

Abstract
During the late phase of HIV-1 replication, newly synthesized retroviral Gag proteins are targeted to lipid raft regions of specific cellular membranes, where they assemble and bud to form new virus particles. Gag binds preferentially to the plasma membrane (PM) of most hematopoietic cell types, a process mediated by interactions between the cellular PM marker phosphatidylinositol-(4,5)-bisphosphate (PI(4,5)P2) and Gag’s N-terminally-myristoylated matrix (MA) domain. We recently demonstrated that PI(4,5)P2 binds to a conserved cleft on MA and promotes myristate exposure, suggesting a role as both a direct membrane anchor and myristyl switch trigger. Here we show that PI(4,5)P2 is also capable of binding to MA proteins containing point mutations that inhibit membrane binding in vitro, and in vivo, including V7R, L8A and L8I. However, these mutants do not exhibit PI(4,5)P2- or concentration-dependent myristate exposure. NMR studies of V7R and L8A MA reveal minor structural changes that appear to be responsible for stabilizing the myristate-sequestered (myr(s)) species and inhibiting exposure. Unexpectedly, the myristyl group of a revertant mutant with normal PM targeting properties (V7R,L21K) is also tightly sequestered and insensitive to PI(4,5)P2 binding. This mutant binds PI(4,5)P2 with two-fold higher affinity compared with the native protein, suggesting a potential compensatory mechanism for membrane binding.
Keywords: Human immunodeficiency virus type-1 (HIV-1); Gag; myristyl (myr); matrix (MA); phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2); analytical ultracentrifugation (AU); nuclear magnetic resonance (NMR)
Introduction
The assembly of retroviruses in infected cells proceeds by complex, multi-stepped mechanisms that are only beginning to become understood. The major structural constituent of retroviruses is called Gag, a multi-domain protein that is post-translationally acylated (acetylated in the alpharetroviruses and myristoylated in most other retroviruses). Several thousand copies of Gag assemble to form a single virus particle, which is enveloped by a lipid bilayer derived from the host cell. Although the Gag proteins of the betaretroviruses assemble in the cytoplasm prior to budding from the plasma membrane1, the Gag proteins of most other retroviruses (including the human immunodeficiency virus type-1, HIV-1) assemble at cellular membranes and require the assistance of host factors associated with the endosomal protein sorting machinery2,3. In most hematopoetic cell types, the HIV-1 Gag proteins assemble and bud from the plasma membrane (PM)4,5. Some studies suggest that assembly in macrophages occurs mainly at the membranes of late endosomes and/or multivesicular bodies (MVBs)6–8, although the significance of endosomal assembly has recently been questioned9. The Gag proteins co-localize at punctate sites10,11, and analysis of the lipid and lipid-bound protein components of isolated virions indicates that the viral membranes are derived from lipid raft microdomains. Lipid rafts are enriched with cholesterol and sphingolipids with saturated fatty acid chains, and form liquid-ordered membrane structures that serve as preferential binding sites for specific cellular proteins. Several animal viruses are known to associate with rafts upon cellular entry and/or egress11.
The trafficking of Gag to membrane assembly sites is mediated by the protein’s N-terminal matrix (MA) domain. Tight binding of the HIV-1 Gag protein to the PM is driven by a bipartite signal that constitutes a myristyl group (myr) and a cluster of basic residues on the surface of the MA protein 12,13. Mutations and/or deletions within the first 6 residues (Gly-2 to Ser-6; myristate group is residue number 1) of MA that affect Gag myristylation 14–18, or within the polybasic region of MA 5,12,19,20, impair membrane binding in vitro and can retarget Gag to the cytoplasm and/or intracellular compartments in vivo. In addition, there is considerable evidence that the affinity of MA binding for membranes is significantly lower than that of the intact Gag molecules, which led to the suggestion that Gag binding may be mediated by a myristyl switch mechanism.21,22 In addition to promoting membrane binding, myristate exposure appears to be required for targeting Gag to lipid rafts. Resh and co-workers showed that substitution of the saturated myristyl group of HIV-1 Gag by unsaturated lipids reduces the affinity of Gag for rafts, but not for membranes in general, leading to particle assembly defect.23
Mutagenesis studies have identified residues in the N-terminus of HIV-1 MA that are critical for proper membrane selection, including point mutations such as V7R, L8A and L8I. 19,24–26 These substitutions give rise to a phenotype similar to that observed for the unmyristoylated protein, although they do not block myristoylation. They severely inhibit virus assembly and release, cause significant reductions in PM binding, and lead to significant increases in cytosolic localization of Gag. 19,24–26 Interestingly, the assembly-defective phenotype imposed by the V7R mutation is reversed by second-site compensatory mutations elsewhere in the MA domain. Biochemical studies have shown that L21K and K98E mutations reversed the membrane-binding defect caused by V7R and significantly increased Gag binding to the PM. 24 These findings suggested that mutations at positions 7 and 8 may block myristate exposure, and that compensatory substitutions at positions 21 and 98 might restore exposure.
