

Abstract
Autologous hematopoietic stem cell transplantation (HSCT) has resulted in improved event-free survival for patients with advanced neuroblastoma, but the majority of these patients still relapse. We have shown that transient transfection of mouse neuroblastoma cells with plasmid DNA vectors encoding immune co-stimulatory molecules generates cell-based vaccines capable of inducing potent anti-tumor T cell immunity. Here, we explore the effectiveness of tumor vaccine administration early after HSCT. Early after transplantation, only vaccinated mice that had received an adoptive transfer of syngeneic T cells survived tumor challenge. Tumor protective immunity in the transplant recipients was dependent upon both CD4+ and CD8+ T cells, and tumor-reactive T cells in the spleens of vaccinated mice could be detected in IFN-γ ELISPOT assays. Our data indicates that the adoptive transfer of T cells was absolutely required for induction of protective immunity by the tumor vaccine. Adoptive transfer of T cells accelerated T cell reconstitution, but it also resulted in increased percentages of CD4+CD25+Foxp3+ cells early after HSCT. Treatment of HSCT recipients with an anti-CD25 monoclonal antibody prior to tumor vaccination inhibited anti-tumor immunity and significantly decreased the number of IFN-γ-secreting tumor-specific CD4 T cells. However, physical depletion of CD25+ cells from the adoptively transferred splenocytes appeared to increase the efficacy of tumor vaccination. Collectively, these results demonstrate that anti-neuroblastoma immunity can be induced early after HSCT using a novel cell-based cancer vaccine. However, sufficient numbers of T cells must be added to the graft to achieve protective anti-tumor immunity, and depletion of CD25+ T cells from adoptively transferred T cells might provide some additional benefit. These translational studies will aid in our development of post-HSCT vaccines for neuroblastoma.
INTRODUCTION
Neuroblastoma is the most common extracranial solid tumor in children accounting for 8% of all childhood cancer (1). It still has one of the highest death rates of all pediatric cancers and is responsible for approximately 15% of childhood cancer deaths. Despite aggressive treatment, the outcome remains poor for many neuroblastoma patients. Autologous hematopoietic stem cell transplantation (HSCT) has resulted in improved event-free survival for patients with severe disease. However, >50% of patients develop recurrent neuroblastoma, either from residual disease or from contaminating tumor cells in autologous grafts. Because of the high incidence of relapse, more effective treatments targeting minimal residual disease are required.
It has been demonstrated that neuroblastoma cells express tumor antigens that could potentially be recognized by cytotoxic T cells (2–4). However, in vivo the tumor may be poorly immunogenic due to an absence of critical immune co-stimulatory molecules. Neuroblastoma cells that have been genetically modified to express various immune stimulatory molecules have been employed to induce anti-tumor immunity in experimental animal models. Because individual immune stimulatory molecules might trigger diverse patterns of cellular activation that contribute differentially to the induction and effector phases of the immune response, combinations of immune stimulatory molecules could provide synergistic or additive effects to tumor immune responses. Our experiments have demonstrated synergistic anti-tumor effects by combining the co-stimulatory molecules CD80 and CD86 or CD80 and CD137L (4-1BB ligand) (5, 6). We also showed that high-level transfection of neuroblastoma cells to express a panel of four immune co-stimulatory molecules (CD54, CD80, CD86 and CD137L) transformed the tumor cells into a tumor vaccine capable of stimulating a potent T cell response (7). This vaccine increased the numbers of detectable tumor-specific splenic CTL in treated animals, and it induced a more effective anti-tumor response than tumor cells expressing only CD80 and CD86.
In this study we explored the effectiveness of tumor vaccine administration early after high-dose therapy and HSCT since this may be an ideal setting to induce effective tumor immunity due to reduced disease burden (8), altered immune regulation (9), and altered T cell homeostasis (10). Accelerated lymphoid reconstitution of either donor or host origin may overcome inherent defects in T cell signaling (11), defects in antigen-presenting cell (APC) function including antigen processing and/or presentation, or defective T cell co-stimulation by APCs (12, 13). Manipulation of the T cell repertoire by immunization during post-HSCT immune reconstitution might skew the T cell repertoire towards particular antigen specificities (14). Results of animal studies have also suggested that vaccination during homeostatic proliferation could facilitate an immune response to weak self-antigens and enhance T cell-mediated anti-tumor immunity (15–18).
The immediate post-HSCT period is accompanied by immune deficiency as a result of reduced immune effector cell numbers and impaired lymphocyte function (10). We hypothesized that efficient anti-tumor immune responses could be induced early after HSCT only if syngeneic T cells were adoptively transferred at the time of transplant. In this study, we show that the combination of high-dose total body irradiation (TBI), HSCT, T cell add-back, and early post-transplant tumor vaccination generates potent anti-neuroblastoma immunity in a manner that could be exploited in future clinical trials.
METHODS
Mice
The following strains of mice (6–8 weeks of age) were purchased from Jackson Laboratories (Bar Harbor, ME): A/J, C57BL/6 (B6) (CD45.2+; Thy1.2+), congenic B6.PL-Thy1a (CD45.2+; Thy1.1+) and B6-45.1 (CD45.1+; Thy1.2+). The animals were housed in the Medical College of Wisconsin Biomedical Resource Center (Milwaukee, WI), which has been accredited by the American Association for Accreditation of Laboratory Animal Care (AALAC).
Tumor Cells
Neuro-2a, a mouse neuroblastoma of strain A origin, was obtained from the American Type Culture Collection (ATCC) (Manassas, Virginia). The tumor cells express major histocompatibility (MHC) class I antigens, but are MHC class II-negative. An aggressive subclone, designated AGN2a, was derived through sequential in vivo and in vitro passaging (5). An MHC class II+ AGN2a cell line (designated AGN2a-CIITA) was derived by stably transfecting AGN2a with a plasmid expression vector (pcDNA3.1(−); Invitrogen, Carlsbad, CA) encoding the major histocompatibility class II transactivator (CIITA) gene (provided by Dr. Suzanne Ostrand-Rosenberg at The University of Maryland, Baltimore County). SaI, a fibrosarcoma cell line derived from an A/J mouse, was obtained from ATCC.
Antibodies
The following monoclonal antibodies (mAbs), with or without a fluorescent label, were obtained from BD Biosciences (BD Biosciences Pharmingen, San Diego, CA): anti-CD4 (clones GK1.5 and RM4-5), anti-CD8 (clone 53-6.7), anti-CD16/CD32 (clone 2.4G2), anti-CD25 (clones 7D4 and PC61), anti-CD45.1 (clone A20), anti-CD45.2 (clone 104), anti-CD54 (clone 3E2), anti-4-1BBL (clone TKS-1), anti-CD80 (clone 16-10A1), anti-CD86 (clone 37.51), anti-CD90.2 (Thy1.2; clone 53-2.1), and anti-rat IgG2a (clone RG7/1.30). Control antibodies included purified mouse IgG2b and rat IgG2b. Anti-CD90.1 (Thy1.1, clone HIS51) and anti-Foxp3 (clone FJK-16s) mAbs were obtained from eBioscience (eBioscience, San Diego, CA).
Hybridomas producing anti-CD25 mAb (clone PC61), anti-CD4 mAb (clone GK1.5), and anti-CD8 mAb (clone 2.43) were obtained from ATCC. These mAbs were produced in our laboratory using Integra CL 1000 bioreactors (Chur, Switzerland). Anti-Thy1.2-, anti-CD4-, anti-CD8- and anti-PE-conjugated microbeads used for immunomagnetic cell separation were purchased from Miltenyi Biotec (Miltenyi Biotec, Auburn, CA).
Flow cytometry was used to analyze gene modified AGN2a cells for cell surface expression of immune stimulatory molecules as described previously (7). Antibody-stained cells were analyzed with a Becton Dickinson FACScan flow cytometer, and the resulting data were analyzed using Flow-Jo software (Tree Star, Inc, San Carlos, CA).
T Cell Enrichment
A/J spleens were collected and processed into single-cell suspensions. The splenocytes were incubated with anti-Thy1.2-, anti-CD8- or anti-CD4-conjugated microbeads (Miltenyi Biotec), and the T cells were positively selected using a Miltenyi automated immunomagnetic sorter (autoMACS). The level of T cell enrichment was determined by flow cytometric analysis. The Thy1.2-enriched cells typically consisted of >95% T cells, and the purity of CD4- and CD8-enriched T cells was >98%. Alternatively, CD25+ cells were physically depleted from splenocytes by sequential incubation with PE-conjugated anti-CD25 mAb (clone PC61) and anti-PE-conjugated microbeads (Miltenyi Biotec), followed by negative selection using an autoMACS cell sorter.
Syngeneic HSCT and Tumor Vaccination
Femurs and tibiae were obtained from donor A/J mice, and bone marrow (BM) was harvested by flushing the bones with DMEM. For HSCT, A/J recipient mice were lethally irradiated (1100 cGy) and 24 hr later given a single intravenously injection of 107 BM cells with or without 2-4x107 added splenocytes or 5x106 Thy1.2-enriched T cells. In some experiments, the Thy1.2-enriched T cells were administered 3 days after HSCT. In other experiments, lethally irradiated (1000 cGy) C57BL/6 mice were transplanted by intravenous injection with 107 BM cells from B6.PL-Thy1a mice plus 2x107 splenocytes from B6-CD45.1 mice.
