

Abstract
The survival motor neuron (SMN) protein is part of a macromolecular complex that functions in the biogenesis of small nuclear ribonucleoproteins (snRNPs)—the essential components of the pre-messenger RNA splicing machinery—as well as probably other RNPs. Reduced levels of SMN expression cause the inherited motor neuron disease spinal muscular atrophy (SMA). Knowledge of the composition, interactions and functions of the SMN complex has advanced greatly in recent years. The emerging picture is that the SMN complex acts as a macromolecular chaperone of RNPs to increase the efficiency and fidelity of RNA–protein interactions in vivo, and to provide an opportunity for these interactions to be regulated. In addition, it seems that RNA metabolism deficiencies underlie SMA. Here, a dual dysfunction hypothesis is presented in which two mechanistically and temporally distinct defects—that are dependent on the extent of SMN reduction in SMA—affect the homeostasis of specific messenger RNAs encoding proteins essential for motor neuron development and function.
Keywords: neurodegeneration, RNA metabolism, small nuclear ribonucleoproteins, spinal muscular atrophy, survival motor neuron protein
Introduction
The pathway of messenger RNA (mRNA) biogenesis is essential for the temporal and spatial regulation of eukaryotic gene expression. Most post-transcriptional events of mRNA metabolism require ribonucleoprotein (RNP) complexes and occur within dynamic macromolecular assemblies. However, the elaborate networks of RNA–protein and protein–protein interactions that control mRNA metabolism at many levels are prone to errors. It is becoming clear that cells rely on the activity of protein chaperones to provide efficiency and fidelity to these macromolecular transactions in vivo. The survival motor neuron (SMN) protein has emerged as a model molecular chaperone of RNPs, and reduced levels of SMN expression cause the inherited motor neuron disease spinal muscular atrophy (SMA). This review focuses on the role of SMN in RNP biogenesis and the implications of its deficiency for SMA pathophysiology.
The SMN complex
SMN is a ubiquitously expressed protein that localizes to both the cytoplasm and the nucleus, where it accumulates in distinct nuclear bodies termed Gems and Cajal bodies. Gems were first identified in a strain of HeLa cells and are nuclear structures that contain high local concentrations of SMN (Liu & Dreyfuss, 1996). Gems are often associated with Cajal bodies, which are implicated in the assembly and maturation of RNPs. Later studies have shown that Gems coincide with Cajal bodies in the nuclei of most cell lines and adult tissues, but are separate from Cajal bodies in fetal tissues (Carvalho et al, 1999; Young et al, 2001). Importantly, methylation of the Cajal body-marker protein coilin regulates its interaction with SMN and the association of Gems with Cajal bodies (Hebert et al, 2002). The presence of SMN in Cajal bodies is consistent with a role for SMN in RNA metabolism.
SMN is tightly associated with several proteins that are considered to be integral components of a macromolecular complex referred to as the SMN complex (Fig 1). These proteins are called Gemins because they display a subcellular distribution similar to that of SMN, including localization in Gems (see Carissimi et al, 2006a and references therein). SMN interacts with Gemin2, Gemin3, Gemin5 and Gemin7, whereas Gemin4 and Gemin6 are brought to the SMN complex through their interactions with Gemin3 and Gemin7, respectively. Gemin8 binds the Gemin6–Gemin7 heterodimer as well as SMN, and mediates the association of Gemin6, Gemin7 and another component known as unrip with the SMN complex (Carissimi et al, 2006a,b). Unrip is an atypical component of the SMN complex that binds Gemin6 and Gemin7 but does not localize in Gems or Cajal bodies (Carissimi et al, 2005; Grimmler et al, 2005). Additional contacts among Gemins might further contribute to the structural organization of the SMN complex (Otter et al, 2007).
Figure 1.