NMR studies confirmed that the HIV-1 MA myristyl group is capable of adopting sequestered (myr(s)) and exposed (myr(e)) conformations, and demonstrated that myristate exposure can be promoted by factors that increase protein self-association27. More recently, the myristyl switch equilibrium was shown to be sensitive to binding by phosphatidyl-4,5-bisphosphate (PI(4,5)P2), a cellular factor that serves as a membrane marker for proteins that need to associate specifically with the plasma membrane28–31. PI(4,5)P2 plays a major role in directing HIV-1 Gag to the PM32, and appears to function by acting as both a trigger for myristate exposure and as a direct membrane anchor31. To determine the mechanism(s) by which the position-7 and –8 mutations affect in vivo Gag localization and virus assembly, we conducted NMR and thermodynamic studies on the HIV-1 MA mutants V7R, L8A, L8I, L21K, and V7R/L21K (Figure 1), and studied interactions between these mutants and PI(4,5)P2. Our findings reveal that the single-residue mutations do not inhibit PI(4,5)P2 binding, but do inhibit PI(4,5)P2- and concentration-dependent myristate exposure and suggest a novel potential compensatory mechanism for membrane selection and binding by the V7R/L21K revertant mutant.
Figure 1.
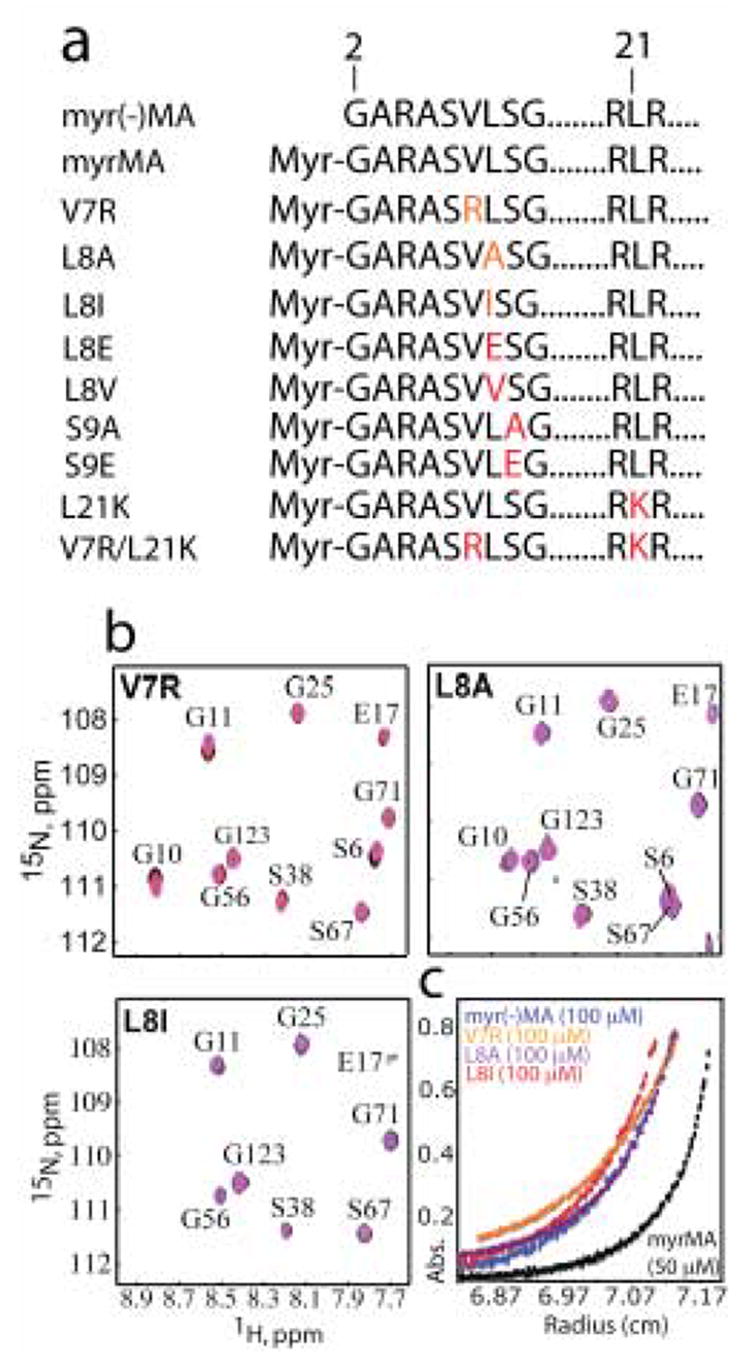
(a) A representation of the MA sequence showing positions of the mutations in red. (b) Overlay of 2D 1H-15N HSQC spectra collected at different concentrations [600–900 μM (black), 300 μM red, 150 μM (blue), 50 μM (magenta)] for V7R, L8A, and L8I samples. (c) Representative sedimentation profiles obtained for myr(-)MA, WT and mutant myrMA proteins [(26,000 rpm, 20°C, 100 μM)]. For WT myrMA, best fits were obtained for a monomer-trimer equilibrium affording association constant (Kassoc) of 2.5 ± 0.6 × 108 M−2.27 Sedimentation equilibrium curves for myr(-)MA, V7R, L8A, and L8I fit best to monomeric species.