For vaccination, on days 7 and 14 after HSCT, recipient A/J mice were given subcutaneous injections of 2x106 irradiated (5,000 cGy) AGN2a cells. Experimental groups of mice were vaccinated with AGN2a cells that had been transfected 24 hr earlier with separate plasmids containing gene inserts for CD54 (pcDNA3.1/Hygro vector; Invitrogen, Carlsbad, CA), CD80, CD86, and CD137L (each in pCI-neo vectors; Promega, Madison, WI) using nucleofection (Amaxa Biosystems, Koeln, Germany) as described previously (7). Control mice were vaccinated with AGN2a cells nucleofected with unmodified pcDNA3.1/Hygro and pCI-neo plasmids. One to three weeks after the last vaccination, the mice were challenged subcutaneously with 103 to 5x106 viable AGN2a cells and followed for tumor development. Mice were considered moribund and killed when tumor size exceeded 250 mm2.
In some experiments, mice were treated with 250 μg of anti-CD25 mAb 4 days after HSCT. In other experiments, mice were depleted of T cell subsets in vivo by treating them with subset-specific mAbs during the induction or effector phases of the vaccine-induced immune response. Mice were injected intraperitoneally with either 250 μg anti-CD4 (GK1.5) or anti-CD8 (2.43) mAbs on days 4, 7, 10, and 14 after HSCT (immune induction phase experiments), or on days 32 and 35 after HSCT (immune effector phase experiments). To assess the involvement of T cell subsets in the induction phase of the vaccine-induced immune response, the mice were challenged with viable tumor cells one week after the second tumor vaccination. For the effector phase experiments, the mice were challenged with viable tumor cells three weeks after the second tumor vaccination.
IFN-γ ELISPOT Assays
To assess numbers of tumor-specific IFN-γ-secreting CD8+ or CD4+ T cells, ELISPOT assays were done using the Mouse IFN-γ ELISPOT Kit from BD Biosciences (San Diego, CA). The numbers of spots were quantitated with an ImmunoSpot Analyzer using included acquisition and analysis software (CTL Analyzers, LLC, Cleveland, OH).
Regulatory T (T-reg) Cell Suppression Assays
To verify the suppressive activity of CD4+CD25+ T cells, 5 × 104 CD4+CD25– responder T cells were plated in microtiter wells alone or with varying numbers of CD4+CD25+ cells. CD4+CD25+ cells and CD4+CD25- responder T cells were isolated from splenocytes using the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec) following the manufacturer’s instructions. The levels of CD4+CD25+ T cell enrichment were determined by flow cytometric analysis. The enriched CD4+CD25+ T cells were typically >96% pure (see Fig. 7). Responder cells were stimulated with anti-CD3/anti-CD28 mAb-coated Dynabeads (DYNAL Biotech ASA, Oslo, Norway) at a bead-to-responder T cell ratio of 1:4. After 72 hours of culture, 3H-thymidine was added to each well for an additional 18 hours. 3H-Thymidine incorporation was measured on a β-scintillation counter, and the results were expressed as mean counts per minute (CPM) of triplicate wells.
Figure 7.
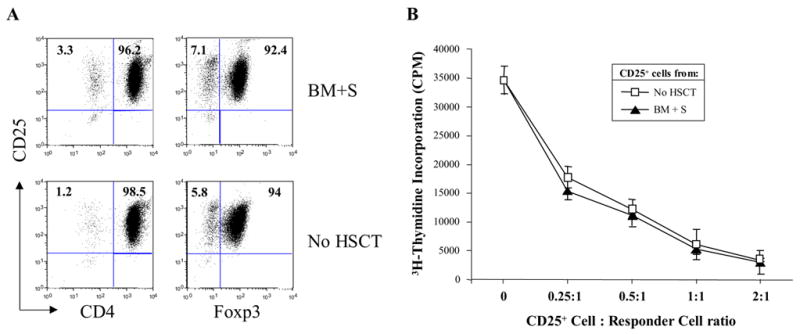
CD4+CD25+ cells isolated from mice early after HSCT exhibited regulatory function in vitro. CD4+CD25+ T cells were isolated from the spleens of normal mice (No HSCT) or from transplanted mice 14 days after HSCT with syngeneic bone marrow cells plus 2x107 added splenocytes (BM+S). Purity of the isolated cells was assessed by flow cytometry, where CD4, CD25, and Foxp3 expression are shown in 2-color histograms (A). In vitro suppressor assays were used to assess regulatory cell function (B). Briefly, 5x104 CD4+CD25− responder T cells were co-cultured with 1.25x104 anti-CD3/anti-CD28 mAb-coated Dynal beads plus the indicated ratios of purified CD4+CD25+ cells. 3H-thymidine incorporation is shown as mean CPM ± the standard deviation of triplicate wells.
Statistics
Survival curves were compared by log-rank analysis. Student’s t test was used to analyze the number of T cells. ‘P values’ <0.05 were considered as significant.
RESULTS
Generation of Effective Anti-Tumor Immunity Early Post-HSCT Requires Adoptive Transfusion of T Cells
We have recently shown that transient transfection of mouse neuroblastoma cells with plasmid DNA vectors encoding a panel of immune stimulatory molecules (CD54, CD80, CD86 and CD137L) generates a cell-based vaccine capable of inducing potent anti-tumor T cell immunity (7). Here, we sought to evaluate whether this tumor vaccine could generate a protective anti-tumor immune response in syngeneic HSCT recipients if administered immediately after transplantation. Studies of T cell reconstitution in mice that had been treated with lethal TBI and a syngeneic HSCT revealed that the immediate post-transplant phase was accompanied by a significant T cell deficiency (data not shown). We hypothesized that adoptive transfer of T cells would be required for the success of tumor vaccination in the early post-transplant period. To test this hypothesis, A/J mice were lethally irradiated one day before HSCT (day -1), infused with 107 unmanipulated syngeneic BM cells on day 0, and infused with or without 5x106 purified T cells (Thy1.2-enriched) at day 3. Mice were vaccinated subcutaneously with 2x106 irradiated AGN2a tumor cells nucleofected to express CD54, CD80, CD86 and CD137L (designated as 4P-AGN2a cells) on days 7 and 14 after HSCT. Control mice were vaccinated with AGN2a cells that were nucleofected with equal amounts of “empty” plasmids (i.e., containing no transgene inserts). Other controls consisted of non-vaccinated BM recipients. One week after the last vaccination, mice were challenged s.c. with 104, 105, or 106 viable AGN2a cells and followed for tumor development. Non-vaccinated mice were unable to resist even the lowest challenge dose of 104 AGN2a cells (Fig. 1), and T cell transfer failed to protect these mice from tumor challenge. Tumor vaccination without T cell adoptive transfer also failed to protect mice from tumor challenge. In contrast, mice given adoptive T cell transfer and vaccination with nucleofected 4P-AGN2a cells were significantly protected from tumor challenge at all three challenge doses (100%, 78.5% and 78.5% survival of mice challenged with 104, 105 and 106 AGN2a cells, respectively; p<0.001 as compared to non-vaccinated mice). AGN2a cells nucleofected with empty vectors also significantly enhanced the survival of adoptive T cell-transferred mice challenged with the lower doses of 104 or 105 tumor cells (88.9% and 33.3% of mice, respectively; p<0.01) (Fig. 1A and Fig. 1B). Similar results had been previously obtained when empty vector/nucleofected AGN2a cells were tested as a vaccine in normal non-transplanted mice (7). These results confirmed our hypothesis that T cell adoptive transfer is required for tumor vaccination to be effective in mice early post-HSCT.
Figure 1.
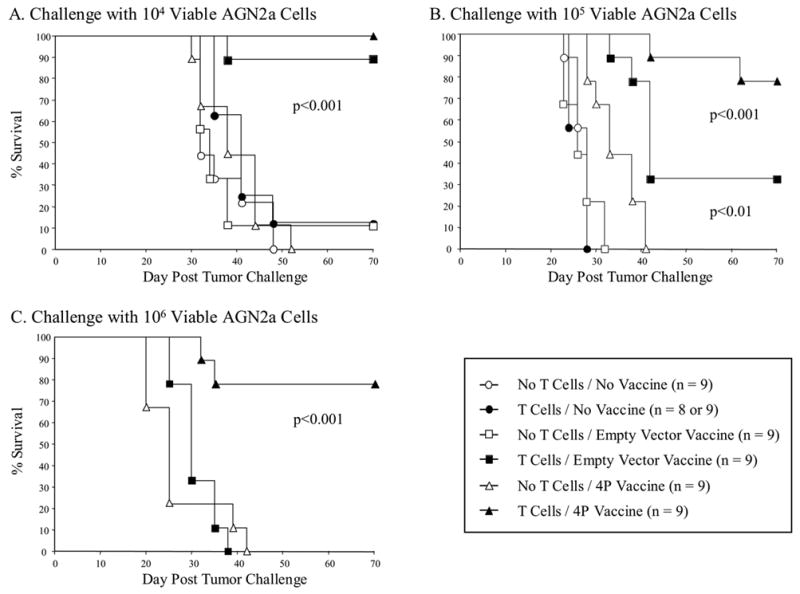
Generation of an effective anti-tumor immune response early after HSCT required adoptive transfer of T cells to facilitate immune recovery. Lethally irradiated A/J mice were transplanted with 107 syngeneic BM cells, and 3 days later given or not given 5x106 Thy1.2-enriched T cells. One week after HSCT, recipients were vaccinated twice weekly with 2x106 irradiated AGN2a cells that had been nucleofected to express CD54, CD80, CD86, and CD137L molecules (4P-AGN2a Vaccine), AGN2a cells that had been nucleofected with control empty plasmid vectors (Empty Vector Vaccine), or not vaccinated (No Vaccine). One week after the last vaccination, all groups of mice were challenged with 104 (A), 105 (B), or 106 (C) viable AGN2a cells and followed for tumor development. The data represents the combined results of two separate experiments.
