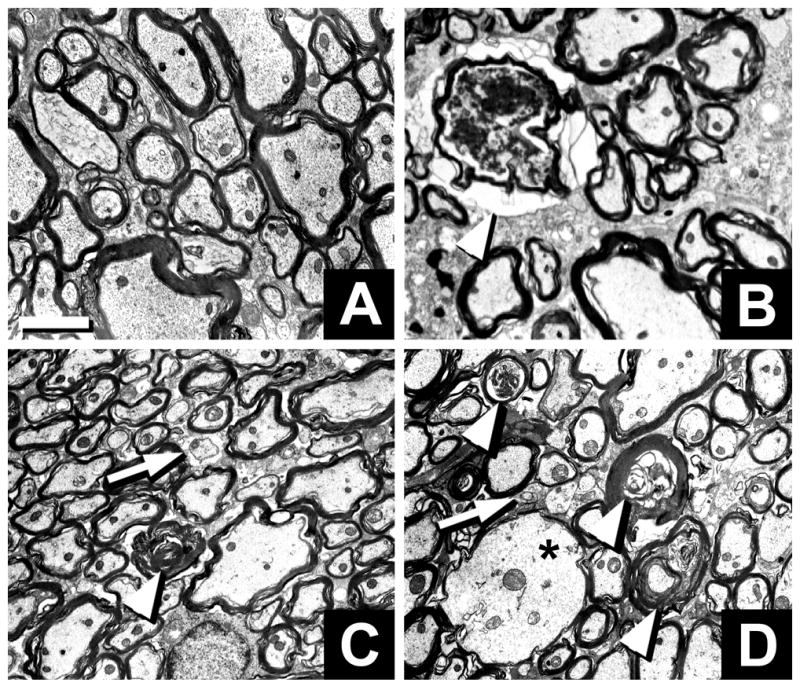
Neuropathology in cervical spinal cords of Mag-, Galgt1- and double-null mice. Low magnification electron microscopic images of transverse sections of the dorsal funiculus of wild type (A), Galgt1-null (B), Mag-null (C), and double-null (D) mice. Wallerian degeneration (arrowheads), dysmyelinated axons (arrows) and axon swelling (asterisk) are noted. Bar = 2 μm.
Axon caliber and neurofilament spacing
Axon calibers were determined from cross sections of sciatic nerves of wild type and mutant mice (Fig. 5). Histograms reveal a distinct shift to smaller axon calibers in all mutant mice compared to wild type, with Mag-null, Galgt1-null and double null histograms nearly overlapping. The number of axons ≥5 μm diameter was significantly reduced (to about half that in wild type mice) in all of the mutant strains, whereas axons ≤2.5 μm diameter were about double those in wild type (Fig. 5). There were no significant differences among the three mutant strains.
Fig. 5.
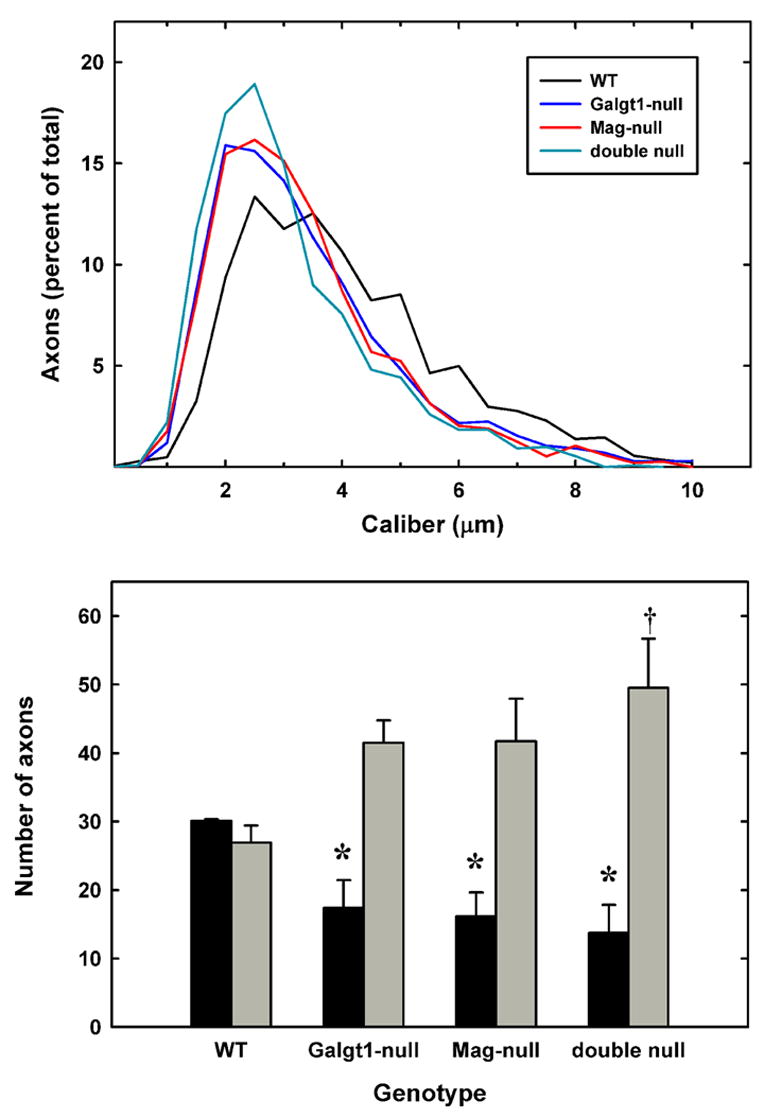
Decreased axon caliber in sciatic nerves of Mag-, Galgt1- and double-null mice. Micrographs of sciatic nerve cross sections were subjected to image analysis to determine the diameters of myelinated axons from the following mice (average age): Mag-null (6.2 months) Galgt1-null (7.4 months), double-null (6.3 months), and wild type (6 months). Diameters of ~500 axons each from 3 mice of each genotype were determined. Top panel: Histograms of pooled axon diameters for each genotype. Bottom panel: Combined data representing the percent of axons ≥5 μm diameter (black bars) and ≤2.5 μm diameter (gray bars). The data are presented as mean ± SE (n = 3). One-way ANOVA for the number of axons >5 μm diameter revealed a significant difference based on genotype (p < 0.05). Post-hoc analysis (Fisher’s PLSD) revealed significant decreases (*) in axon caliber for Galgt1-null mice (p < 0.05), Mag-null mice (p < 0.05) and double-null mice (p < 0.01) compared to wild type mice. AlthoughGalgt1-null and Mag-null mice demonstrated a trend toward greater numbers of axons ≤2.5 μm diameter (p = 0.08), only double-null mice expressed significantly more of these smaller myelinated axons (†, p < 0.05).