Results
V7R, L8A and L8I point mutations inhibit myristate exposure and protein self-association
Single point mutations in the HIV-1 MA protein (V7R, L8A and L8I) were made by site-directed mutagenesis of a co-expression vector harboring the yeast N-terminal myristyl transferase (yNMT) and HIV-1 MA gene. 27 These substitutions did not adversely affect myristylation efficiencies (~100%, compared to ~90/10% myr/myr(-) for the wild-type MA protein; Mmeas:Mcalc = 15801.4:15801.7, 15701.9:15702.5, and 15744.9:15744.6 for V7R, L8A and L8I MA, respectively). 1H and 15N NMR signals observed in two dimensional (2D) [1H–15N] HSQC spectra obtained for V7R, L8A and L8I were insensitive to concentration over the range of 50 μM to 0.9 mM (Figure 1). In contrast, for the wild type (WT) myrMA protein, a subset of signals shift progressively toward the frequencies observed for myr(-)MA upon increasing the protein concentration due to a concomitant shift in the monomer-trimer equilibrium toward the trimeric species and exposure of the myristyl group.27 The absence of concentration-dependent shifts for V7R, L8A, and L8I indicates that these mutants exist in a unique conformation over the range of concentrations tested. Sedimentation equilibrium (SE) data obtained for V7R, L8A and L8I are similar in appearance to SE data obtained for myr(-)MA, and confirm that the myrMA mutants remain monomeric at concentrations as high as 1 mM.
NMR analysis of V7R-, L8A- and L8I-myrMA
To elucidate the conformational changes that occur upon substituting a single amino acid in the N-terminus of myrMA, a combination of 15N-, 13C-, 15N/13C-, 13C/13C- and 13C-edited/12C-double-half-filtered NOE experiments were collected for V7R, L8A, and L8I proteins. In all spectra obtained for the three mutants, minor changes were observed in the pattern of NMR shifts and intensities for residues located in close proximity to the mutation site (Ser-6 – Gly-10). For residues 11–132, no new intraprotein NOEs were detected that would be indicative of an altered protein conformation. Representative portions of the 4D 15N,13C-edietd NOE data obtained for V7R are presented in Figure 2. NOE cross-peaks between protein residues and myristate group are not observed in this dataset because the myristate group is not 13C-labeled. However, 2D NOESY, 3D 13C-edited NOE, and 3D 13C-edited/12C-double-half-filtered NOE data obtained for V7R, L8A, and L8I show numerous unambiguous NOEs between the myristate group and the side chains of Val-/Arg-7, Leu-/Ala-/Ile-8, Trp-16, ILe-34, Val-35, Ser-38, Gly-49, Leu-51, Glu-52, and Leu-85 (Figure 2 and 3). For V7R, NOEs between the myristate group and both Arg-7 (Hβ2/3, Hγ2/3, and Hδ2/3) and Leu-8 (Hβ2/3, Hγ, and Hδ1/2) side chains are clearly observed in the 3D 13C-edited/12C-double-half-filtered NOE data (Figure 2), indicating that the myristate group is sequestered and that the side chains of Arg-7 and Leu-8 are packed against the myristate group. Similar NOEs between the myristate group and side chains of Val-7 and Ala/Ile-8 were also observed for L8A and L8I (Figure 3). NOEs between the myristate group and the side chains of Val-7 and Leu-8 were also observed for the WT myrMA protein. 27 These NMR observations indicate that mutation of Arg-7 and Leu-8 does not greatly alter the conformation of the loop and that residues 7 and 8 play an important role into the stabilization of the myr(s) form.