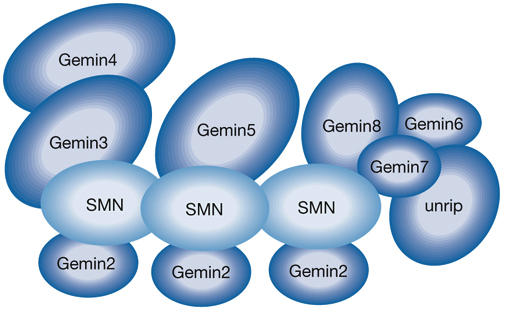
Schematic illustration of the survival motor neuron (SMN) complex. The survival motor neuron (SMN) protein binds Gemin2, Gemin3, Gemin5, Gemin7 and Gemin8, whereas Gemin4 and Gemin6 associate with SMN through interactions with Gemin3 and Gemin7, respectively. Gemin8 also binds the Gemin6–Gemin7 heterodimer, and mediates the association of Gemin6, Gemin7 and unrip with SMN. The SMN protein oligomerizes and, for simplicity, is depicted as a trimer although it can form much larger oligomeric structures (Pellizzoni et al, 1999). The representation of the SMN complex is an arbitrary simplification because the stoichiometry of its components is unknown.
Human SMN is a 294 amino-acid protein and it is unlikely that all the interactions with Gemins occur on the same SMN molecule. Instead, SMN oligomerization, which allows the formation of higher-order complexes ranging from 20S to 80S and is impaired in SMN mutants of SMA patients (Lorson et al, 1998; Pellizzoni et al, 1999; Paushkin et al, 2002; Carissimi et al, 2005, 2006a), probably has a central role in the architecture of the SMN complex through bringing together the different components within the same macromolecular entity. Interestingly, several SMN complex components are also found outside the SMN complex (Mourelatos et al, 2002; Carissimi et al, 2005, 2006b; Grimmler et al, 2005), suggesting that they might perform additional cellular functions and be recruited by SMN to carry out specific tasks within the complex.
The SMN complex chaperones snRNP biogenesis
The best-characterized targets of SMN complex function are the spliceosomal small nuclear RNPs (snRNPs; reviewed by Meister et al, 2002; Paushkin et al, 2002). Each snRNP contains one snRNA molecule, seven common Sm proteins and a set of specific proteins (Will & Luhrmann, 2001). Although snRNPs carry out pre-mRNA splicing in the nucleus, snRNP biogenesis occurs in both the nucleus and the cytoplasm of higher eukaryotes (Fig 2). The SMN complex mediates the assembly of the Sm core, which is a heptameric ring of Sm proteins around a conserved sequence of snRNAs called the Sm site. Sm core formation occurs in the cytoplasm through a stepwise pathway, including the association of Sm proteins with both the chloride-conductance regulatory protein (pICln) and the protein arginine methyltransferase 5 (PRMT5) complex prior to their association with the SMN complex. Symmetrical dimethylation of a subset of Sm proteins by the PRMT5 complex enhances their affinity for SMN and is thought to be important for snRNP assembly (Meister et al, 2002; Paushkin et al, 2002). Surprisingly, however, knockout of dart5 and valois—two fly orthologues of PRMT5 complex components—abolishes symmetrical dimethylation of Sm proteins but has no effect on snRNP biogenesis (Gonsalvez et al, 2006). The SMN complex bound to the seven Sm proteins interacts with newly exported spliceosomal snRNAs and mediates the ATP-dependent assembly of the Sm core (Meister et al, 2001; Meister & Fischer, 2002; Pellizzoni et al, 2002). Interestingly, distinct SMN complexes form Sm cores on U7 snRNA and small viral RNAs (Pillai et al, 2003; Golembe et al, 2005a). Hence, the SMN complex is the macromolecular machine used by cells for snRNP assembly.
Figure 2.