Optimal Tumor Vaccine-Induced Immunity was Obtained using 2x107 Non-Separated Splenocytes as the Source of T Cell Adoptive Transfer
Next we sought to determine whether “untouched” T cells, in the form of non-separated splenocytes, would be as effective at facilitating tumor immunity as purified T cells that had been positively-selected by immunomagnetic bead sorting. Mice were subjected to HSCT as before, but purified T cells (5x106) or splenocytes (2x107 or 4x107) were co-infused with the BM, instead of given 3 days after transplant. A dose of 5x106 T cells approximates the T cell content in 2x107 splenocytes (data not shown). On days 7 and 14 after HSCT, the mice were vaccinated s.c. with 2x106 irradiated 4P-AGN2a cells. A control group of mice given HSCT, but no T cell transfer or vaccine, was included in the experiments. All mice were challenged subcutaneously on day 21 post-HSCT with 5x106 viable AGN2a cells. Adoptive transfer of 2x107 splenocytes appeared to provide a better vaccine-induced anti-tumor response than 5x106 purified T cells, as indicated by increased, although not statistically different, survival (56% vs. 20% survival; p=0.058) (Fig. 2). These results suggested that either the immunomagnetic positive selection had an impact on survival and/or function of the adoptively transferred T cells, or the splenocytes contained non-T cells capable of increasing the vaccine-induced anti-tumor effect. Increasing the dose of splenocytes given at the time of HSCT from 2 x107 to 4x107 cells did not further improve survival after tumor challenge (Fig. 2). Based on these results, a simplified protocol using 2x107 non-separated splenocytes as the source of T cell add-back was chosen for the remaining experiments in this study.
Figure 2.
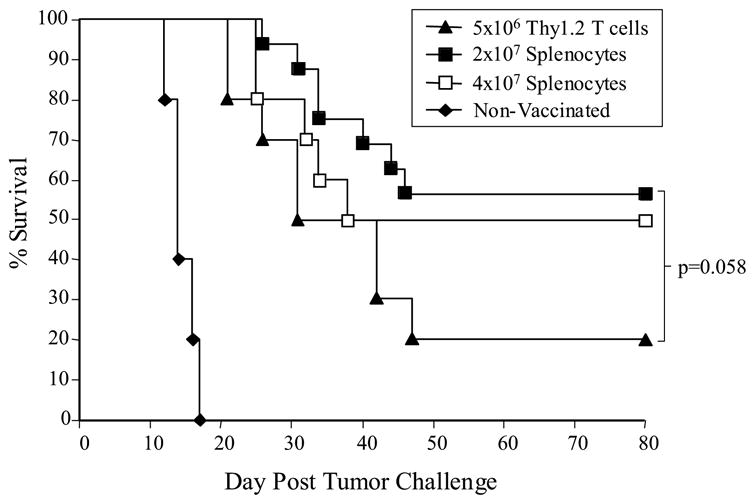
Adoptive transfer of non-separated splenocytes provided a better vaccine-induced anti-tumor effect than T cells purified by positive selection using immunomagnetic sorting. Lethally irradiated A/J mice were transplanted with 107 syngeneic BM cells with or without (a) 5x106 positively-selected Thy1.2+ T cells, (b) 2x107 non-separated splenocytes, or (c) 4x107 non-separated splenocytes. One week after HSCT, the mice given adoptive T cell/spleen cell transfer were vaccinated twice weekly with 2x106 irradiated 4P-AGN2a cells. One week after the second vaccination (day 21 after HSCT) all mice were challenged with 5x106 viable AGN2a cells. The data represents the combined results of two or three separate experiments, and the groups consisted of 10-16 total mice per group.
Adoptive Transfer of Splenocytes and Bone Marrow from Donors with Established Tumors Generates Vaccine-Induced Anti-Tumor Immunity
To more closely mimic the clinical setting, and to test if the presence of established AGN2a tumors results in tolerized tumor-reactive immune cells, mice with established neuroblastoma were used as donors for BM transplantation and adoptive spleen cell transfer. A/J donor mice were inoculated s.c. with 1x106 AGN2a cells 12 days prior to BM and splenocyte harvest. Lethally irradiated A/J recipients were transplanted with BM and splenocytes harvested from the tumor-bearing or non-tumor-bearing A/J donors. At the time of tissue harvest, the tumor cell-inoculated mice had palpable tumors that were greater than 100 mm2 in size; the percentages of CD25+Foxp3+CD4+ T-reg cells in the spleens of tumor-bearing mice were not significantly increased as compared with tumor-free mice (data not shown). Seven days after HSCT, the recipient mice were vaccinated with irradiated 4P-AGN2a cells twice weekly. Non-vaccinated mice were included as a control group. All mice were then challenged subcutaneously with 106 AGN2a cells 7 days after the second vaccination. The non-vaccinated control mice rapidly died from tumor progression between 2-3 weeks post tumor challenge (Fig. 3). In contrast, 81% of mice given adoptive transfer of splenocytes from non-tumor bearing mice survived the tumor challenge. Although recipients of splenocytes from tumor-bearing donors had decreased survival as compared to recipients of splenocytes from non-tumor-bearing donors (70% vs. 81%), the decrease in survival was not significant. These results indicated that adoptive transfer of splenocytes from tumor bearing-bearing mice did not significantly compromise the ability to generate vaccine-induced tumor immunity. Therefore, in the remaining experiments we chose to use splenocytes from non-tumor-bearing mice as the source of adoptive cell transfer in order to simplify the experimental design.
Figure 3.
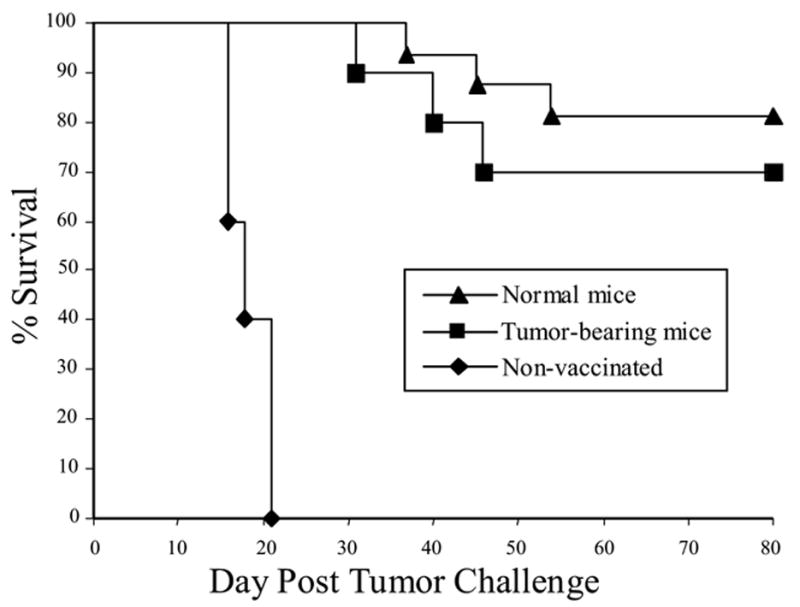
Anti-tumor immunity could be generated in HSCT recipients given adoptive transfer of splenocytes from donors with established neuroblastoma. A/J mice were transplanted with 107 syngeneic BM cell and 2x107 splenocytes from either normal mice or mice with established neuroblastomas (see Materials and Methods for specifics). Briefly, donor mice were inoculated with 106 AGN2a cells subcutaneously, and 12 days later BM and spleen cells were collected and used for HSCT. The recipients were vaccinated on days 7 and 14 after HSCT with 2x106 irradiated 4P-AGN2a cells. The mice were then challenged with 106 viable AGN2a tumor cells subcutaneously and followed for survival. A group of non-vaccinated controls was included in the experiments. The data represents the combined results of 2–3 separate experiments, and the groups consisted of 10–16 total mice per group.
The Anti-Neuroblastoma Response Induced by Vaccination with 4P-AGN2a Cells Early Post-HSCT is Dependent upon Both CD4 and CD8 T Cells
To determine which T cells are responsible for the protective anti-tumor immune responses, mice were depleted of CD4 or CD8 cells in vivo using mAbs. For HSCT, lethally irradiated A/J mice were transplanted with BM cells admixed with 2x107 splenocytes from normal A/J donors. The transplanted mice were vaccinated with nucleofected 4P-AGN2a cells on days 7 and 14 after HSCT. Anti-CD4 or anti-CD8 mAb was then administered either during the induction phase (day 4, 7, 10, and 14 post-HSCT) or during the effector phase (18 and 21 days after last vaccination and 32 and 35 days post-HSCT). More then 99% of CD8+ and 95% of CD4+ T cells were depleted by the mAb treatment (data not shown). Mice were challenged with 106 live AGN2a cells 21 days (induction phase) after HSCT or 5x105 live AGN2a 35 days (effector phase) after HSCT.
Approximately 80% of non-antibody treated controls survived the tumor challenge (Fig. 4A and 4B). Depletion of either CD4 or CD8 T cells during the ‘induction phase’ of the immune response significantly reduced survival of the vaccinated mice (p<0.001) (Fig. 4A), indicating that both T cell subsets are important for generating the anti-tumor response.
Figure 4.