Figure 2.
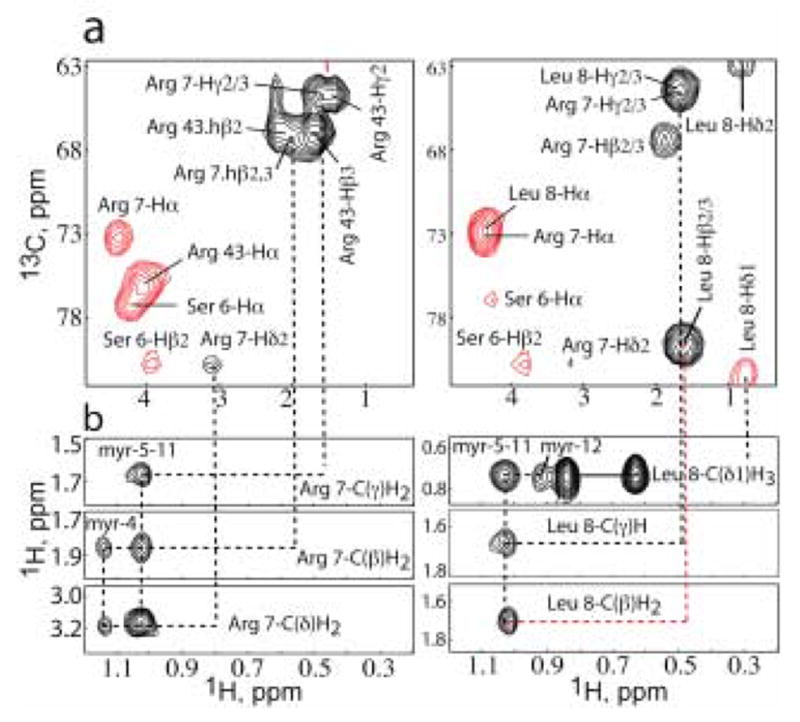
Representative NMR data obtained for V7R. (a) 4D 15N,13C-edited NOE data showing intramolecular and intermolecular NOEs for residues Arg-7 (left) and Leu-8 (right). NOE cross-peaks to the myristate group are not observed because the myristate group is not 13C-labeled. (b) 13C-edited/12C-double-half-filtered NOE data showing unambiguously assigned intermolecular NOEs between unlabeled myristyl group and 13C-labeled protein. Solid lines denote 1H-12C breakthrough doublets NOE peaks.
Figure 3.

(a) Representative 13C-edited/12C-double-half-filtered NOE data showing unambiguously assigned intermolecular NOEs between unlabeled myristyl group and 13C-labeled V7R (a), L8A (b), and L8I (c). Solid and dashed lines denote 1H-12C breakthrough doublets and intermolecular NOE peaks, respectively.
For V7R and L8I, the terminal methyl group (myr-C14H3) exhibited an upfield chemical shift comparable to that observed for the WT myrMA protein (~0.5 ppm). 27 Structural analysis of the WT myrMA protein revealed that the terminal myr-C14H3 group packs against the side chain of Trp-16. For L8A, the myr-C14H3 group exhibited a larger upfield shift (~0.3 ppm), indicating that the methyl group is packed even closer to the aromatic ring of Trp-16.
Structures of V7R- and L8A-myrMA
We chose V7R and L8A proteins as representative models to study the structural changes that led to the stabilization of myristyl-sequestered form. Superpositions of 20 structures for V7R and L8A generated from NMR data (500–800 μM and 35°C) are shown in Figure 4. The backbone atoms of Val-7–Ile-104 are well defined and are very similar to the coordinates of the WT myrMA protein (Figure 5 and Table 1),27,31 indicating that single mutations in the N-terminal loop had no effect on the globular fold of the protein. The myristyl group adopts an extended conformation with the myr-C14H3 group packing in close proximity to the side chains of core residues Trp-16, Ile-34, and Leu-85, and other myristate methylene groups interacting with the side chains of Ala-5, Ser-6, Val-7, Ile-34, Ser-38, Pro-48, Val-35, Leu-51, and Glu-52. In all spectra obtained, NOEs to the myristate were observed, but no new intraprotein NOEs were detected that would be indicative of an altered protein conformation. In addition, the relative NOE intensities and cross-peak patterns matched the data obtained for the WT myr(s)MA protein. 27,31
Figure 4.