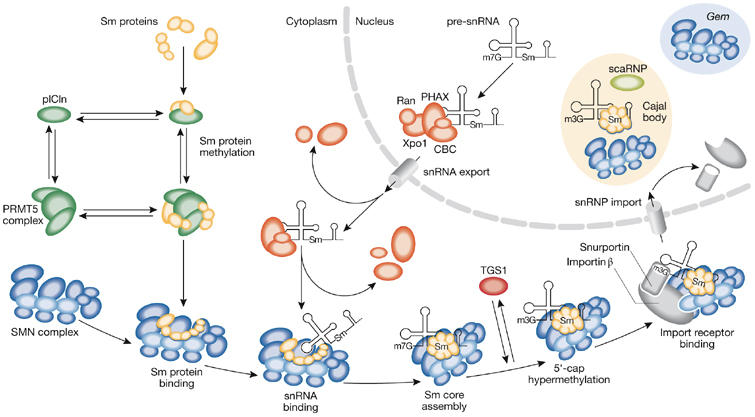
The small nuclear ribonucleoprotein biogenesis pathway. RNA polymerase II transcribes snRNAs as precursors containing an m7G cap and short 3′-end extensions in the nucleus. The cap-binding complex (CBC) binds nascent snRNAs and mediates the recruitment of the phosphorylated adaptor for RNA export (PHAX), exportin 1 (Xpo1) and ras-related nuclear protein GTP (RanGTP) for nuclear export. In the cytoplasm, the seven Sm proteins bind first to the chloride conductance regulatory protein (pICln) and the protein arginine methyltransferase 5 (PRMT5) complex—which symmetrically dimethylates SmB, SmD1 and SmD3—and then to the survival motor neuron (SMN) complex. The SMN complex bound to the Sm proteins interacts with snRNAs and mediates Sm core assembly. Hypermethylation of the 5′ cap of snRNAs by trimethylguanosine synthase 1 (TGS1) occurs after Sm core formation; both the SMN complex and snurportin, which associates with the 5′ cap of snRNAs, then bind to importin-β and mediate snRNP nuclear import. The SMN complex and snRNPs transiently localize to Cajal bodies, in which snRNPs undergo further maturation steps including methylation and pseudo-uridylation by small Cajal body-specific ribonucleoproteins (scaRNPs), before functioning in pre-messenger RNA splicing. Depending on the cell type and developmental stage, the SMN complex also localizes to Gems. snRNP, small nuclear ribonucleoprotein.
Recent studies are beginning to shed light on the contribution of Gemins to SMN complex activity (Feng et al, 2005; Shpargel & Matera, 2005; Battle et al, 2006; Carissimi et al, 2006a,b; Otter et al, 2007). To carry out snRNP assembly, the SMN complex must bring together Sm proteins and snRNAs. SMN and several Gemins bind to specific domains and distinct subsets of Sm proteins (Meister et al, 2002; Paushkin et al, 2002). Interestingly, Gemin6 and Gemin7 have an Sm protein-like structure that might facilitate Sm protein recruitment to the SMN complex (Ma et al, 2005). Consistently, loss of Gemin6, Gemin7 and unrip interaction with SMN after Gemin8-knockdown impairs Sm protein association with the SMN complex and snRNP assembly (Carissimi et al, 2006b). The SMN complex also interacts with snRNAs. Specific structural features of snRNAs and their correct spatial arrangement are crucial for SMN complex binding and snRNP assembly (Yong et al, 2002, 2004; Golembe et al, 2005b). A recent study showed that Gemin5 is the snRNA-binding protein of the SMN complex (Battle et al, 2006).
The SMN complex has additional functions in snRNP biogenesis beyond Sm core formation. SMN interacts with the trimethylguanosine synthase TGS1, and might contribute to cap hypermethylation of snRNAs after Sm core assembly (Mouaikel et al, 2003). The SMN complex, together with snurportin, binds importin-β and is required for nuclear import of snRNPs (Narayanan et al, 2004). Further characterization of the SMN complex-dependent intermediates of snRNP biogenesis will probably highlight other new aspects of the mechanisms by which this multifaceted macromolecular machine builds cellular RNPs.