Axon caliber is dependent, in part, on the spacing of axon neurofilaments. Electron micrographs of sciatic nerve cross sections revealed closer neurofilament spacing in Mag-null, Galgt1-null and double-null mice compared to wild type mice (Fig. 6). Quantitative analysis revealed a significant 25% reduction in neurofilament spacing in all mutant strains compared to wild type mice, with no significant difference among the mutant strains.
Fig. 6.
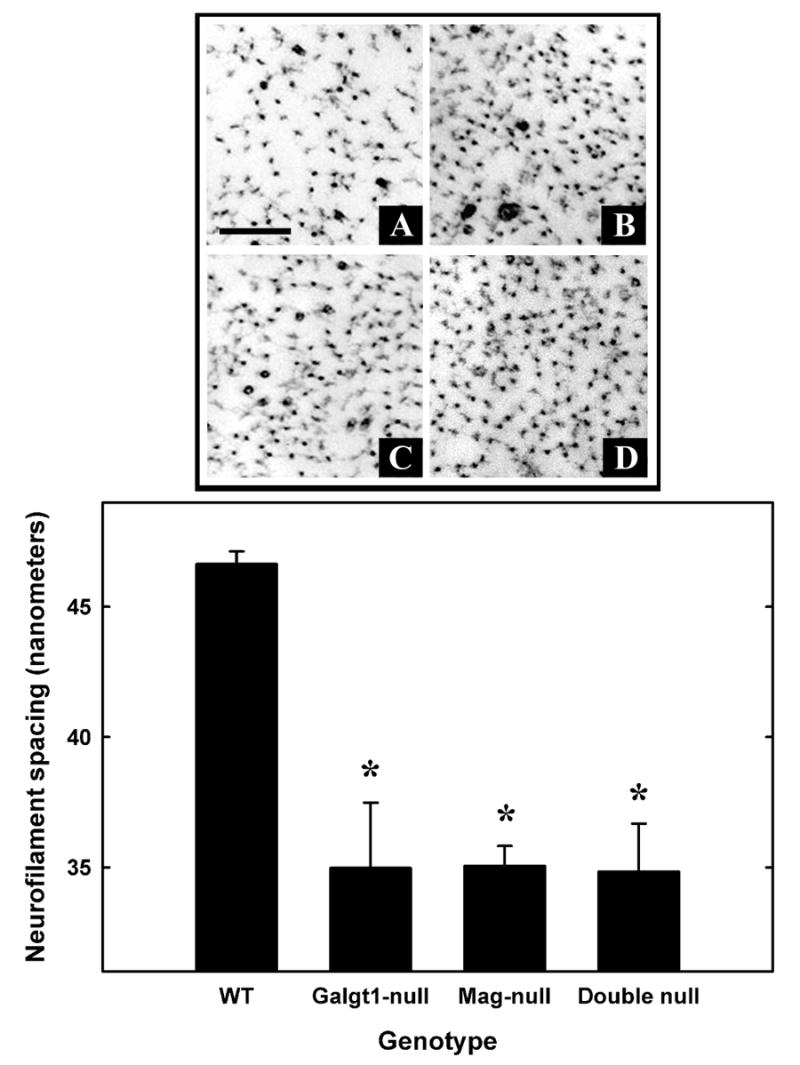
Decreased neurofilament spacing in sciatic nerve axons of Mag-, Galgt1- and double-null mice. Top panel: Electron micrographic images of cross sections of sciatic nerves, showing individual neurofilaments and their spacing, in wild type (A), Galgt1-null (B), Mag-null (C), and double-null (D) mice. Bar = 0.2 μm. Bottom panel: Nearest neighbor spacing was determined for ≥15 axons from each of three mice of each genotype (age): Mag-null (6.2 months), Galgt1-null (7.4 months), double-null (6.3 months), and wild type C57BL/6 (6 months). Values are presented as mean ± SE. One-way ANOVA for neurofilament spacing was highly significant (p < 0.01). Post-hoc analysis (Fisher’s PLSD) revealed that neurofilaments of Galgt1-, Mag- and double-null mice had significantly reduced spacing compared to wild type mice (*, p ≤ 0.001).
Dysmyelination
Dysmyelination was seen in both PNS and CNS of all mutant mouse strains (Figs. 2 & 3), similar to the changes documented previously in Galgt1-null and Mag-null single-null mice (Sheikh et al., 1999; Montag et al., 1994; Li et al., 1994). These changes appeared to be more prominent in the double null animals.
Behavior of Mag-null, Galgt1-null, and double-null mice
Motor behavior, reflexes, and tremor
Consistent with their histopathology, mutant mice displayed motor and behavioral deficits compared to their wild type counterparts. Rotarod testing, in which mice are timed for their ability to remain on a rotating cylinder, is a combined measure of motor balance, coordination, and muscle control (Kuhn et al., 1995). Compared to wild type mice, all mutant strains were less capable of remaining on a cylinder rotating at 10 rpm, with double-null mice significantly more impaired than Mag-null or Galgt1-null mice (Fig. 7). This differential in motor coordination was even more apparent when the cylinder was rotating at 5 rpm, a speed at which only double-null mice were significantly impaired compared to wild type mice.
Fig. 7.