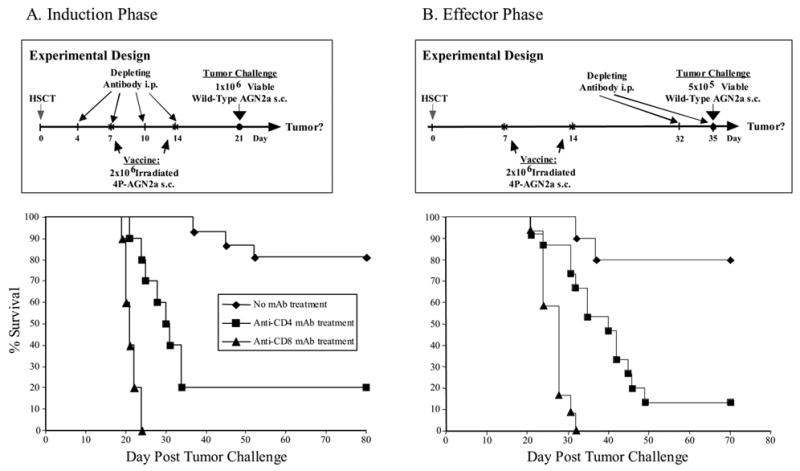
The anti-neuroblastoma immune response induced by early post-transplant vaccination is dependent upon both CD4+ and CD8+ T cells. A/J mice were transplanted with syngeneic 107 BM cells and 2x107 splenocytes, and then vaccinated on days 7 and 14 after HSCT with 2x106 irradiated 4P-AGN2a cells. In vivo-depleting anti-CD4 or anti-CD8 mAbs were: (A) given on days 4, 7, 10, and 14 after HSCT (Induction Phase) and the mice challenged with 106 live AGN2a cells on day 21, or (B) given on days 32 and 35 after HSCT (Effector Phase) and the mice challenged with 5x105 live AGN2a cells on day 35. The data represents the combined results of two separate experiments, and the groups consisted of 10–15 total mice per group. Anti-CD4 or anti-CD8 mAb treatment significantly reduced the survival of vaccinated mice during the induction (A; p<0.001 for both anti-CD4 and anti-CD8 treated mice) and effector phases (B; p<0.01 or p<0.001, respectively) of the immune response as compared to untreated mice.
As expected, CD8+ T cells were found to be important effectors of the anti-tumor immune response (Fig. 4B; p=0.0001 as compared to non-antibody treated mice). However, CD4+ T cells also appear to play as a role in the effector phase of the immune response, as the ability to resist tumor challenge was almost completely abrogated by treatment with anti-CD4 mAb (Fig. 4B p=0.002 as compared to non-antibody treated mice).
Immunization with 4P-AGN2a Cells Elicits Tumor-Reactive IFN-γ-Producing CD4+ and CD8+ T Cells in HSCT Recipients
T cell tumor reactivity elicited early after HSCT by 4P-AGN2a vaccination was assessed in vitro using IFN-γ ELISPOT assays. Lethally irradiated mice were transplanted with BM ± splenocytes ± tumor vaccination (days 7 and 14 post-HSCT). An additional control group consisted of non-transplanted mice given two weekly vaccinations with 4P-AGN2a cells. Five days after the last vaccination, spleens were harvested and CD4+ and CD8+ T cells purified by immunomagnetic sorting. The purified cells were then analyzed for the presence of tumor-reactive, IFN-γ -producing cells in ELISPOT assays. Results of these assays, plotted as the number of INF-γ spots per 105 T cells, are shown in Figure 5. Mice given the combination of BM, spleen cells, and vaccination had significantly increased frequencies of IFNγ -producing, tumor-specific CD8+ T cells as compared to mice given BM and vaccination (no spleen cells), mice given BM and splenocytes without vaccination, or non-transplanted/vaccinated mice (p< 0.001 or p<0.01) (Fig. 5A). Post-HSCT vaccination without spleen cell adoptive transfer also had significantly increased CD8+ IFN-γ spot frequencies as compared to cells from non-vaccinated mice (p< 0.01) (Fig. 5A). Relatively low CD8+ IFN-γ spot frequencies were observed in response to the control MHC-matched SaI tumor cells. The creation of MHC class II+ AGN2a cells by stable transfection of the human MHC class II transactivator (CIITA) gene allowed us to also assess tumor-reactive, IFN-γ-producing CD4+ T cells by ELISPOT. We found that CD4+ IFN-γ spot frequencies in response to AGN2a-CIITA cells were significantly higher in cells from mice given BM, spleen cells, and vaccination as compared to all other groups of mice, including the non-transplanted/vaccinated group (p< 0.001) (Fig. 5B). Together, results of the ELISPOT assays confirmed the involvement of both tumor-reactive CD4+ and CD8+ T cells in the post-transplant induction of anti-tumor immunity by the 4P-AGN2a tumor vaccine.
Figure 5.
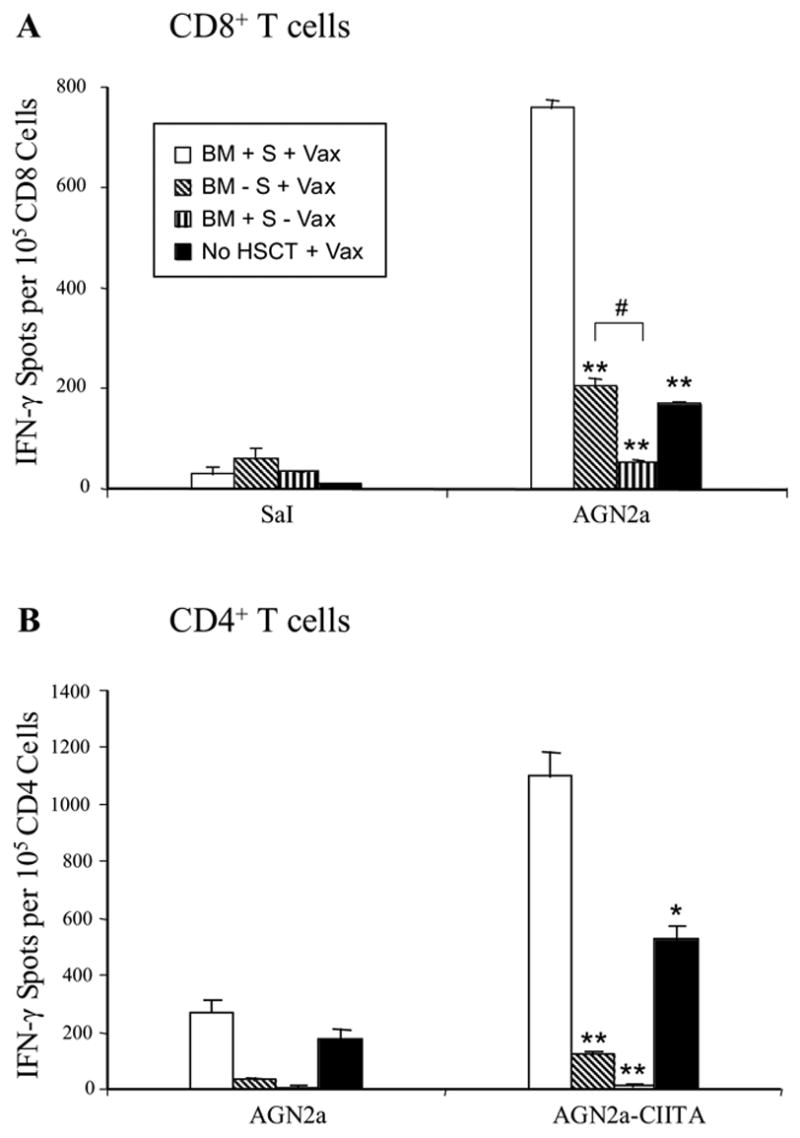
Vaccination with 4P-AGN2a cells early after HSCT elicits tumor-reactive IFN-γ-producing CD4+ and CD8+ T cells. A/J mice were transplanted with 107 syngeneic BM cells plus 2x107 splenocytes (BM + S) or BM alone (BM - S). The mice were then vaccinated on days 7 and 14 after HSCT with CD54/80/86/137L-AGN2a cells (+ Vax) or not vaccinated (- Vax). A group of normal non-transplanted mice given two weekly tumor vaccinations (No HSCT + Vax) was also included in the experiments for comparison. Five days after the second vaccination, all mice were killed, splenic T cell subsets isolated by immunomagnetic sorting, and the CD8+ (A) and CD4+ (B) cells assayed for tumor-reactive IFN-γ-secreting cell frequencies by ELISPOT. The data are from 1 of 2 replicate experiments.
** p<0.001 and * p<0.01 as compared to BM + S + Vax
#p<0.01.
Total spleen cell numbers and percentages of splenic T cell subsets were also analyzed to compare levels of T cell reconstitution between the experimental groups. Adoptive transfer of splenocytes with HSCT accelerated T cell reconstitution as reflected by significantly increased splenic CD4 and CD8 T cell numbers as compared to mice given HSCT only (Table 1). Furthermore, HSCT recipients given adoptive T cell transfer and vaccination had 10-20 times more tumor-reactive CD4 and CD8 T cells in their spleens than vaccinated mice transplanted with bone marrow cells only (data not shown).
Table 1.