Biological significance of an RNP chaperone
Early studies have shown that purified Sm proteins could form thermodynamically stable Sm cores on snRNAs without the assistance of additional proteins (Raker et al, 1999). The discovery that Sm core formation requires the SMN complex raised the question of the biological significance of having such a large macromolecular machine to carry out an apparently spontaneous process. An explanation for this paradox came from the realization that the Sm site—a short single-stranded and uridine-rich sequence—does not contain sufficient information to be effectively distinguished from other RNAs by the Sm proteins (Pellizzoni et al, 2002). Through direct binding to Sm proteins and snRNAs, the SMN complex ensures that Sm cores are assembled only on the correct RNA targets (Pellizzoni et al, 2002; Yong et al, 2002, 2004; Golembe et al, 2005a,b). The SMN complex therefore functions as a chaperone of RNPs by increasing the efficiency and specificity of snRNP assembly, and by preventing the promiscuous association of Sm proteins with other RNAs. Furthermore, the SMN complex provides an opportunity for snRNP biogenesis to be regulated.
Although these findings have allowed a conceptual advance in understanding the physiological role of the SMN complex, several questions remain. It is unclear whether low levels of SMN cause an illicit association of Sm proteins with other cellular RNAs in vivo, and, if so, whether this potentially deleterious situation contributes to SMA pathophysiology. It is possible that pICln and the PRMT5 complex exert additional protective roles in cells by restraining the RNA-binding propensity of Sm proteins when SMN levels are reduced. According to this scenario, non-specific RNA association of Sm proteins should be observed only when both chaperone systems are impaired. Furthermore, if the SMN complex imparts quality control to RNP assembly, what are the checkpoints? Is there a discard pathway for by-products of unproductive RNP assembly?
Links and gaps between snRNP dysfunction and SMA
The loss of SMN is incompatible with life, and reduced levels cause the inherited motor neuron disease SMA (reviewed by Monani, 2005), which is the leading genetic cause of infant mortality. It is unclear why deficiencies in snRNP biogenesis should selectively cause motor neuron degeneration whereas other cells are unaffected. This is mostly due to the common belief that snRNP synthesis is uniformly required in all cells because pre-mRNA splicing is essential. The observation that SMN activity in snRNP assembly does not necessarily correlate with its expression levels, but rather varies markedly among tissues and during development, challenged this assumption (Gabanella et al, 2005). In the spinal cord, SMN activity is high during embryonic and early postnatal development, and decreases sharply at the onset of myelination when it reaches a low steady level that is maintained throughout life (Gabanella et al, 2005). These findings, which are schematically presented in Fig 3, suggest a prominent requirement for snRNP synthesis during neural development. Remarkably, in zebrafish embryos, motor axon degeneration caused by knockdown of SMN, Gemin2 or pICln is rescued by injection of snRNPs (Winkler et al, 2005). Together with evidence of defective Sm protein binding and snRNP assembly activity of SMN mutants of SMA patients (Buhler et al, 1999; Pellizzoni et al, 1999; Shpargel & Matera, 2005; Wan et al, 2005), these results support the view that developmental deficiencies in snRNP biogenesis trigger motor neuron degeneration in SMA.
Figure 3.
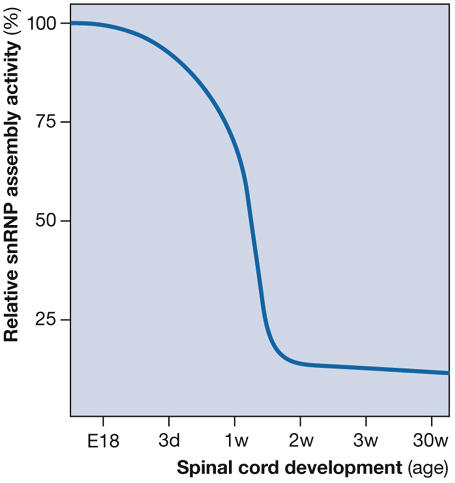
Developmental regulation of the survival motor neuron complex activity in the spinal cord. The graph shows the relative levels of small nuclear ribonucleoprotein (snRNP) assembly activity during spinal cord development. Spinal cords were isolated from embryos at embryonic day 18 (E18) as well as from mice at the indicated postnatal ages, SMN activity was then analysed in snRNP assembly experiments with in vitro-transcribed radioactive U1 snRNA and tissue extracts normalized for equal SMN content (Gabanella et al, 2005). The levels of U1 snRNP assembled in these reactions were quantified and expressed as a percentage of that of embryos at E18 arbitrarily set as 100%. d, day; SMN, survival motor neuron; w, week.