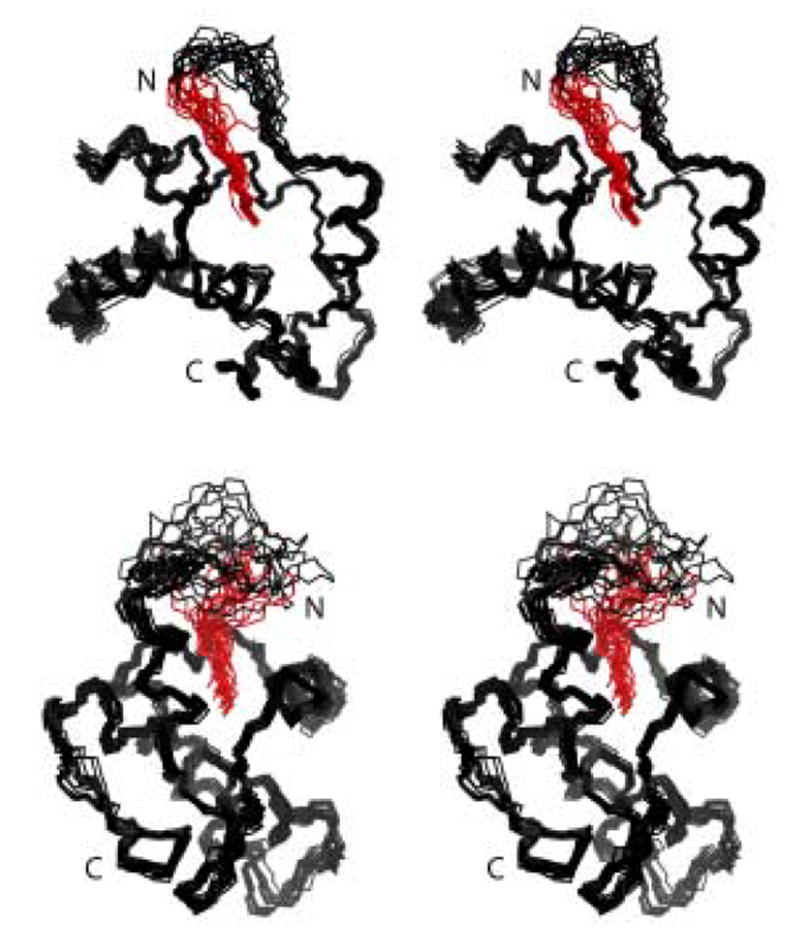
Stereoviews showing the best-fit backbone superpositions of the 20 refined structures calculated for V7R (top) and L8A (bottom) proteins. Myristate group is shown in red.
Figure 5.
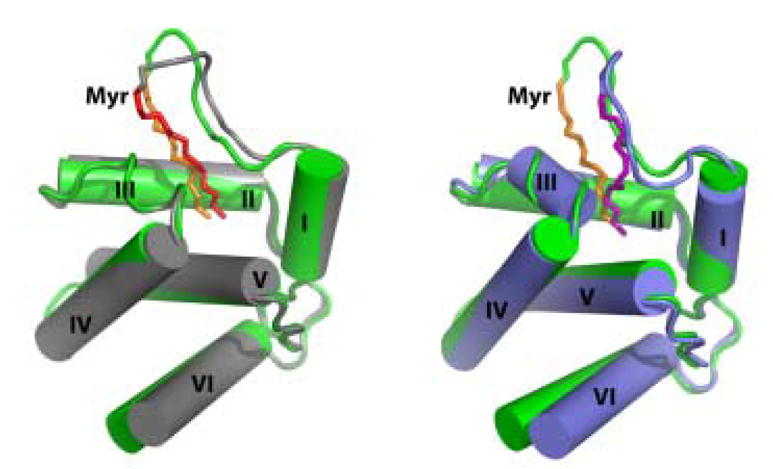
Representative structures of V7R (grey) and L8A (slate) superimposed with WT myrMA protein (green). Myristate groups of WT myrMA, V7R- and L8A proteins are shown in orange, red and purple, respectively. NMR data revealed that Helix VI is flexible for native and mutant myrMA proteins.
Table 1.