T cell reconstitution after HSCT
| Group a |
|||
|---|---|---|---|
| Cells per spleen (x 105) | BM - S + Vax | BM + S + Vax | No HSCT + Vax |
| CD8 T cells | 3.9 ±4.1 | 12.2 ±4.1 † | 57.6 ±11.1 |
| CD4 T cells | 12.1 ±6.5 | 32.4 ±5.4 † | 99.7 ±16.4 |
Mice were transplanted with 107 syngeneic BM cells plus 2x107 splenocytes (BM + S) or BM alone (BM - S). The mice were then vaccinated on days 7 and 14 after HSCT with CD54/80/86/137L-AGN2a cells (+ Vax). A group of normal non-transplanted mice given two weekly tumor vaccinations (No HSCT + Vax) was included in the experiments for comparison. Five days after the second vaccination, spleens were collected and the absolute T cell numbers were assessed. The data for each group represents the mean absolute numbers of cells ± SEM from 6–8 total mice (combined from two independent experiments).
p < 0.01 for comparisons between BM + S + Vax and BM – S + Vax
Effects of T-reg Inhibition in the HSCT Recipients or Physical Depletion of T-reg Cells from the Transferred Donor-Derived Lymphocytes
CD4+CD25+Foxp3+ regulatory T (T-reg) cells have been found to be important suppressors of immune reactivity in a variety of disease settings including cancer (19, 20). Experiments in our laboratory have found that a single treatment with anti-CD25 mAb before tumor vaccination augments the anti-tumor response to AGN2a (manuscript in press). Since the persistence/presence of CD4+CD25+Foxp3+ T-reg cells could have a negative impact on our ability to effectively vaccinate mice early after HSCT, we wanted to: (a) examine whether these cells were able to survive lethal TBI or expand early post-HSCT from donor-derived cells, and (b) test whether anti-CD25 mAb treatment prior to early post-HSCT tumor vaccination could enhance the vaccine-induced immune response.
First, we assessed the effect of lethal TBI and HSCT on host-derived, donor BM-derived, and donor spleen cell-derived CD4+CD25+ and CD4+Foxp3+ cells. Lethally irradiated C57BL/6 recipient mice (Thy1.2+/CD45.2+) were transplanted with 107 BM cells from B6.PL-Thy1a mice (Thy1.1+/CD45.2+) plus 2x107 splenocytes from B6-CD45.1 mice (Thy1.2+/CD45.1+). Mice were killed on days 3, 7, 14, 21, 28 and 35 after HSCT, spleen cells isolated and counted, and the cells analyzed by flow cytometry to determine percentages of CD4+ cells co-expressing CD25 or Foxp3 and absolute numbers of CD4+CD25+ and CD4+Foxp3+ cells. Even though the mice had been infused with 2x107 splenocytes, they remained lymphopenic until 3 weeks after transplant (Fig. 6A). The lymphopenia was reflected by low splenic CD4+ T cell numbers (Fig. 6B). Only small numbers of CD4+CD25+ and CD4+Foxp3+ cells could be detected in the first two weeks after HSCT, and these cells were primarily host-derived on day 3 after HSCT, but they switched to being mostly derived from the adoptively-transferred T cells by day 7 (Fig. 6C and 6D). Interestingly, residual host-derived CD4+CD25+ and CD4+Foxp3+ cells appeared to expand and/or accumulate in the spleen 3-5 weeks after HSCT. BM-derived CD4+CD25+ and CD4+Foxp3+ cells could not be detected until 3 weeks after HSCT, and they expanded in number, but still constituted only a minority of the total CD4+CD25+ and CD4+Foxp3+ cells at 5 weeks. This implies that no single time point in the HSCT procedure can be assumed to be free of Treg influences.
Figure 6.
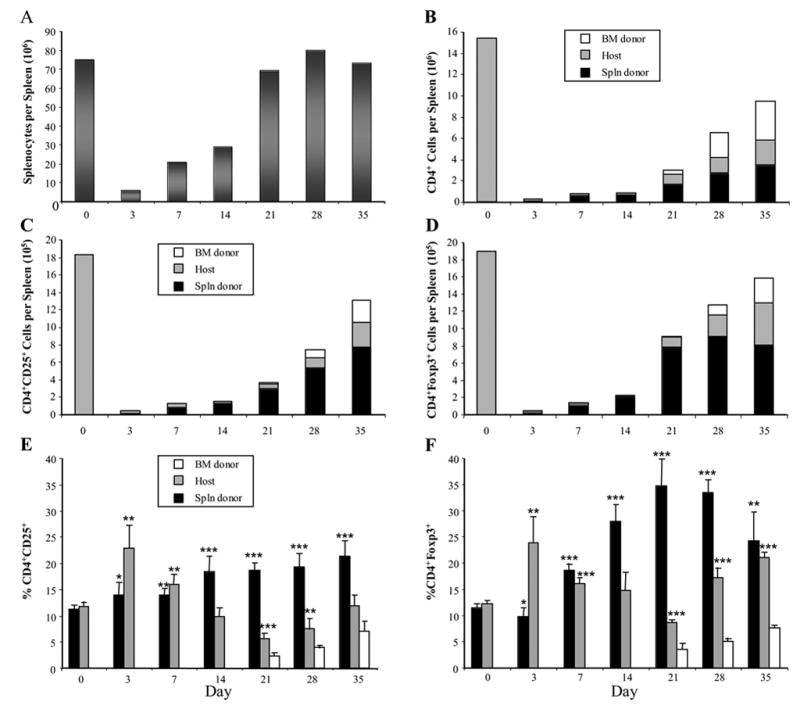
CD4+CD25+Foxp3+ T cells primarily derived from the T cell (splenocyte) adoptive transfer can be detected early after syngeneic HSCT. C57BL/6 (Thy1.2+CD45.2+) mice were lethally irradiated and 24 hours later injected with 107 BM cells from congenic B6.PL-Thy1a mice (Thy1.1+CD45.2+) plus 2x107 splenocytes from congenic B6-CD45.1 mice (Thy1.2+CD45.1+). Mice were killed at the indicated time points and the spleens collected for analysis. Nucleated cell numbers were counted (A), and the remaining splenocytes were analyzed by flow cytometry to assess absolute numbers of CD4+ cells (B), numbers of CD4+ cells co-expressing CD25 (C), numbers of CD4+ cells co-expressing Foxp3 (D), percentages of CD4+ cells co-expressing CD25 (E), and percentages of CD4+ cells co-expressing Foxp3 (F). Due to the allelic differences in Thy1 and CD45 expression, cells of donor BM origin, host origin, or cells derived from the donor splenocytes could be distinguished from one another. The bars in E and F represent the percentages of CD4+ cells in each individual cell compartment (i.e., donor bone marrow-derived (BM donor)), host-derived (Host), or donor splenocyte-derived (Spln donor). The data represent the mean values of 6 individual spleens from 2 experiments (3 individual spleens per experiment).
*** p<0.001; ** p<0.01; * p<0.05 as compared with values from normal donor or host mice (Day 0).
In normal non-transplanted mice, it has been shown that approximately 10% of splenic CD4+ cells co-express CD25 or Foxp3 (21, 22). We observed similar percentages in the spleens of the BM donors or recipients (Fig. 6E and 6F; Day 0). During the 35-day time period after HSCT, the percentages of residual host CD4+ cells that co-expressed CD25 or Foxp3 showed a biphasic increase. The adoptively transferred CD4+ cells showed a gradual increase in CD25 expression over time, but they had a much more pronounced increase in Foxp3 expression that peaked at approximately 35% on day 21 after HSCT. These results indicated that despite the state of profound lymphopenia during the first 2 weeks after HSCT, elevated percentages of splenic CD4+ cells co-expressed CD25 and Foxp3, representing an apparent skewing towards increased percentages of T-reg cells in the residual host and adoptively transferred T cells. To test if the CD4+CD25+ cells present in mice early after HSCT were capable of exhibiting regulatory (or suppressive) function, CD25+ cells were isolated by immunomagnetic sorting from normal mice (Fig. 7; No HSCT) or from mice given HSCT 14 days earlier with syngeneic bone marrow cells plus 2x107 added splenocytes (Fig. 7; BM+S). When the purified CD25+ cells were analyzed by flow cytometry, the cells were >99% CD25+, >96% of the cells expressed CD4, and >92% of the cells were Foxp3+ (Fig. 7A). In functional assays, CD25+ from the mice given syngeneic HSCT (BM+S) suppressed the polyclonal proliferation of CD4+CD25− responder cells as effectively as CD25+ cells from normal mice (No HSCT) (Fig. 7B), indicating that the CD4+CD25+Foxp3+ T-reg cells in mice given HSCT were functional.
Based on our observations, we hypothesized that treatment of the HSCT recipients with anti-CD25 mAb prior to tumor vaccination would result in increased anti-tumor immunity. To test this hypothesis, lethally irradiated A/J mice were given HSCT with BM cells plus splenocytes. Some of the transplanted mice were then treated with 250 μg of anti-CD25 mAb 3 days before the first of two weekly vaccinations with CD54/80/86/137L-AGN2a cells. The goal of this strategy was to inhibit both endogenous and infused CD25+ T-reg cells. Seven days after the second tumor vaccination, the HSCT recipients were challenged with either 106 (Fig. 8A) or 5x106 (Fig. 8B) live AGN2a cells. Tumor vaccination once again significantly protected mice from tumor challenge (all vaccinated groups vs. the non-vaccinated group; p<0.001). In contrast to previous results in normal mice, where treatment with anti-CD25 mAb improved anti-tumor immunity (manuscript in press), anti-CD25 mAb treatment early after HSCT diminished the tumor vaccine-induce immune response as indicated by decreased survival (Fig. 8A and 8B). Although the decreases in survival did not reach statistical significance, the results suggested that our hypothesis is incorrect, and they further suggested that the anti-CD25 mAb treatment might have inhibited CD25+ effector T cells generated in the vaccinated hosts. To address this possibility, phenotypic and functional studies were done on tissues of anti-CD25 mAb treated and vaccinated mice. Flow cytometric analysis of tissues (blood, lymph nodes and spleen) from anti-CD25 mAb treated mice 10 days after antibody treatment indicated that CD25 expression was significantly decreased on CD4+ cells (Table 2). The percentages of Foxp3+CD4+ T cells were also decreased in anti-CD25 mAb treated mice as compared to non-anti-CD25 treated mice, but to a lesser degree (Table 2). Results of IFN-γ ELISPOT assays indicated that early post-HSCT anti-CD25 mAb treatment significantly decreased the frequency of tumor-reactive CD4+ T cells (Fig. 8C).