Nevertheless, several gaps need to be filled. Although snRNP assembly is reduced in extracts of SMA fibroblasts (Wan et al, 2005), these data are a measure of the capacity for snRNP assembly—that is, the amount of SMN complexes competent for Sm core formation—rather than ongoing snRNP synthesis. If these SMN complexes are present in excess, cells might still meet the in vivo demand for snRNP synthesis. Notably, there has been no evidence of reduced snRNP levels in SMA cells. Reduced SMN expression and impaired snRNP synthesis do not affect snRNP levels in DT40 cells (Wan et al, 2005). In addition, it is unclear whether SMN-knockdown decreases the amount of snRNPs in zebrafish (Winkler et al, 2005). To support the conclusion that defective snRNP biogenesis underlies motor neuron degeneration in SMA, it is crucial to show that snRNP levels are affected in SMA cells. Assuming that levels of snRNPs are reduced in SMA, the expected consequence would be a weakening of the splicing machinery, rather than a complete inactivation. As introns are spliced with different efficiencies in vivo, processing of some mRNAs might be greatly affected even if global splicing activity is not. So far, however, there is no evidence of splicing defects in SMA, and the identification of mRNAs for which processing might be altered in motor neurons will represent a fundamental step forward for the SMA field.
A chaperone of RNPs for motor neuron survival
Why do low levels of SMN affect motor neurons but not other similarly large and metabolically active neurons? Are motor neurons more sensitive to deficiencies in snRNP biogenesis? Does SMN have a motor neuron-specific function? These are some of the most pressing yet unresolved questions in SMA biology. SMN is widely but unevenly expressed in the central nervous system (CNS), and high levels of SMN are present in motor neurons throughout life (see Giavazzi et al, 2006 and references therein). Hence, motor neurons probably have a strong requirement for SMN at all ages. Motor neuron dysfunction occurs early during spinal cord development and death follows shortly after birth in severe SMA mouse models (Monani, 2005). SMN activity in snRNP assembly is prominent during this phase (Gabanella et al, 2005) and developmental deficiencies in snRNP biogenesis correlate well with the accumulation of motor neuron defects in severe SMA; however, this is less true in mild forms of SMA, in which disease onset occurs a long time after the maturation of the neuromuscular system when SMN activity in snRNP assembly is down-regulated. It is tantalizing to speculate that down-regulation of snRNP assembly activity might reflect a switch to a different SMN function. Consistently, SMN changes subcellular localization and progressively accumulates in the axons of motor neurons during human CNS maturation (Giavazzi et al, 2006). Furthermore, a remarkable study in zebrafish indicated that SMN has a crucial function in motor axons that is independent of snRNP biogenesis and is relevant to SMA (Carrel et al, 2006). In neural tissues, SMN associates with Gemins and is a component of large macromolecular complexes throughout CNS ontogenesis (Gabanella et al, 2005). Importantly, SMN and Gemins co-localize in granules that are actively transported into axons and growth cones of cultured motor neurons (Zhang et al, 2006). These SMN granules do not contain Sm proteins and it is unlikely that snRNP assembly occurs in neuronal processes (Sharma et al, 2005; Zhang et al, 2006). Therefore, neurons probably contain SMN complexes of different molecular compositions that perform specialized functions in distinct cellular compartments. So, what is the function of SMN in axons? There is evidence that SMN associates with the heterogeneous nuclear RNP (hnRNP) R protein and β-actin mRNA in neuronal processes (Rossoll et al, 2003). Importantly, cultured motor neurons from SMA mice have reduced levels of β-actin mRNA in distal axons and growth cones, indicating that SMN is involved in the axonal transport of localized mRNAs in neurons (Rossoll et al, 2003). Consistent with a role for the SMN complex as a chaperone of RNPs and with the findings of fluorescence-microscopy studies, SMN might assist the assembly of specific mRNAs into mRNP particles as well as their targeting to the axonal transport system and their localized translational regulation in motor neurons. To address these possibilities, future studies focusing on the thorough molecular characterization of axonal SMN complexes are required. As localized synthesis of β-actin is important for growth cone motility and axon guidance, its impairment correlates with reduced axon outgrowth in cultured SMA motor neurons (Rossoll et al, 2003). A defect in β-actin mRNA localization, rather than in snRNP biogenesis, could therefore account for motor neuron degeneration in SMA; at present, however, both functions have similar limitations in explaining the preferential sensitivity of motor neurons to reduced SMN levels, as neither is restricted to these cells.