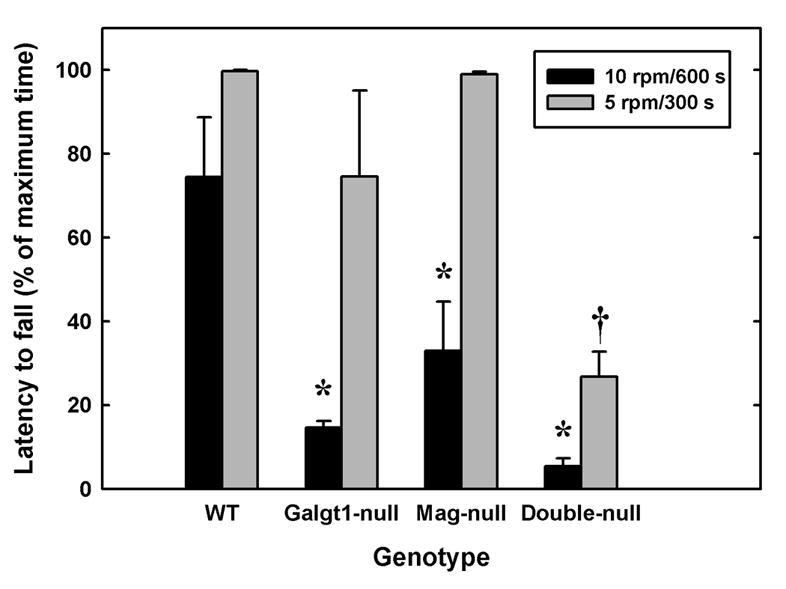
Impaired motor coordination and balance in Mag-null, Galgt1-null and double-null mice. Motor coordination was evaluated by measuring the latency to fall using a rotarod apparatus (5 cm diameter drum) at two fixed speeds, 10 rpm (black bars, maximum 600s), and 5 rpm (gray bars, maximum 300 s). Data are presented as mean ± SE. Mice tested included (number, average age): Mag-null (n=5, 5.4 months); Galgt1-null (n=3, 6.9 months); double-null (n=5, 4.7 months); and wild type C57BL/6 (n=4, 6 months). At 10 rpm, one-way ANOVA for latency to fall was highly significant (p < 0.001). Post-hoc analysis (Fisher’s PLSD) revealed that Galgt1-null mice (p < 0.01), Mag-null mice (p < 0.01) and double-null mice (p < 0.001) were significantly impaired (*) compared to wild type mice, and that double-null mice were significantly impaired (p < 0.05) compared to Mag-null mice. Under less stringent conditions (5 rpm) one-way ANOVA for latency to fall was also highly significant (p < 0.001), although post-hoc analysis revealed that only double-null mice were significantly impaired in comparison to each of the other genotypes (†, p ≤ 0.001).
Hindlimb reflex extension was also clearly impaired in all mutant mouse strains (Fig. 8). When a wild type mouse is gently lifted by the tail, it typically extends its hindlimbs outward in a steady 90–120° angle. By comparison, mutant mice tended to retract their hindlimbs, holding them close to the body, often with inversion and flexion of the paws (Chiavegatto et al., 2000). As with motor coordination, double-null mice had a more severely impaired reflex response than did Mag-null or Galgt1-null mice.
Fig. 8.
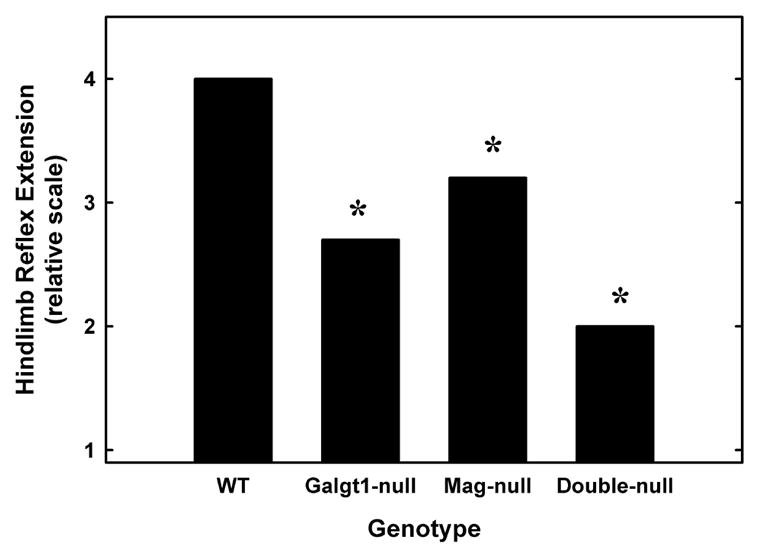
Impaired hindlimb reflexes in Mag-null, Galgt1-null and double-null mice. Wild type mice typically hold their hindlimbs extended steadily at an angle of ≥90° when lifted by the tail. The position of the hindlimbs of wild type and mutant mice were scored on a scale from 0 (most impaired, no reflex) to 4 (normal). The best performance in three trials was averaged for 3–5 mice of each genotype. Data were analyzed using the Kruskal-Wallis nonparametric test and are shown as means for clarity. There was a significant effect of genotype (*, p < 0.01) with all mutant strains impaired compared to wild type mice. Ages and numbers of mice used were as detailed in Fig. 7, legend. [Note: non-parametric statistical analysis was used, precluding the use of error bars.]
A marked whole body tremor was recorded in double-null mice (Fig. 9). Wild type mice showed no signs of tremor, whereas double-null mice showed tremors in nearly all trials. Although tremor was detected in a proportion of Mag-null and Galgt1-null mice, only double-null mice were uniformly affected.
Fig. 9.