Statistics for HIV-1 V7R and L8A-myrMA Structures1
| NMR-derived restraints | V7R | L8A |
|---|---|---|
| 1H-13H distance restraints | 834 | 818 |
| Intraresidue | 81 | 80 |
| Sequential(| i − j| = 1) | 165 | 166 |
| Medium & Long range(| i − j | > 1) | 579 | 565 |
| Backbone H-bonds (4/H-bond) | 0 | 0 |
| Side-chain H-bonds (4/H-bond) | 20 | 16 |
| Dipolar coupling restraints | 87 | 88 |
| Intermolecular | 0 | 0 |
| Average restraints/refined residue | 14.8 | 14.1 |
| Target function (Å2) | ||
| Mean (± Std Dev) | 1.29 (0.23) | 1.56 (0.20) |
| Minimum | 0.79 | 0.98 |
| Maximum | 1.58 | 1.73 |
| Restraint violations | ||
| Av. max. upper dist. viol. (Å) | 0.36 | 0.39 |
| (± Std Dev) | 0.06 | 0.09 |
| Av. max. VDW viol. (Å) | 0.30 | 0.28 |
| (± Std Dev) | 0.07 | 0.08 |
| Av. max RDC viol. (Hz) | 0.40 | 0.36 |
| (± Std Dev) | 0.16 | 0.11 |
| Structure Convergence (Å)2 | ||
| Main chain atoms | ||
| Mean (± Std Dev) | 0.48 (0.11) | 0.58 (0.08) |
| All heavy atoms | ||
| Mean (± Std Dev) | 0.95 (0.10) | 1.02 (0.09) |
A total of 20 refined structures per ensemble (see Materials and Methods for structure refinement details).
RMSDs calculated relative to mean atom coordinates for residues 10 to 30 and 35 to 100
The main differences between the structures are localized within the disordered loop formed by residues Gly-2 – Ser-9. For V7R, the side chain of Arg-7 is poised to make a salt bridge with the side chain of Glu-52, and the mythelene groups of Arg-7 contribute to the hydrophobic interactions with the methylene groups of the myristate (Figure 6). For L8A, the smaller size of Ala-8 creates a better cavity for the myristate group, which packs closer to Trp-16 as evidenced by the upfield shift of the terminal myr-C14H3 (Figure 6). Thus, structural changes implicated by the chemical shift data involve only residues located near the mutation site, which may have only average intrinsic conformational mobility.
Figure 6.
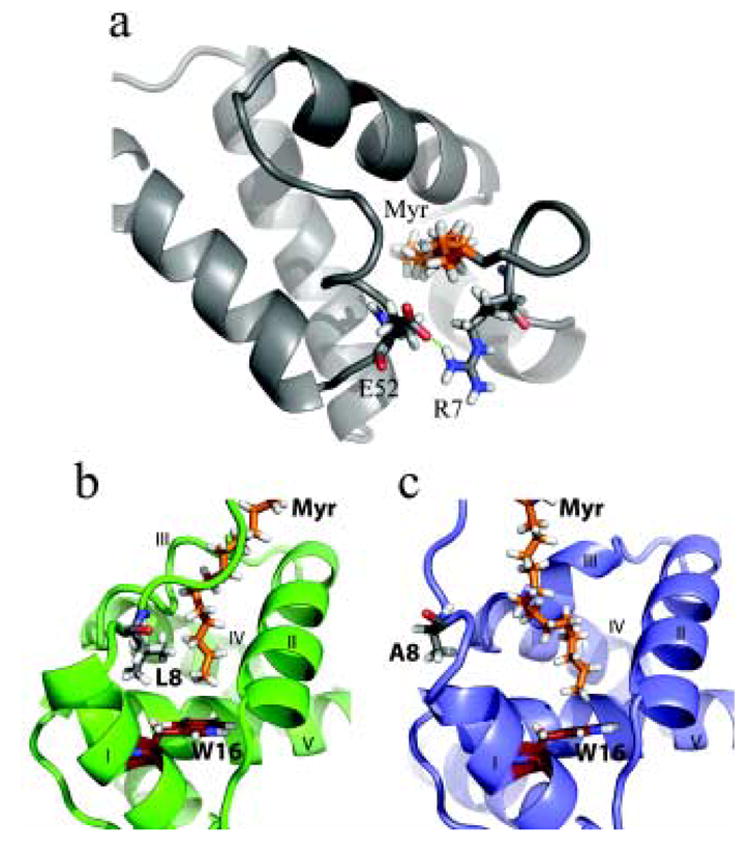
Cartoon and stick representation showing minor conformational adjustments in the N terminus of V7R and L8A proteins compared with WT myrMA. (a) For V7R, a salt bridge between Arg-7 and Glu-52 stabilizes the myr(s) form. (b) When compared with WT myrMA (green), substitution of Leu-8 with Ala (slate, c) creates a better cavity for the myristate group (orange) and allows for the terminal methyl of myristate group to pack closer to Trp-16 (firebrick).