Here I propose a dual dysfunction hypothesis, which suggests that two mechanistically and temporally distinct defects—both dependent on the extent of SMN reduction—are responsible for motor neuron degeneration in SMA. According to this view, marked reduction of SMN expression preferentially affects snRNP biogenesis, resulting in early developmental defects and severe SMA. By contrast, more modest yet pathological decreases substantially impair axonal mRNP metabolism but not snRNP assembly, giving rise to comparatively milder forms of SMA. An overlap between these situations might explain the wide spectrum of clinical severity of SMA patients, as well as the range of phenotypes observed owing to decreased SMN expression in both vertebrate and invertebrate animal models (Monani, 2005); these include global developmental arrest, motor neuron-pathfinding defects, synapse dysfunction and dying-back axonal degeneration. The unifying theme of both mechanisms is that the pathological consequence of low SMN levels in SMA is a qualitative and quantitative imbalance of the homeostasis of mRNAs that are uniquely expressed or required in motor neurons. Defects in RNA processing, localization or translation of specific mRNAs would temporally and spatially alter the expression of one or more proteins required for the development and function of the neuromuscular system.
Conclusions
Studies of SMN biology have revealed an unexpected link between deficiencies in RNP metabolism and SMA pathology. However, there is a need for a more detailed understanding of the neuronal functions of SMN, the molecular consequences of impaired snRNP biogenesis and why alterations in these processes have particularly profound effects on motor neuron physiology. Furthermore, it is still unclear whether motor neuron dysfunction is a cell-autonomous process, and whether it can be rescued by increasing SMN levels after onset of the disease. The availability of motor neurons differentiated from embryonic stem cells will allow several long-standing questions in the SMA field to be addressed with biochemical and cell-biological tools. Beyond providing fundamental insights into motor neuron biology, future efforts will help to unravel the molecular defects of SMA and will help the design of appropriate therapeutic strategies.

Livio Pellizzoni
Acknowledgments
I apologize to those authors whose work could not be discussed here owing to space limitations. L.P. is an Assistant Telethon Scientist.