Figure 8.
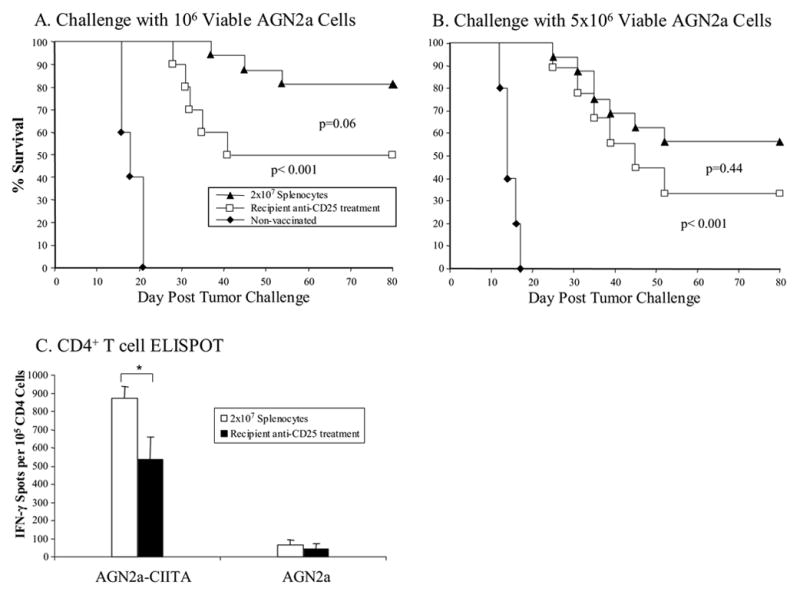
Treatment of HSCT recipients with anti-CD25 mAb prior to early post-transplant tumor vaccination negatively influenced the anti-tumor response. A/J mice were transplanted with 107 syngeneic BM cells plus 2x107 splenocytes. The transplanted mice were vaccinated on days 7 and 14 after HSCT with irradiated 4P-AGN2a cells. Some of the recipient mice were treated with anti-CD25 mAb 3 days before the first vaccination. A group on non-vaccinated control mice was included. The mice were then challenged on day 21 after HSCT with 106 (A) or 5x106 (B) viable AGN2a cells. The survival data represents the combined results of 2-3 separate experiments, including a total of 9-16 mice in each group. In panel C, CD4+ T cells were isolated from vaccinated mice seven days after the initial vaccination (day 14 after HSCT) and tested in ELISPOT assays to determine frequencies of INF-γ-producing cells in response to MHC class II+ (AGN2a-CIITA) tumor cells. The ELISPOT data are from 1 of 2 replicate experiments.
* p<0.05
Table 2.
T-reg cell reconstitution after HSCT
| Group a |
|||||
|---|---|---|---|---|---|
| T-reg Subset | Tissue | BM + S | BM + S (CD25 depl.) | BM + S + anti CD25 mAb | Normal Controls |
| CD25+ CD4+ | Blood | 42.9 ±4.1 | 40.7 ±4.7 | 1.4 ±1.2 ‡‡ | 7.6 ±1.1 |
| LN | 22.0 ±1.4 | 22.0 ±1.7 | 8.4 ±3.4 ‡‡ | 12.5 ±0.1 | |
| Spleen | 44.0 ±1.1 | 38.4 ±1.3 † | 1.2 ±0.2 ‡‡ | 13.5 ±1.4 | |
|
| |||||
| Foxp3+ CD4+ | Blood | 43.0 ±5.4 | 40.0 ±6.5 | 36.7 ±4.0 | 7.9 ±1.0 |
| LN | 21.6 ±0.9 | 20.3 ±0.6 | 17.6 ±0.3 ‡ | 12.1 ±0.2 | |
| Spleen | 43.0 ±1.0 | 35.2 ±0.8 †† | 33.0 ±0.1‡‡ | 13.4 ±0.9 | |
Mice were transplanted with 107 syngeneic BM cells plus 2x107 splenocytes (BM + S) or BM plus CD25-depleted splenocytes ((BM + S (CD25 depl.)). Some mice transplanted with BM cells and splenocytes were treated with 250 μg anti-CD25 mAb 4 days after HSCT (BM + S + anti CD25 mAb). Normal non-transplanted mice were included as a control group. Tissue samples were collected 14 days after HSCT. The data for each each group represents the mean percentages of CD4+ cells that co-expressed CD25 or Foxp3 ± SEM. 6–8 mice were analyzed for each group (combined from two independent experiments).
p<0.05;
p< 0.01 for comparisons between BM + S and BM + S (CD25 depl.)
p<0.05;
‡ p<0.01 for comparisons between BM + S and BM + S + anti CD25 mAb
As an alternative strategy to address the possible influence of CD25+ T-reg cells on early post-HSCT tumor vaccination, we physically depleted CD25+ cells from the adoptively transferred splenocytes using immunomagnetic sorting. This approach was based on the results in Fig. 6C and Fig. 6D showing that the majority of CD4+CD25+Foxp3+ cells early post-HSCT were derived from the adoptively transferred T cells. Immunomagnetic sorting eliminated more than 95% of the CD25+ T cells in the transferred population, and approximately 70% of Foxp3+CD4+ T cells (data not shown). Lethally irradiated A/J mice were given HSCT with BM cells admixed with non-depleted splenocytes or splenocytes depleted of CD25+ cells. Fourteen or twenty-one days after two weekly vaccinations with 4P-AGN2a cells, the HSCT recipients were challenged with 5x106 live AGN2a cells. Recipients of CD25-depleted splenocytes had better survival than recipients of non-depleted splenocytes (77% vs. 50%) (Fig. 9), but the increased survival did not reach statistical significance (p=0.14).
Figure 9.
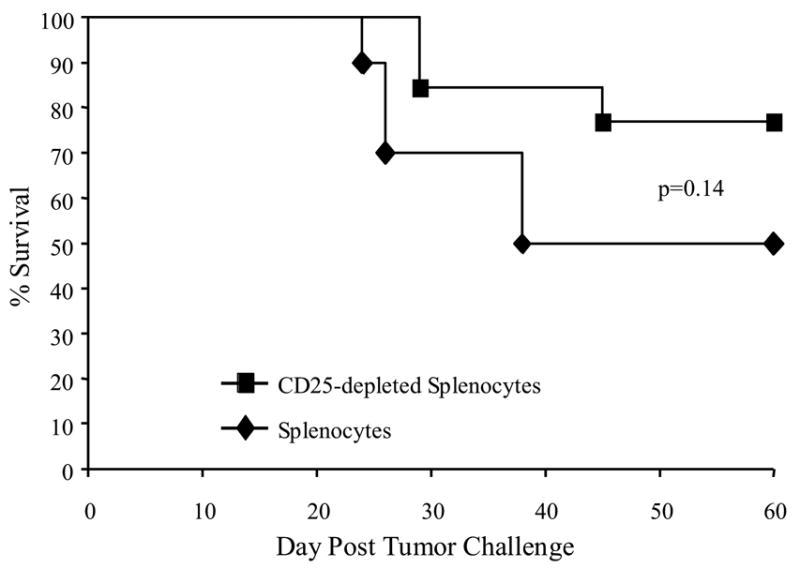
Adoptive transfer of CD25-depleted splenocytes at the time of HSCT resulted in increased tumor vaccine efficacy. A/J mice were transplanted with 107 syngeneic BM cells plus 2x107 non-separated splenocytes or splenocytes depleted of CD25 positive cells by immunomagnetic depletion. The transplanted mice were vaccinated on days 7 and 14 after HSCT with irradiated 4P-AGN2a cells. All mice were then challenged on day 14 or 21 after last vaccination with 5x106 viable AGN2a cells. The data represents the combined results of 2 separate experiments. The groups consisted of 10-13 total mice per group.
To determine the influence of CD25 depletion on CD25+Foxp3+CD4+ cell reconstitution, the percentages of CD25+CD4+ and Foxp3+CD4+ in blood, lymph nodes, and spleen were examined (Table 2). Two weeks after HSCT and adoptive transfer of splenocytes, both groups of mice ((BMS + S (CD25 depl.) vs. BM + S) contained similar percentages of Foxp3+CD4+ and CD25+CD4+ T cells in the blood and lymph nodes, but decreased percentages of Foxp3+CD4+ and CD25+CD4+ T cells were detected in the spleens of mice given CD25-depleted splenocytes. These results suggested that CD25-Foxp3+CD4+ cells in the transferred splenocytes expanded and up-regulated expression of CD25. This also indicates that optimization of T-reg cell depletion from a transferred T cell population awaits the development of cell surface markers others than CD25.
Discussion
Adoptive cellular therapy in the context of HSCT has the potential to boost the effectiveness of cell-based cancer vaccines. In this report, we tested whether vaccination with genetically modified neuroblastoma cells could generate anti-neuroblastoma immunity when administered early (i.e., during the first 2 weeks) after lethal irradiation and syngeneic HSCT. We discovered that potent anti-tumor immunity could be generated early after HSCT; however, the transplant recipients also required an adoptive transfer of T cells for the vaccine to be effective.