PI(4,5)P2 binds to myrMA mutants but does not trigger myristate exposure
Binding studies were conducted with di-C4-phosphatidylinositol phosphate (di-C4-PI(4,5)P2), a soluble form of PI(4,5)P2 containing truncated lipids. Representative 1H-15N HSQC NMR data obtained upon titration of V7R with di-C4-PI(4,5)P2 are shown in Figure 7. Titration of di-C4-PI(4,5)P2 led to significant changes in the backbone 1H and 15N NMR chemical shifts of residues Arg-22, Lys-26, Lys-27, His-33, Glu-73, Glu-74, Leu-75 and Ser-77, (Group 1; ΔδHN ((Δδ1H)2+(Δδ15N)2)1/2 = 0.1–0.8 ppm) (Figure 7). These residues reside on the β-II-V cleft, and were previously shown to contribute to the PI(4,5)P2 binding site. Non-linear least squares fits of the titration data afforded a dissociation constant (Kd) value (261 ± 18 μM), similar to those observed for WT myr(-)MA and myrMA (Figure 7 and Table 2). Titration of di-C4-PI(4,5)P2 into L8A and L8I mutants also led to signal shifts and Kd values that are very similar to those obtained for V7R (Table 2). NMR data obtained for V7R (Figure 7), L8A, and L8I proteins confirmed that di-C4-PI(4,5)P2 binds to these mutants in a manner identical to that observed for the WT myr(-) and myrMA proteins 31.
Figure 7.
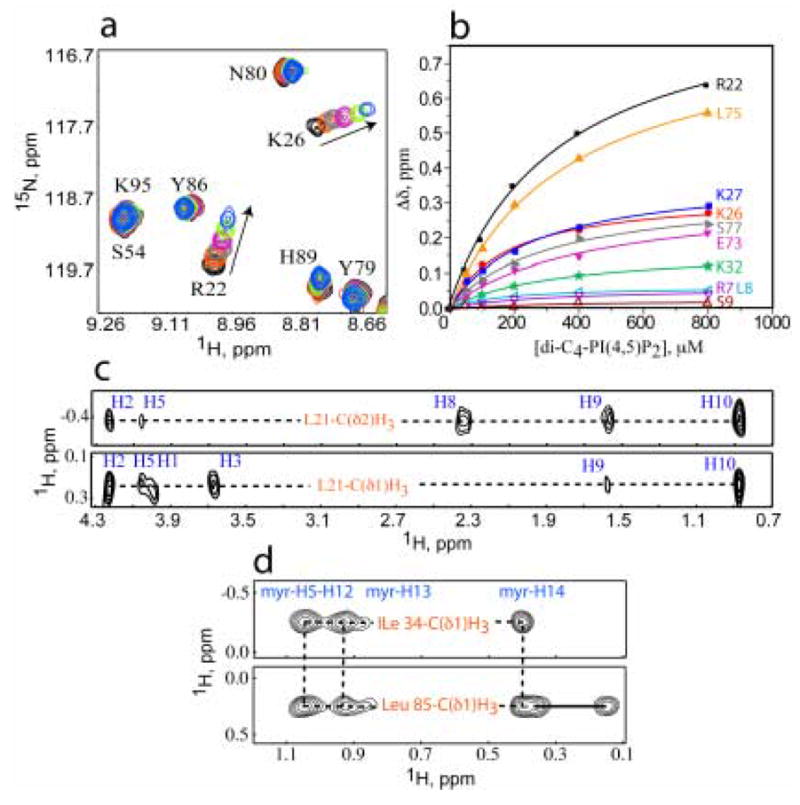
(a) Overlay of 2D 1H-15N HSQC spectra upon titration of V7R with di-C4-PI(4,5)P2 [50 μM, 35 °C; di-C4-PI(4,5)P2:MA = 0:1 (black), 1:1 (red) , 2:1 (grey), 4:1 (magenta), 8:1 (green), 16:1 (blue)]. (b) 15N NMR chemical shift titration data, which fit to 1:1 binding isotherms (Kd = 261 ± 18 μM). Representative 13C-edited/12C-double-half-filtered NOE data showing unambiguously assigned intermolecular NOEs between (c) Leu-21 and di-C4-PI(4,5)P2, and (d) Ile-34 and Leu-85 to the myristate group. Solid and dashed lines denote 1H-12C breakthrough doublets and intermolecular NOE peaks, respectively.
Table 2.