References
- Battle DJ, Lau CK, Wan L, Deng H, Lotti F, Dreyfuss G (2006) The Gemin5 protein of the SMN complex identifies snRNAs. Mol Cell 23: 273–279 [DOI] [PubMed] [Google Scholar]
- Buhler D, Raker V, Luhrmann R, Fischer U (1999) Essential role for the tudor domain of SMN in spliceosomal U snRNP assembly: implications for spinal muscular atrophy. Hum Mol Genet 8: 2351–2357 [DOI] [PubMed] [Google Scholar]
- Carissimi C, Baccon J, Straccia M, Chiarella P, Maiolica A, Sawyer A, Rappsilber J, Pellizzoni L (2005) Unrip is a component of SMN complexes active in snRNP assembly. FEBS Lett 579: 2348–2354 [DOI] [PubMed] [Google Scholar]
- Carissimi C, Saieva L, Baccon J, Chiarella P, Maiolica A, Sawyer A, Rappsilber J, Pellizzoni L (2006a) Gemin8 is a novel component of the survival motor neuron complex and functions in snRNP assembly. J Biol Chem 281: 8126–8134 [DOI] [PubMed] [Google Scholar]
- Carissimi C, Saieva L, Gabanella F, Pellizzoni L (2006b) Gemin8 is required for the architecture and function of the survival motor neuron complex. J Biol Chem 281: 37009–37016 [DOI] [PubMed] [Google Scholar]
- Carrel TL, McWhorter ML, Workman E, Zhang H, Wolstencroft EC, Lorson C, Bassell GJ, Burghes AH, Beattie CE (2006) Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci 26: 11014–11022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho T, Almeida F, Calapez A, Lafarga M, Berciano MT, Carmo-Fonseca M (1999) The spinal muscular atrophy disease gene product, SMN: A link between snRNP biogenesis and the Cajal (coiled) body. J Cell Biol 147: 715–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Gubitz AK, Wan L, Battle DJ, Dostie J, Golembe TJ, Dreyfuss G (2005) Gemins modulate the expression and activity of the SMN complex. Hum Mol Genet 14: 1605–1611 [DOI] [PubMed] [Google Scholar]
- Gabanella F, Carissimi C, Usiello A, Pellizzoni L (2005) The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum Mol Genet 14: 3629–3642 [DOI] [PubMed] [Google Scholar]
- Giavazzi A, Setola V, Simonati A, Battaglia G (2006) Neuronal-specific roles of the survival motor neuron protein: evidence from survival motor neuron expression patterns in the developing human central nervous system. J Neuropathol Exp Neurol 65: 267–277 [DOI] [PubMed] [Google Scholar]
- Golembe TJ, Yong J, Battle DJ, Feng W, Wan L, Dreyfuss G (2005a) Lymphotropic herpesvirus saimiri uses the SMN complex to assemble Sm cores on its small RNAs. Mol Cell Biol 25: 602–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golembe TJ, Yong J, Dreyfuss G (2005b) Specific sequence features, recognized by the SMN complex, identify snRNAs and determine their fate as snRNPs. Mol Cell Biol 25: 10989–11004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalvez GB, Rajendra TK, Tian L, Matera AG (2006) The Sm-protein methyltransferase, dart5, is essential for germ-cell specification and maintenance. Curr Biol 16: 1077–1089 [DOI] [PubMed] [Google Scholar]
- Grimmler M, Otter S, Peter C, Muller F, Chari A, Fischer U (2005) Unrip, a factor implicated in cap-independent translation, associates with the cytosolic SMN complex and influences its intracellular localization. Hum Mol Genet 14: 3099–3111 [DOI] [PubMed] [Google Scholar]
- Hebert MD, Shpargel KB, Ospina JK, Tucker KE, Matera AG (2002) Coilin methylation regulates nuclear body formation. Dev Cell 3: 329–337 [DOI] [PubMed] [Google Scholar]
- Liu Q, Dreyfuss G (1996) A novel nuclear structure containing the survival of motor neurons protein. EMBO J 15: 3555–3565 [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, Le T, Burghes AH, Androphy EJ (1998) SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet 19: 63–66 [DOI] [PubMed] [Google Scholar]
- Ma Y, Dostie J, Dreyfuss G, Van Duyne GD (2005) The Gemin6–Gemin7 heterodimer from the survival of motor neurons complex has an Sm protein-like structure. Structure 13: 883–889 [DOI] [PubMed] [Google Scholar]
- Meister G, Fischer U (2002) Assisted RNP assembly: SMN and PRMT5 complexes cooperate in the formation of spliceosomal UsnRNPs. EMBO J 21: 5853–5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Buhler D, Pillai R, Lottspeich F, Fischer U (2001) A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat Cell Biol 3: 945–949 [DOI] [PubMed] [Google Scholar]