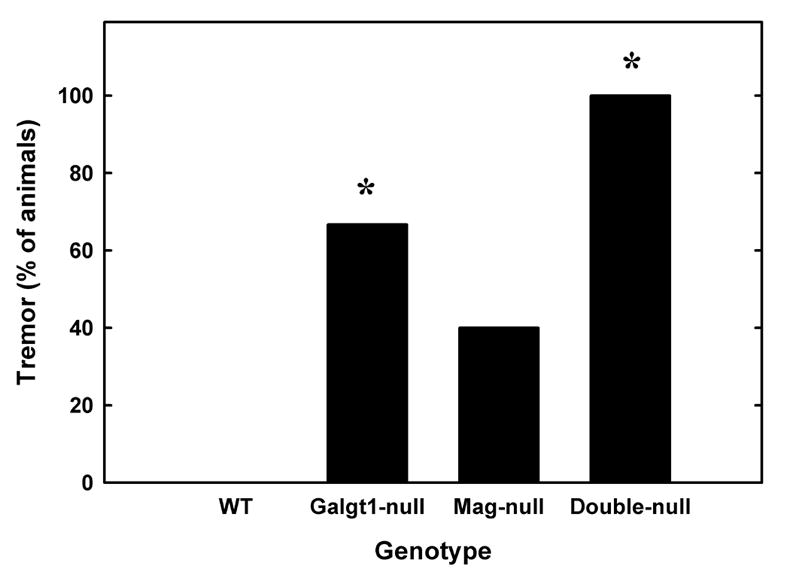
Tremor in Mag/Galgt1 double-null mice. All double-null mice displayed severe episodes of body tremor while resting and during movement, whereas no wild type mice displayed tremor under any circumstance. To quantify this observation, whole body tremor was scored (as present or absent) in wild type, Mag-null, Galgt1-null and double-null mice. Data were analyzed using logistic regression and are shown as a percentage of mice exhibiting tremor for each genotype (n = 3–5). There was a significant effect of genotype withGalgt1-null and double-null mice exhibiting significantly more tremor than wild type mice (*, p < 0.05 and p < 0.001 respectively). Although tremor was recorded in a portion of the single-null mice, tremor was a consistent behavior in double-nulls. Ages and numbers of mice used were as detailed in Fig. 7, legend. [Note: non-parametric statistical analysis was used, precluding the use of error bars].
In the beam balance, platform, forelimb grip strength, and catalepsy tests, the mutant mouse strains were not significantly different from wild type mice (data not shown).
Locomotor hyperactivity
Despite motor behavioral deficits, all of the mutant mouse strains were hyperactive, with total spontaneous locomotor activity over a 24-h period increased 60–130% above that of wild type mice (Fig. 10). During the latter half of the dark period, when spontaneous locomotor activity of wild type mice decreased compared to the first half of the dark period (open bar, Fig. 10, top), the activity of mutant strains remained high, resulting in significant hyperactivity of each mutant strain compared to wild type mice during that period (p < 0.03, Student’s t-test). When summed over the entire 24-h test period, Galgt1-null were 2.3-fold more active than wild type mice, whereas Mag-null and double-null mice displayed ~1.6-fold higher activity than wild type mice (Fig. 10, bottom).
Fig. 10.
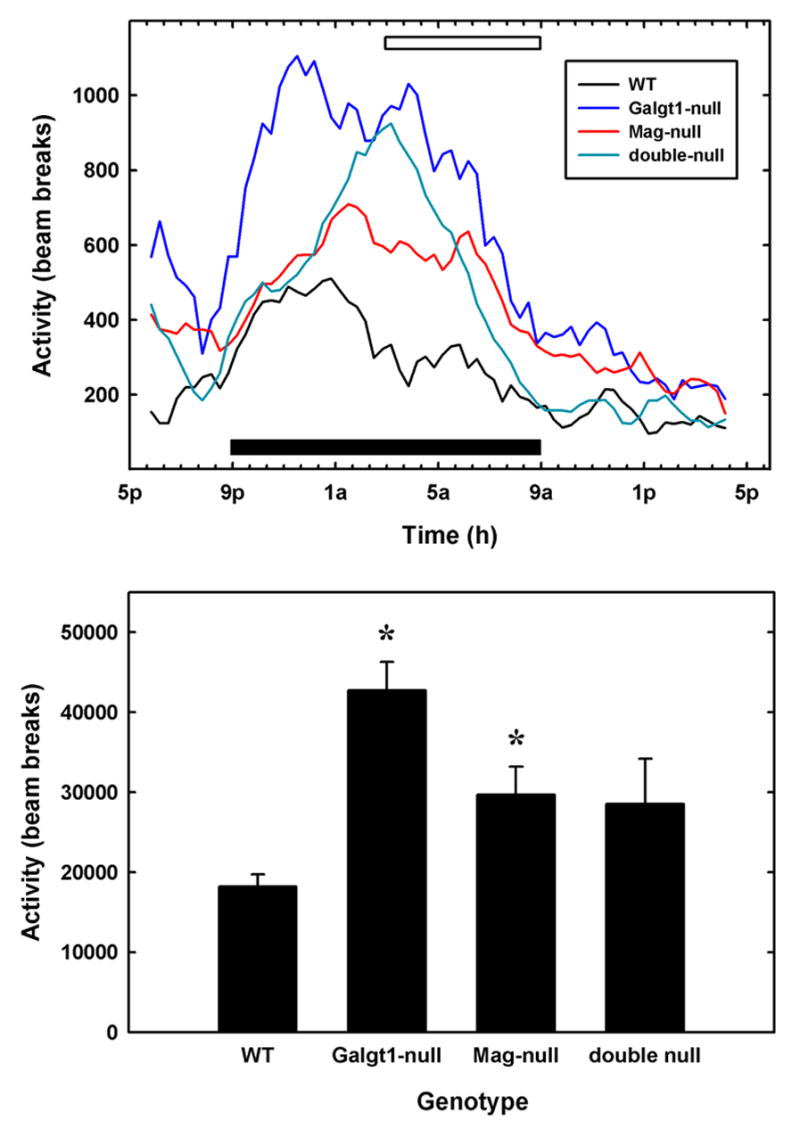
Hyperactivity in Mag-null, Galgt1-null and double-null mice. Spontaneous locomotor activity was determined for 3–6 individual mice of each genotype in two 24-h sessions in 29 x 50 cm Plexiglas boxes, each fitted with a grid of infrared beams. Top panel: The number of beam breaks per interval was averaged for mice of each genotype as a function of time. Data were smoothed over five data points (100 min). The solid bar designates the 12-h dark cycle. Over the time period constituting the latter half of the dark cycle (open bar), each mutant mouse strain was significantly more active than wild type mice (Mag-null, p < 0.01; Galgt1-null, p < 0.001; double-null, p < 0.03, pairwise Student’s t-tests). Bottom panel: The average total beam breaks over the entire 24-h test period was determined for mice of each genotype. Data are presented as mean ± SE. One-way ANOVA for total activity was highly significant (p < 0.01). Post-hoc analysis (Fisher’s PLSD) revealed that both Galgt1-null mice (p < 0.001) andMag-null mice (p < 0.05) were significantly more active (*) than wild type mice over the entire test period, with double-null mice trending in the same direction (p = 0.06). The following mice were used (number, average age): Mag-null (n=6, 5.4 months); Galgt1-null (n=3, 6.9 months); double-null (n=5, 4.9 months); and wild type C57BL/6 (n=4, 6.5 months).