Tumor vaccination 3–6 weeks after HSCT in mice had been shown by others to induce a superior anti-tumor immune response as compared with vaccination of non-transplanted animals (15). In contrast, immediate post-transplant vaccination (1-2 weeks after HSCT) in those experiments was unable to induce significant tumor rejection due to a state of profound immune deficiency. Another report showed that anti-tumor immune responses could be elicited by a tumor lysate–pulsed dendritic cell vaccine initiated on day 7 post-BMT without adoptive transfusion of T cells (17). However, success of the vaccine was dependent upon the presence of mature T cells in the transplanted bone marrow and complete tumor elimination required 3 weekly vaccinations. Our results show that adoptive transfer of syngeneic T cells plus tumor vaccination facilitates the generation of a protective anti-tumor response (Fig. 1). The adoptively transferred T cells accelerated T cell reconstitution (Table 1), and the frequencies of tumor-reactive CD4+ and CD8+ T cells in the spleens of transplanted/vaccinated mice were significantly increased as compared to non-transplanted mice (Fig. 5), suggesting that the immune response was more effectively driven towards tumor reactivity by early post-transplant vaccination. Similar to our results in non-transplanted mice (7), vaccination with tumor cells that had been nucleofected with “empty” plasmid vectors also protected mice from lower tumor challenge doses (Fig. 1A and 1B). Nucleofection with empty vectors did not induce any change in MHC class I or class II, CD54, CD80, CD86, or CD137L molecule expression on the tumor cell surface (data not shown). One explanation for the increased tumor immunity is that the bacterial plasmid backbones contain immunostimulatory CpG motifs. It has been shown that one of the plasmid vectors used in our experiments (pCDNA3 vector) contains immunostimulatory CpG motifs (23), and CpG motifs have been previously shown by investigators to facilitate the activation of both innate and acquired immune responses towards tumors (24). As previously documented, the nucleofection process results in the immediate introduction of thousands of plasmid DNA copies into each cell (7), making this explanation plausible.
Our results are clinically relevant since autologous HSCT in patients is similarly accompanied by a period of pronounced immune deficiency. After autologous HSCT, patients typically have low lymphocyte counts and impaired T cell immune function during the first 3 months following transplant (10), and attempts to vaccinate early after transplant are likely hampered due to the immune deficient state. Early post-transplant immune deficiency may also contribute to increased risk of relapse from minimal residual disease. Retrospective studies suggest that superior clinical survival may be associated with more rapid lymphocyte recovery after transplantation. The rate of immune reconstitution has been shown to be an independent marker of time to progression in advanced ovarian cancer patients after peripheral blood stem cell transplantation (25). A recently published randomized clinical study showed that a single early post-transplant infusion of vaccine-primed and co-stimulated autologous T cells in conjunction with immunization improved post-transplant immunodeficiency and was able to facilitate the generation of pneumococcal immunity (26). The results of these two studies suggest that a rapid post-transplant recovery of the immune system in a situation of minimal residual disease will be a prerequisite for effective anti-tumor immunotherapy. Furthermore, they demonstrate the feasibility of T cell adoptive transfer either at the time of, or early after, HSCT.
Immune tolerance is a major barrier to effective immunization against tumor, and tumor establishment has been associated with tolerance to tumor antigens (27, 28). Our results demonstrated that immune cells from mice with established tumors were capable of facilitating vaccine-induced neuroblastoma immunity early post-HSCT (Fig. 3). The splenocytes used for adoptive transfusion were collected from mice with 100 mm2 12-day AGN2a tumors. Although it has been shown in other mouse cancer models that tumor progression can be accompanied by increased frequencies of T-reg cells in the peripheral lymphoid tissues (29, 30), we did not detect a significant increase in CD25+Foxp3+CD4+ T-reg cells in the spleens of mice bearing 12-day AGN2a tumors. Furthermore, we have also been unable to detect increased percentages of CD25+Foxp3+CD4+ T-reg cells in the spleens of mice bearing larger AGN2a tumors of ~250 mm2 in size (unpublished data). Although our data suggests that tumor-reactive T cells in mice with established neuroblastoma are not eliminated or irreversibly tolerized in the peripheral lymphoid tissues, we cannot rule out the possibility results would be different in mice bearing even larger tumor burdens. We currently hypothesize that immunization with our cell-based vaccine during the process of homeostatic expansion early post-HSCT facilitates activation and proliferation of low-affinity tumor-reactive T cells (31–35), and that this allows “tolerized” T cells to react against tumor-self antigens. This data has important implications as we tailor our future studies towards a model of established neuroblastoma, where we plan to test treatment of tumor-bearing mice with the combination of high-dose therapy, HSCT, adoptive immunotherapy (with cells from tumor-bearing donors), and tumor vaccination.
Both CD8 and CD 4 T cells are involved in the effector phase of the vaccine-induced immune response early after HSCT (Fig. 4B). This was surprising since the AGN2a tumor cells do not express MHC class II molecules. We speculate that the involvement of CD4+ T cells in the effector response could be either: (a) by providing helper activity to aid in the prompt activation of CD8+ cytotoxic effector cells or tissue macrophages, or (b) by directly serving as effector cells through tumoricidal cytokine production. Mechanisms that have been previously implicated in CD4 anti-tumor effector function include secretion of IFN-γ, which has been shown to mediate tumor rejection by its anti-tumor and anti-angiogenic properties (36, 37), or activation of macrophages capable of suppressing tumor growth (38, 39).
Animal data from Antony et al. suggested that diminished numbers of T-reg cells are in part responsible for the enhanced efficacy of tumor immunotherapy in lymphodepleted hosts (40). It has been shown that depletion or inhibition of CD4+CD25+ cells by anti-CD25 mAb administration can enhance tumor rejection (41–43), and that tumor vaccination combined with T-reg cell depletion/inhibition can increase vaccine efficacy (44, 45). Recently, we found that anti-CD25 mAb treatment can improve vaccine-induced anti-tumor responses to AGN2a (manuscript in press). In the current report, we investigated the impact of lethal TBI, HSCT, and adoptive T cell transfer (splenocytes) on CD4+CD25+ and CD4+Foxp3+ cells of host and donor origin. As far as we aware, this is the first time this type of detailed study has been done in mice early after HSCT. During the period of T cell lymphopenia early after HSCT, increased percentages of splenic CD4+CD25+Foxp3+ cells capable of eliciting functional T-reg activity (Fig. 7) were observed in mice transplanted with BM and splenocytes, and most of these cells appeared to be derived from the transferred T cells (Fig. 6). However, anti-neuroblastoma immunity could still be efficiently induced early after HSCT. Due to the increased frequency of CD4+CD25+Foxp3+ cells, we wished to determine if inhibition of residual T-reg cells could further influence vaccine efficacy. To do this, we tried two different strategies. First, we treated the HSC recipients with anti-CD25 mAb before the initial tumor vaccination, since this same strategy had increased vaccine-induced immunity in normal mice (manuscript in press). In the second approach, CD25 positive cells in the adoptively transferred splenocytes were physically depleted before infusion. Treatment of the HSC recipients with anti-CD25 mAb negatively influenced vaccine-induced anti-tumor immunity (Fig. 8A, B), suggesting that the antibody treatment inhibited effector cell function in this setting. To support this conclusion, IFN-γ ELISPOT analysis demonstrated that in vivo anti-CD25 mAb treatment resulted in decreased numbers of tumor-reactive CD4+ T cells in vaccinated HSCT recipients (Fig 8C). It is unclear why pre-vaccination anti-CD25 treatment inhibits immune effector cells in the early post-HSCT setting but enhances tumor reactivity in the normal setting. Perhaps the difference is related to increased expression of IL-2 receptors on T cells in a lymphopenic environment (46).
Different results were seen in experiments testing the second strategy, as infusion of CD25-depleted splenocytes increased survival of tumor-vaccinated mice (Fig. 9). While depletion of the CD25 positive cells from adoptively transferred cells did not influence the percentages of Foxp3+CD4+ cells in the peripheral blood or lymph nodes 14 days after HSCT, percentages of Foxp3+CD4+ cells were significantly decreased in the spleen (Table 2). Unfortunately, we did not evaluate the tissues at earlier time points, as we may have missed more profound differences early on that could influence tumor vaccination. As a cautionary note, the increased survival of mice receiving CD25-depleted splenocytes (27% increase over mice given non-depleted splenocytes) was not statistically significant. Since approximately 30% of Foxp3+CD4+ cells do not co-express CD25, it is possible that elimination of both these cells and CD25+Foxp3+CD4+ cells from the adoptively transferred T cells by some other means would further enhance tumor immunity. More specific T-reg cell reagents are clearly needed to better target these cells for elimination/inhibition.
In summary, our results demonstrate that anti-neuroblastoma immunity can be induced early after HSCT by tumor vaccination, but sufficient numbers of T cells must be included in the graft to achieve protective anti-tumor immunity. Both CD4+ and CD8+ T cells are involved in the induction and effector phases of the vaccine-induced immune response, and early post-HSCT vaccination resulted in elevated frequencies of tumor-reactive T cells as compared to non-transplanted mice that had been vaccinated. Even though percentages of Foxp3+CD25+CD4+ T cells were elevated early after HSCT, protective tumor immunity could still be induced. However, elimination of CD25+ cells from splenocytes (as a source of T cells) added to the graft appears to enhance vaccine efficacy. These results provide experimental data in support of a multi-facetted clinical approach for the treatment of neuroblastoma combining high-dose therapy, autologous HSCT, adoptive immunotherapy (T cell add-back), and post-transplant tumor vaccination.