- Meister G, Eggert C, Fischer U (2002) SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol 12: 472–478 [DOI] [PubMed] [Google Scholar]
- Monani UR (2005) Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron 48: 885–896 [DOI] [PubMed] [Google Scholar]
- Mouaikel J, Narayanan U, Verheggen C, Matera AG, Bertrand E, Tazi J, Bordonne R (2003) Interaction between the small-nuclear-RNA cap hypermethylase and the spinal muscular atrophy protein, survival of motor neuron. EMBO Rep 4: 616–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G (2002) miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev 16: 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U, Achsel T, Luhrmann R, Matera AG (2004) Coupled in vitro import of U snRNPs and SMN, the spinal muscular atrophy protein. Mol Cell 16: 223–234 [DOI] [PubMed] [Google Scholar]
- Otter S, Grimmler M, Neuenkirchen N, Chari A, Sickmann A, Fischer U (2007) A comprehensive interaction map of the human SMN–complex. J Biol Chem 282: 5825–5833 [DOI] [PubMed] [Google Scholar]
- Paushkin S, Gubitz AK, Massenet S, Dreyfuss G (2002) The SMN complex, an assemblyosome of ribonucleoproteins. Curr Opin Cell Biol 14: 305–312 [DOI] [PubMed] [Google Scholar]
- Pellizzoni L, Charroux B, Dreyfuss G (1999) SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci USA 96: 11167–11172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L, Yong J, Dreyfuss G (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science 298: 1775–1779 [DOI] [PubMed] [Google Scholar]
- Pillai RS, Grimmler M, Meister G, Will CL, Luhrmann R, Fischer U, Schumperli D (2003) Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm11, in histone RNA processing. Genes Dev 17: 2321–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raker VA, Hartmuth K, Kastner B, Luhrmann R (1999) Spliceosomal U snRNP core assembly: Sm proteins assemble onto an Sm site RNA nonanucleotide in a specific and thermodynamically stable manner. Mol Cell Biol 19: 6554–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossoll W, Jablonka S, Andreassi C, Kroning AK, Karle K, Monani UR, Sendtner M (2003) Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 163: 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lambrechts A, Hao le T, Le TT, Sewry CA, Ampe C, Burghes AH, Morris GE (2005) A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res 309: 185–197 [DOI] [PubMed] [Google Scholar]
- Shpargel KB, Matera AG (2005) Gemin proteins are required for efficient assembly of Sm-class ribonucleoproteins. Proc Natl Acad Sci USA 102: 17372–17377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Battle DJ, Yong J, Gubitz AK, Kolb SJ, Wang J, Dreyfuss G (2005) The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol Cell Biol 25: 5543–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Luhrmann R (2001) Spliceosomal UsnRNP biogenesis, structure and function. Curr Opin Cell Biol 13: 290–301 [DOI] [PubMed] [Google Scholar]
- Winkler C, Eggert C, Gradl D, Meister G, Giegerich M, Wedlich D, Laggerbauer B, Fischer U (2005) Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev 19: 2320–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong J, Pellizzoni L, Dreyfuss G (2002) Sequence-specific interaction of U1 snRNA with the SMN complex. EMBO J 21: 1188–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong J, Golembe TJ, Battle DJ, Pellizzoni L, Dreyfuss G (2004) snRNAs contain specific SMN-binding domains that are essential for snRNP assembly. Mol Cell Biol 24: 2747–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young PJ, Le TT, Dunckley M, Nguyen TM, Burghes AH, Morris GE (2001) Nuclear gems and Cajal (coiled) bodies in fetal tissues: nucleolar distribution of the spinal muscular atrophy protein, SMN. Exp Cell Res 265: 252–261 [DOI] [PubMed] [Google Scholar]
- Zhang H, Xing L, Rossoll W, Wichterle H, Singer RH, Bassell GJ (2006) Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci 26: 8622–8632 [DOI] [PMC free article] [PubMed] [Google Scholar]