Discussion
Qualitative and quantitative similarities in the histopathological and behavioral deficits of Mag-null and Galgt1-null mice, along with prior data indicating that MAG selectively and efficiently binds to the terminal saccharide sequence on gangliosides GD1a and GT1b (Yang et al., 1996; Collins et al., 1997b; Collins et al., 1997a), support the conclusion that MAG (on myelin) and gangliosides GD1a/GT1b (on axons) function as complementary receptor and ligands enhancing axon-myelin stability. The distribution of MAG on myelin (Trapp et al., 1989) and gangliosides GD1a and/or GT1b on axolemma (DeVries and Zmachinski, 1980) is consistent with this conclusion. The potential relevance of MAG-ganglioside binding to human axon-myelin stability is supported by the observations that human and mouse MAG share >94% amino acid identity, and that the structures of major brain gangliosides (including GD1a and GT1b) are essentially identical among mammals (Tettamanti et al., 1973).
In addition to mild dysmyelination (Li et al., 1994; Montag et al., 1994; Bartsch et al., 1997; Fujita et al., 1998), prior studies demonstrated axon degeneration in the PNS of aging Mag-null mice (Fruttiger et al., 1995; Weiss et al., 2001), but notably reported an absence of axon degeneration in the CNS (Lassmann et al., 1997; Uschkureit et al., 2000; Quarles, 2002). In contrast, the current study found clear evidence of CNS, as well as PNS, axon degeneration in Mag single-null mice (Figs. 3 & 4). We infer that the genetic background impacts the CNS phenotype. A widely used Mag-null mouse strain (Li et al., 1994) (distributed by The Jackson Laboratory), is maintained on a mixture of C57BL/6 and 129 inbred strains and the CD1 random bred strain. A second widely used strain (Montag et al., 1994) was derived by injecting 129 strain ES cells into B6CBAF1 hybrid blastocysts. In the current study, extensive back-crossing of the former strain to C57BL/6 mice resulted in 99.5% strain purity, resulting in clear indications of CNS (as well as PNS) axon degeneration in aging Mag-null mice.
The functional relationship between MAG and gangliosides is supported by comparisons of axon degeneration and axon architecture in Mag-null and Galgt1-null mice. In addition to supporting axon survival, MAG signals increased neurofilament phosphorylation in myelinated axons (Yin et al., 1998), resulting in increased neurofilament spacing and larger axon caliber. The findings that, compared to Mag-null mice, Galgt1-null mice have similar occurrence of axon degeneration (Fig. 4) and essentially identical decreases in axon caliber and neurofilament spacing (Figs. 5 & 6), strongly implicate gangliosides in this intercellular signaling pathway.
The observation of close similarities in the behavioral phenotypes of Mag- and Galgt1-null mice further support a functional relationship between MAG and complex gangliosides. In nearly every behavioral measure where a deficit was detected, Mag- and Galgt1-null mice had qualitatively and quantitatively similar responses.
MAG is a member of the Siglec family of sialic acid binding lectins, and binds preferentially to the terminal glycan sequence “NeuAc α2-3 Gal β1-3 GalNAc” (Crocker et al., 1996). In the brain and peripheral nerves, unlike in other tissues, gangliosides (rather than glycoproteins) are the predominant sialoglycoconjugates, representing >75% of the bound sialic acid in both rodents and humans (Tettamanti et al., 1973; Schnaar, 2000). Furthermore, an abundant terminal sequence on brain gangliosides is the preferred MAG-binding determinant, NeuAc α2-3 Gal β1-3 GalNAc (Schnaar, 2000). Based on these observations, it is reasonable to propose that MAG and gangliosides GD1a and GT1b are functional receptor and ligand. The Galgt1 gene product, UDP-N-acetylgalactosamine:GM2/GD2 β-N-acetylgalactosaminyltransferase, is specific for biosynthesis of glycolipids and does not transfer GalNAc to glycoproteins (Furukawa et al., 2002). Deleting this gene results in loss of all complex gangliosides and a concomitant increase in the simple gangliosides, GM3 and GD3 ((Liu et al., 1999), see Fig. 1). Galgt1-null mice express similar concentrations of brain gangliosides and ganglioside sialic acid as their wild type counterparts, only differing in complexity of the structures expressed (Sun et al., 2004; Wu et al., 2001). We conclude that the phenotypes reported in Galgt1-null mice are due to the specific depletion of complex gangliosides.
It should be noted that the Galgt1 mutation results in loss of all complex gangliosides, not just GD1a and GT1b ((Sun et al., 2004), and see Fig. 1). Although a role for GD1a and GT1b in axon-myelin stability is inferred based on their selective interaction with MAG, one cannot rule out a role for other complex gangliosides in the phenotypes detected. A directed mutation in the sialyltransferase(s) responsible for adding the key terminal sialic acid residue on GD1a and GT1b responsible for MAG binding would provide stronger support for the role of these particular gangliosides in axon-myelin stability. However, there are at least six sialyltransferases capable of transferring sialic acid to the 3-position of galactose (Takashima et al., 2000), and the one(s) responsible for transfer of the terminal sialic acid on GD1a and GT1b has(have) not yet been identified by gene deletion. Until a more directed mutation is achieved, the conclusion that the loss of GD1a and GT1b, in particular, are responsible for the pathologies Galgt1-null mice remains a working hypothesis.
If MAG and GD1a/GT1b act via complementary binding to one another, Mag-Galgt1 double-null mice would be expected to have a similar phenotype to the single null mice. To a significant extent, this appears to be the case, at least qualitatively. Double-null mice had similar axon degeneration, similarly decreased axon caliber, and similarly decreased neurofilament spacing compared to the single null strains. When behavior was tested, the double-null mice showed similar qualitative deficits in motor coordination, reflexes, and tremor, although to a greater extent than in single-null mice. These observations, combined with prior MAG binding specificity studies (Collins et al., 1997a), are consistent with the conclusion that MAG and complex gangliosides function, at least in part, via complementary interaction.