Acknowledgments
We would like to thank M. Eric Kohler for providing the AGN2A-CIITA tumor cells and Natalia Natalia for expert technical assistance. This work was supported by US Public Health Service Grant CA100030 and the Midwest Athletes Against Childhood Cancer (MACC Fund, Inc., Milwaukee, WI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carachi R. Perspectives on neuroblastoma. Pediatr Surg Int. 2002;18:299–305. doi: 10.1007/s00383-002-0793-4. [DOI] [PubMed] [Google Scholar]
- 2.Sarkar AK, Nuchtern JG. Lysis of MYCN-amplified neuroblastoma cells by MYCN peptide-specific cytotoxic T lymphocytes. Cancer Res. 2000;60:1908–13. [PubMed] [Google Scholar]
- 3.Rodolfo M, Luksch R, Stockert E, et al. Antigen-specific immunity in neuroblastoma patients: antibody and T-cell recognition of NY-ESO-1 tumor antigen. Cancer Res. 2003;63:6948–55. [PubMed] [Google Scholar]
- 4.Oberthuer A, Hero B, Spitz R, Berthold F, Fischer M. The tumor-associated antigen PRAME is universally expressed in high-stage neuroblastoma and associated with poor outcome. Clin Cancer Res. 2004;10:4307–13. doi: 10.1158/1078-0432.CCR-03-0813. [DOI] [PubMed] [Google Scholar]
- 5.Johnson BD, Yan X, Schauer DW, Orentas RJ. Dual expression of CD80 and CD86 produces a tumor vaccine superior to single expression of either molecule. Cell Immunol. 2003;222:15–26. doi: 10.1016/s0008-8749(03)00079-0. [DOI] [PubMed] [Google Scholar]
- 6.Yan X, Johnson BD, Orentas RJ. Murine CD8 lymphocyte expansion in vitro by artificial antigen-presenting cells expressing CD137L (4-1BBL) is superior to CD28, and CD137L expressed on neuroblastoma expands CD8 tumour-reactive effector cells in vivo. Immunology. 2004;112:105–16. doi: 10.1111/j.1365-2567.2004.01853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson BD, Gershan JA, Natalia N, et al. Neuroblastoma cells transiently transfected to simultaneously express the co-stimulatory molecules CD54, CD80, CD86, and CD137L generate antitumor immunity in mice. J Immunother. 2005;28:449–60. doi: 10.1097/01.cji.0000171313.93299.74. [DOI] [PubMed] [Google Scholar]
- 8.Helson L. Autologous bone marrow transplantation. A maximal therapy design for disseminated neuroblastoma. Am J Pediatr Hematol Oncol. 1985;7:45–50. [PubMed] [Google Scholar]
- 9.Sondak VK, Wagner PD, Shu S, Chang AE. Suppressive effects of visceral tumor on the generation of antitumor T cells for adoptive immunotherapy. Arch Surg. 1991;126:442–6. doi: 10.1001/archsurg.1991.01410280040005. [DOI] [PubMed] [Google Scholar]
- 10.Guillaume T, Rubinstein DB, Symann M. Immune reconstitution and immunotherapy after autologous hematopoietic stem cell transplantation. Blood. 1998;92:1471–90. [PubMed] [Google Scholar]
- 11.Finke JH, Zea AH, Stanley J, et al. Loss of T-cell receptor zeta chain and p56lck in T-cells infiltrating human renal cell carcinoma. Cancer Res. 1993;53:5613–6. [PubMed] [Google Scholar]
- 12.Dong R, Cwynarski K, Entwistle A, et al. Dendritic cells from CML patients have altered actin organization, reduced antigen processing, and impaired migration. Blood. 2003;101:3560–7. doi: 10.1182/blood-2002-06-1841. [DOI] [PubMed] [Google Scholar]
- 13.Pockaj BA, Basu GD, Pathangey LB, et al. Reduced T-cell and dendritic cell function is related to cyclooxygenase-2 overexpression and prostaglandin E2 secretion in patients with breast cancer. Ann Surg Oncol. 2004;11:328–39. doi: 10.1245/aso.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 14.Mackall CL, Bare CV, Granger LA, Sharrow SO, Titus JA, Gress RE. Thymic-independent T cell regeneration occurs via antigen-driven expansion of peripheral T cells resulting in a repertoire that is limited in diversity and prone to skewing. J Immunol. 1996;156:4609–16. [PubMed] [Google Scholar]
- 15.Borrello I, Sotomayor EM, Rattis FM, Cooke SK, Gu L, Levitsky HI. Sustaining the graft-versus-tumor effect through posttransplant immunization with granulocyte-macrophage colony-stimulating factor (GM-CSF)-producing tumor vaccines. Blood. 2000;95:3011–9. [PubMed] [Google Scholar]
- 16.Hu HM, Poehlein CH, Urba WJ, Fox BA. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002;62:3914–9. [PubMed] [Google Scholar]
- 17.Asavaroengchai W, Kotera Y, Mule JJ. Tumor lysate-pulsed dendritic cells can elicit an effective antitumor immune response during early lymphoid recovery. Proc Natl Acad Sci U S A. 2002;99:931–6. doi: 10.1073/pnas.022634999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma J, Urba WJ, Si L, Wang Y, Fox BA, Hu HM. Anti-tumor T cell response and protective immunity in mice that received sublethal irradiation and immune reconstitution. Eur J Immunol. 2003;33:2123–32. doi: 10.1002/eji.200324034. [DOI] [PubMed] [Google Scholar]
- 19.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–52. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 20.Orentas RJ, Kohler ME, Johnson BD. Suppression of anti-cancer immunity by regulatory T cells: back to the future. Semin Cancer Biol. 2006;16:137–49. doi: 10.1016/j.semcancer.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 22.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Boccaccio GL, Mor F, Steinman L. Non-coding plasmid DNA induces IFN-gamma in vivo and suppresses autoimmune encephalomyelitis. Int Immunol. 1999;11:289–96. doi: 10.1093/intimm/11.2.289. [DOI] [PubMed] [Google Scholar]
- 24.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 25.Ferrandina G, Pierelli L, Perillo A, et al. Lymphocyte recovery in advanced ovarian cancer patients after high-dose chemotherapy and peripheral blood stem cell plus growth factor support: clinical implications. Clin Cancer Res. 2003;9:195–200. [PubMed] [Google Scholar]
- 26.Rapoport AP, Stadtmauer EA, Aqui N, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med. 2005;11:1230–7. doi: 10.1038/nm1310. [DOI] [PubMed] [Google Scholar]
- 27.Ye X, McCarrick J, Jewett L, Knowles BB. Timely immunization subverts the development of peripheral non-responsiveness and suppresses tumor development in simian virus 40 tumor antigen-transgenic mice. Proc Natl Acad Sci U S A. 1994;91:3916–20. doi: 10.1073/pnas.91.9.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staveley-O'Carroll K, Sotomayor E, et al. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc Natl Acad Sci U S A. 1998;95:1178–83. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghiringelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336–44. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- 30.Larmonier N, Marron M, Zeng Y, et al. Tumor-derived CD4(+)CD25 (+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol Immunother. 2006 Apr 13; doi: 10.1007/s00262-006-0160-8. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wrzesinski C, Restifo NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Curr Opin Immunol. 2005;17:195–201. doi: 10.1016/j.coi.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kieper WC, Jameson SC. Homeostatic expansion and phenotypic conversion of naive T cells in response to self peptide/MHC ligands. Proc Natl Acad Sci U S A. 1999;96:13306–13311. doi: 10.1073/pnas.96.23.13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldrath AW, Bevan MJ. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity. 1999;11:183–190. doi: 10.1016/s1074-7613(00)80093-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 35.Dummer W, Niethammer AG, Baccala R, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110:185–92. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mumberg D, Monach PA, Wanderling S, et al. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN- γ. Proc Natl Acad Sci U S A. 1999;96:8633–8. doi: 10.1073/pnas.96.15.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin Z, Blankenstein T. CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN γ receptor expression by nonhematopoietic cells. Immunity. 2000;12:677–86. doi: 10.1016/s1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- 38.Corthay A, Skovseth DK, Lundin KU, et al. Primary antitumor immune response mediated by CD4(+) T cells. Immunity. 2005;22:371–83. doi: 10.1016/j.immuni.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Hokey DA, Larregina AT, Erdos G, Watkins SC, Falo LD., Jr Tumor cell loaded type-1 polarized dendritic cells induce Th1-mediated tumor immunity. Cancer Res. 2005;65:10059–67. doi: 10.1158/0008-5472.CAN-05-1692. [DOI] [PubMed] [Google Scholar]
- 40.Antony PA, Piccirillo CA, Akpinarli A, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones E, Dahm-Vicker M, Simon AK, et al. Depletion of CD25+ regulatory cells results in suppression of melanoma growth and induction of autoreactivity in mice. Cancer Immun. 2002;2:1. [PubMed] [Google Scholar]
- 42.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–8. [PubMed] [Google Scholar]
- 43.Tanaka H, Tanaka J, Kjaergaard J, Shu S. Depletion of CD4+ CD25+ regulatory cells augments the generation of specific immune T cells in tumor-draining lymph nodes. J Immunother. 2002;25:207–17. doi: 10.1097/00002371-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Hu P, Khawli LA, Epstein AL. Complete regression of experimental solid tumors by combination LEC/chTNT-3 immunotherapy and CD25(+) T-cell depletion. Cancer Res. 2003;63:8384–92. [PubMed] [Google Scholar]
- 45.Sutmuller RP, van Duivenvoorde LM, van Elsas A, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194:823–32. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat Immunol. 2002;3:33–41. doi: 10.1038/ni743. [DOI] [PubMed] [Google Scholar]