The somewhat more marked behavioral deficits in the double-null mice compared to Galgt-null mice may be due, in part, to the presence of GM3 in the Galgt1-null mice. GM3 retains some MAG-binding capability, although significantly less than GD1a or GT1b (Collins et al., 1997a). Any residual MAG-interaction capability in Galgt1-null mice provided by GM3 would be lost in the double-null mice. However, the more marked behavioral deficits of double-null (and to some extent Galgt1-null) mice compared to Mag-null mice (Figs. 7–9) indicate that complex gangliosides also function via mechanisms independent of MAG. This is a reasonable hypothesis since all complex gangliosides are ablated in Galgt1 mice, whereas among the major brain gangliosides only GD1a and GT1b interact with MAG. Therefore, functions of complex gangliosides other than those that interact with MAG (e.g. the major brain gangliosides GM1, GD1b) would be seen only in Galgt1-null and double-null mice.
A previously unanticipated shared phenotype of the mutant mice was marked hyperactivity (Fig. 10). Recently, a null mutation in the ganglioside biosynthetic gene responsible for synthesis of GM3 (SIAT9, see Fig. 1) in humans was found to result in seizures and progressive neurological degeneration (Simpson et al., 2004). Similarly, Siat8a/Galgt1 double-null mutant mice, which express only GM3, also display a seizure disorder (Kawai et al., 2001). Prior studies noted that children with central dysmyelination, specifically periventricular leukomalacia (PVL) resulting from neonatal hypoxia in very low birth weight babies, are prone to hyperactivity (Galli et al., 2004). In a recently reported mouse model of PVL, neonatal hypoxia resulted in decreased myelin markers, including MAG, myelin basic protein, and Nogo (among others), presumably due to selective loss of periventricular white matter (Curristin et al., 2002; Weiss et al., 2004). Adult mice that had been subjected to neonatal hypoxia were hyperactive compared to normoxic controls (Weiss et al., 2004). This was interpreted, at least in part, to be due to inappropriate sprouting of axons due to reduction in myelin-derived axon outgrowth inhibitors. Whether similar inappropriate axon sprouting occurs in Mag- or Galgt1-null mice remains to be tested.
The underlying molecular defects that lead to the documented neuropathological changes in Mag- and Galgt1-null mice have yet to be fully elucidated. MAG participates in defining the distribution of axon molecules at nodes of Ranvier; the distribution of juxtaparanodal K+ channels and the cell recognition marker Caspr2 on axons is altered in MAG mutants (Marcus et al., 2002). Additional studies on the roles of MAG and axonal gangliosides in maintaining nodal molecular expression and architecture may be particularly revealing.
Cell surfaces, including those of axons, are dominated by glycan structures. In the nervous system and elsewhere, evidence is mounting that glycan structures serve as recognition molecules that may interact with complementary carbohydrate binding proteins (lectins) on apposing cell surfaces (Taylor and Drickamer, 2003). The data presented here support the hypothesis that axonal gangliosides serve as functional ligands for myelin-associated glycoprotein, providing recognition required for appropriate axon cytoarchitecture and long-term axon-myelin stability. A more complete understanding of the biosynthesis of the key MAG-binding terminal sialic acid determinant on GD1a and GT1b, and the signaling pathways by which MAG-ganglioside binding is translated into enhanced axon architecture and long-term stability, promise to improve our understanding of axon-myelin interactions.
Acknowledgments
The authors thank Carol Rubright and Alene Carteret, Peripheral Nerve Laboratories, for their excellent assistance in tissue preparation and David Fromholt for his gracious help with behavioral studies. This work was supported by National Institutes of Health Grants NS37096 and NS42888, and a research grant from the National Multiple Sclerosis Society.
Footnotes
Corresponding author: Ronald L. Schnaar, Depts. of Pharmacology and Neuroscience, The Johns Hopkins School of Medicine, 725 N. Wolfe Street, Baltimore, MD 21205 USA, Phone: +1-410-955-8392; Fax: +1-410-955-3023, email: schnaar@jhu.edu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartsch S, Montag D, Schachner M, Bartsch U. Increased number of unmyelinated axons in optic nerves of adult mice deficient in the myelin-associated glycoprotein (MAG) Brain Res. 1997;762:231–234. doi: 10.1016/s0006-8993(97)00484-8. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–171. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Yin X, Trapp BD. Axonal pathology in myelin disorders. J Neurocytol. 1999;28:383–395. doi: 10.1023/a:1007010205037. [DOI] [PubMed] [Google Scholar]
- Bruck W, Stadelmann C. Inflammation and degeneration in multiple sclerosis. Neurol Sci. 2003;24:S265–S267. doi: 10.1007/s10072-003-0170-7. [DOI] [PubMed] [Google Scholar]
- Chiavegatto S, Sun J, Nelson RJ, Schnaar RL. A functional role for complex gangliosides: motor deficits in GM2/GD2 synthase knockout mice. Exp Neurol. 2000;166:227–234. doi: 10.1006/exnr.2000.7504. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Perry VH. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 2002;25:532–537. doi: 10.1016/s0166-2236(02)02255-5. [DOI] [PubMed] [Google Scholar]
- Collins BE, Kiso M, Hasegawa A, Tropak MB, Roder JC, Crocker PR, Schnaar RL. Binding specificities of the sialoadhesin family of I-type lectins. Sialic acid linkage and substructure requirements for binding of myelin-associated glycoprotein, Schwann cell myelin protein, and sialoadhesin. J Biol Chem. 1997a;272:16889–16895. doi: 10.1074/jbc.272.27.16889. [DOI] [PubMed] [Google Scholar]
- Collins BE, Yang LJS, Mukhopadhyay G, Filbin MT, Kiso M, Hasegawa A, Schnaar RL. Sialic acid specificity of myelin-associated glycoprotein binding. J Biol Chem. 1997b;272:1248–1255. doi: 10.1074/jbc.272.2.1248. [DOI] [PubMed] [Google Scholar]
- Crocker PR. Siglecs: sialic-acid-binding immunoglobulin-like lectins in cell-cell interactions and signalling. Curr Opin Struct Biol. 2002;12:609–615. doi: 10.1016/s0959-440x(02)00375-5. [DOI] [PubMed] [Google Scholar]
- Crocker PR, Kelm S, Hartnell A, Freeman S, Nath D, Vinson M, Mucklow S. Sialoadhesin and related cellular recognition molecules of the immunoglobulin superfamily. Biochem Soc Trans. 1996;24:150–156. doi: 10.1042/bst0240150. [DOI] [PubMed] [Google Scholar]
- Curristin SM, Cao A, Stewart WB, Zhang H, Madri JA, Morrow JS, Ment LR. Disrupted synaptic development in the hypoxic newborn brain. Proc Natl Acad Sci U S A. 2002;99:15729–15734. doi: 10.1073/pnas.232568799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries GH, Zmachinski CJ. The lipid composition of rat CNS axolemma-enriched fractions. J Neurochem. 1980;34:424–430. doi: 10.1111/j.1471-4159.1980.tb06613.x. [DOI] [PubMed] [Google Scholar]
- Domeniconi M, Cao Z, Spencer T, Sivasankaran R, Wang K, Nikulina E, Kimura N, Cai H, Deng K, Gao Y, He Z, Filbin M. Myelin-associated glycoprotein interacts with the nogo66 receptor to inhibit neurite outgrowth. Neuron. 2002;35:283–290. doi: 10.1016/s0896-6273(02)00770-5. [DOI] [PubMed] [Google Scholar]
- Edgar JM, Garbern J. The myelinated axon is dependent on the myelinating cell for support and maintenance: molecules involved. J Neurosci Res. 2004;76:593–598. doi: 10.1002/jnr.20063. [DOI] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- Frei R, Motzing S, Kinkelin I, Schachner M, Koltzenburg M, Martini R. Loss of distal axons and sensory Merkel cells and features indicative of muscle denervation in hindlimbs of P0-deficient mice. J Neurosci. 1999;19:6058–6067. doi: 10.1523/JNEUROSCI.19-14-06058.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruttiger M, Montag D, Schachner M, Martini R. Crucial role for the myelin-associated glycoprotein in the maintenance of axon-myelin integrity. Eur J Neurosci. 1995;7:511–515. doi: 10.1111/j.1460-9568.1995.tb00347.x. [DOI] [PubMed] [Google Scholar]
- Fujita N, Kemper A, Dupree J, Nakayasu H, Bartsch U, Schachner M, Maeda N, Suzuki K, Popko B. The cytoplasmic domain of the large myelin-associated glycoprotein isoform is needed for proper CNS but not peripheral nervous system myelination. J Neurosci. 1998;18:1970–1978. doi: 10.1523/JNEUROSCI.18-06-01970.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Takamiya K, Furukawa K. Beta1,4-N-acetylgalactosaminyltransferase--GM2/GD2 synthase: a key enzyme to control the synthesis of brain-enriched complex gangliosides. Biochim Biophys Acta. 2002;1573:356–362. doi: 10.1016/s0304-4165(02)00403-8. [DOI] [PubMed] [Google Scholar]
- Galli KK, Zimmerman RA, Jarvik GP, Wernovsky G, Kuypers MK, Clancy RR, Montenegro LM, Mahle WT, Newman MF, Saunders AM, Nicolson SC, Spray TL, Gaynor JW. Periventricular leukomalacia is common after neonatal cardiac surgery. J Thorac Cardiovasc Surg. 2004;127:692–704. doi: 10.1016/j.jtcvs.2003.09.053. [DOI] [PubMed] [Google Scholar]
- Kawai H, Allende ML, Wada R, Kono M, Sango K, Deng C, Miyakawa T, Crawley JN, Werth N, Bierfreund U, Sandhoff K, Proia RL. Mice Expressing only Monosialoganglioside GM3 Exhibit Lethal Audiogenic Seizures. J Biol Chem. 2001;276:6885–6888. doi: 10.1074/jbc.C000847200. [DOI] [PubMed] [Google Scholar]
- Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, Kamholz J, Shy ME. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. 2000;123:1516–1527. doi: 10.1093/brain/123.7.1516. [DOI] [PubMed] [Google Scholar]
- Kuhn PL, Petroulakis E, Zazanis GA, McKinnon RD. Motor function analysis of myelin mutant mice using a rotarod. Int J Dev Neurosci. 1995;13:715–722. doi: 10.1016/0736-5748(96)81215-9. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Bartsch U, Montag D, Schachner M. Dying-back oligodendrogliopathy: a late sequel of myelin-associated glycoprotein deficiency. Glia. 1997;19:104–110. [PubMed] [Google Scholar]
- Li C, Tropak MB, Gerial R, Clapoff S, Abramow-Newerly W, Trapp B, Peterson A, Roder J. Myelination in the absence of myelin-associated glycoprotein. Nature. 1994;369:747–750. doi: 10.1038/369747a0. [DOI] [PubMed] [Google Scholar]
- Liu BP, Fournier A, GrandPre T, Strittmatter SM. Myelin-Associated Glycoprotein as a Functional Ligand for the Nogo-66 Receptor. Science. 2002;297:1190–1193. doi: 10.1126/science.1073031. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wada R, Kawai H, Sango K, Deng C, Tai T, McDonald MP, Araujo K, Crawley JN, Bierfreund U, Sandhoff K, Suzuki K, Proia RL. A genetic model of substrate deprivation therapy for a glycosphingolipid storage disorder. J Clin Invest. 1999;103:497–505. doi: 10.1172/JCI5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus J, Dupree JL, Popko B. Myelin-associated glycoprotein and myelin galactolipids stabilize developing axo-glial interactions. J Cell Biol. 2002;156:567–577. doi: 10.1083/jcb.200111047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew TM. An efficient sampling scheme for estimating fibre number from nerve cross sections: the fractionator. J Anat. 1988;157:127–134. [PMC free article] [PubMed] [Google Scholar]
- Mayhew TM, Sharma AK. Sampling schemes for estimating nerve fibre size. I Methods for nerve trunks of mixed fascicularity. J Anat. 1984;139:45–58. [PMC free article] [PubMed] [Google Scholar]
- McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE. Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron. 1994;13:805–811. doi: 10.1016/0896-6273(94)90247-x. [DOI] [PubMed] [Google Scholar]
- Montag D, Giese KP, Bartsch U, Martini R, Lang Y, Bluthmann H, Karthingasan J, Kirschner DA, Wintergerst ES, Nave KA, Zielasek J, Toyka KV, Lipp HP, Schachner M. Mice deficient for the myelin-associated glycoprotein show subtle abnormalities in myelin. Neuron. 1994;13:229–246. doi: 10.1016/0896-6273(94)90472-3. [DOI] [PubMed] [Google Scholar]
- Morris RE, Ciraolo GM, Cohen DA, Bubel HC. In situ fixation of cultured mouse peritoneal exudate cells: comparison of fixation methods. In Vitro. 1980;16:136–146. doi: 10.1007/BF02831504. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–767. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- Quarles RH. Myelin sheaths: glycoproteins involved in their formation, maintenance and degeneration. Cell Mol Life Sci. 2002;59:1851–1871. doi: 10.1007/PL00012510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig A, Berry M, Barrett LB, Butt A, Logan A. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: Expression, receptor signaling, and correlation with axon regeneration. Glia. 2004;46:225–251. doi: 10.1002/glia.10315. [DOI] [PubMed] [Google Scholar]
- Schnaar RL. Glycobiology of the nervous system. In: Ernst B, Hart GW, Sinaÿ P, editors. Carbohydrates in Chemistry and Biology, Part II: Biology of Saccharides. Wiley-VCH; Weinheim, Germany: 2000. pp. 1013–1027. [Google Scholar]
- Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, Griffin JW, Schnaar RL. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci U S A. 1999;96:7532–7537. doi: 10.1073/pnas.96.13.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson MA, Cross H, Gurtz K, Priestman DA, Neville DCA, Reinkensmeier G, Wiznitzer M, Proukakis C, Verganelaki A, Pryde A, Patton MA, Dwek RA, Butters TD, Platt FM, Crosby AH. Infantile onset symptomatic epilepsy syndrome caused by homozygous loss of function mutations in GM3 synthase. Nat Genet. 2004;36:1225–1229. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- Sun J, Shaper NL, Itonori S, Heffer-Lauc M, Sheikh KA, Schnaar RL. Myelin-associated glycoprotein (Siglec-4) expression is progressively and selectively decreased in the brains of mice lacking complex gangliosides. Glycobiology. 2004;14:851–857. doi: 10.1093/glycob/cwh107. [DOI] [PubMed] [Google Scholar]
- Svennerholm L. Designation and schematic structure of gangliosides and allied glycosphingolipids. Prog Brain Res. 1994;101:xi–xiv. doi: 10.1016/S0079-6123(08)61935-4. [DOI] [PubMed] [Google Scholar]
- Takashima S, Kono M, Kurosawa N, Yoshida Y, Tachida Y, Inoue M, Kanematsu T, Tsuji S. Genomic organization and transcriptional regulation of the mouse GD3 synthase gene (ST8Sia I): comparison of genomic organization of the mouse sialyltransferase genes. J Biochem (Tokyo) 2000;128:1033–1043. doi: 10.1093/oxfordjournals.jbchem.a022831. [DOI] [PubMed] [Google Scholar]
- Taylor ME, Drickamer K. Introduction to Glycobiology. Oxford University Press; Oxford: 2003. p. 207. [Google Scholar]
- Tettamanti G, Bonali F, Marchesini S, Zambotti V. A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim Biophys Acta. 1973;296:160–170. doi: 10.1016/0005-2760(73)90055-6. [DOI] [PubMed] [Google Scholar]
- Trapp BD. Myelin-associated glycoprotein. Location and potential functions. Ann N Y Acad Sci. 1990;605:29–43. doi: 10.1111/j.1749-6632.1990.tb42378.x. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Andrews SB, Cootauco C, Quarles R. The myelin-associated glycoprotein is enriched in multivesicular bodies and periaxonal membranes of actively myelinating oligodendrocytes. J Cell Biol. 1989;109:2417–2426. doi: 10.1083/jcb.109.5.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uschkureit T, Sporkel O, Stracke J, Bussow H, Stoffel W. Early onset of axonal degeneration in double (plp-/-mag-/-) and hypomyelinosis in triple (plp-/-mbp-/-mag-/-) mutant mice. J Neurosci. 2000;20:5225–5233. doi: 10.1523/JNEUROSCI.20-14-05225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J, Takizawa B, McGee A, Stewart WB, Zhang H, Ment L, Schwartz M, Strittmatter S. Neonatal hypoxia suppresses oligodendrocyte Nogo-A and increases axonal sprouting in a rodent model for human prematurity. Exp Neurol. 2004;189:141–149. doi: 10.1016/j.expneurol.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Weiss MD, Luciano CA, Quarles RH. Nerve conduction abnormalities in aging mice deficient for myelin-associated glycoprotein. Muscle Nerve. 2001;24:1380–1387. doi: 10.1002/mus.1159. [DOI] [PubMed] [Google Scholar]
- Wu G, Xie X, Lu ZH, Ledeen RW. Cerebellar neurons lacking complex gangliosides degenerate in the presence of depolarizing levels of potassium. Proc Natl Acad Sci U S A. 2001;98:307–312. doi: 10.1073/pnas.011523698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61:23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]
- Xu Z, Marszalek JR, Lee MK, Wong PC, Folmer J, Crawford TO, Hsieh ST, Griffin JW, Cleveland DW. Subunit composition of neurofilaments specifies axonal diameter. J Cell Biol. 1996;133:1061–1069. doi: 10.1083/jcb.133.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LJS, Zeller CB, Shaper NL, Kiso M, Hasegawa A, Shapiro RE, Schnaar RL. Gangliosides are neuronal ligands for myelin-associated glycoprotein. Proc Natl Acad Sci U S A. 1996;93:814–818. doi: 10.1073/pnas.93.2.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P, Lee VM, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z. Signaling mechanisms of the myelin inhibitors of axon regeneration. Curr Opin Neurobiol. 2003;13:545–551. doi: 10.1016/j.conb.2003.09.006. [DOI] [PubMed] [Google Scholar]