Summary of Kd (μM) values for di-C4-PI(4,5)P2 binding to MA proteins.
| Sample | Kd (μM) |
|---|---|
| myr(-)MAa | 240 ± 60 |
| myrMAa | 150 ± 30 |
| V7R | 261 ± 18 |
| L8A | 150 ± 24 |
| L8I | 108 ± 5 |
| L21K | 67 ± 5 |
| V7R-L21K | 84 ± 9 |
Values reported previously (ref 31)
Interestingly, 1H and 15N NMR signal corresponding to residues Gly-2-Ser-9, Glu-12-Asp-15, Arg-39, and His 89 “group 2” did not exhibit detectable chemical shift changes upon titration of V7R, L8A, and L8I with di-C4-PI(4,5)P2 (Figure 7). Chemical shift changes of “group 2” were observed for WT myrMA upon addition of di-C4-PI(4,5)P2 and NMR-based structural studies confirmed that they reflect di-C4-PI(4,5)P2-dependent exposure of the myristyl group. 31 In addition, intermolecular NOEs between protein residues and myristate group are clearly observed in the 3D 13C-edited/12C-double-half-filtered NOE experiment obtained for the V7R:di-C4-PI(4,5)P2 complex (Figure 7), indicating that di-C4-PI(4,5)P2 binding does not trigger myristyl exposure.
Compensatory mutant V7R/L21K-myrMA binds PI(4,5)P2 with enhanced affinity
To understand how a compensatory mutation (Leu-21) reversed the membrane-binding defect imposed by V7R, we collected NMR and thermodynamic data for V7R/L21K and L21K protein samples. 2D [1H–15N] HSQC spectra obtained for L21K at concentrations of 50 – 900 μM showed that as protein concentration is increased, several signals (e.g., those corresponding to Ser 6, Gly 10, Gly 11) shifted progressively towards the corresponding frequencies of myr(-)MA. These signal shifts are similar to those observed for WT myrMA, and are indicative of a shift in equilibrium from myr(s) to myr(e) form. SE data obtained for L21K protein confirm that the protein exists in a monomer-trimer equilibrium with Kassoc = 3.7 ± 0.4 × 108 M−2 (data not shown). Interestingly, [1H–15N] HSQC spectra for V7R/L21K show that signals were insensitive to protein concentration and SE data confirm that V7R/L21K is a monomer, indicating that a second mutation (L21K) does not trigger myristyl exposure.
Structural studies on the WT myrMA protein bound to PI(4,5)P2 revealed that the side chains of Leu-21 play a major role in stabilizing the hydrophobic interactions with the 2’-acyl chain of PI(4,5)P2. Thus, we hypothesized that substitution of Leu-21 with lysine may affect the binding affinity of PI(4,5)P2 to the MA protein. For L21K and V7R/L21K, 1H-15N HSQC NMR data obtained upon titration with di-C4-PI(4,5)P2 showed that “group 1” peaks exhibited similar shifts to those observed for V7R, L8A and L8I as well as WT myr(-)MA and myrMA. Non-linear least squares fits of the titration data for L21K and V7R/L21K afforded binding affinity values (Figure 7 and Table 2), which are ~2-fold greater than WT myrMA. For L21K, group 2 signals also exhibited di-C4-PI(4,5)P2-dependent 1H-15N HSQC signals that shift progressively toward values observed for myr(-)MA. Analysis of a combination of 15N-, 15N/13C- and 13C-edited/12C-double-half-filtered NOE experiments confirm that di-C4-PI(4,5)P2 binds to the II-V-β cleft on L21K and V7R/L21K in a manner very similar to the WT myrMA protein (Figure 7). Interestingly, for V7R/L21K the 1H-15N HSQC signals corresponding to “group 2” residues did not exhibit chemical shift changes upon titration with di-C4-PI(4,5)P2, indicating that although di-C4-PI(4,5)P2 binds to V7R/L21K with affinity similar to that observed for L21K, that binding does not trigger myristate exposure.
Point mutations near the amino-terminus of myrMA inhibit PM binding in vivo
Previous studies demonstrated that point mutations near the amino terminus of myrMA can lead to defects in membrane association. 19,24–26 We have now determined the in vivo localization patterns of HIV-1 Gag proteins containing the following point mutations: L8E, L8V, S9A, S9E, V7R, and V7R/L21K (Figure 1). As expected from our membrane binding studies, all of the single mutants show an increase in hazy cytosolic staining relative to WT (Figure 8), consistent with their membrane-binding defect. This hazy, non-punctate staining pattern is typical of HIV-1 Gag mutants that are defective in membrane binding33,34. This Gag localization phenotype is distinct from that induced by mutations in the MA "80's domain” or the MA basic domain, which show retargeting to the MVB 5,12,19,35. Thus, consistent with earlier biochemical data24, these mutations appear to induce defects in membrane binding rather than targeting. Interestingly, V7R/L21K displays a highly punctate, non-diffuse cell-surface Gag localization pattern, consistent with the ability of the L21K mutation to correct the membrane-binding defect imposed by V7R24.
Figure 8.
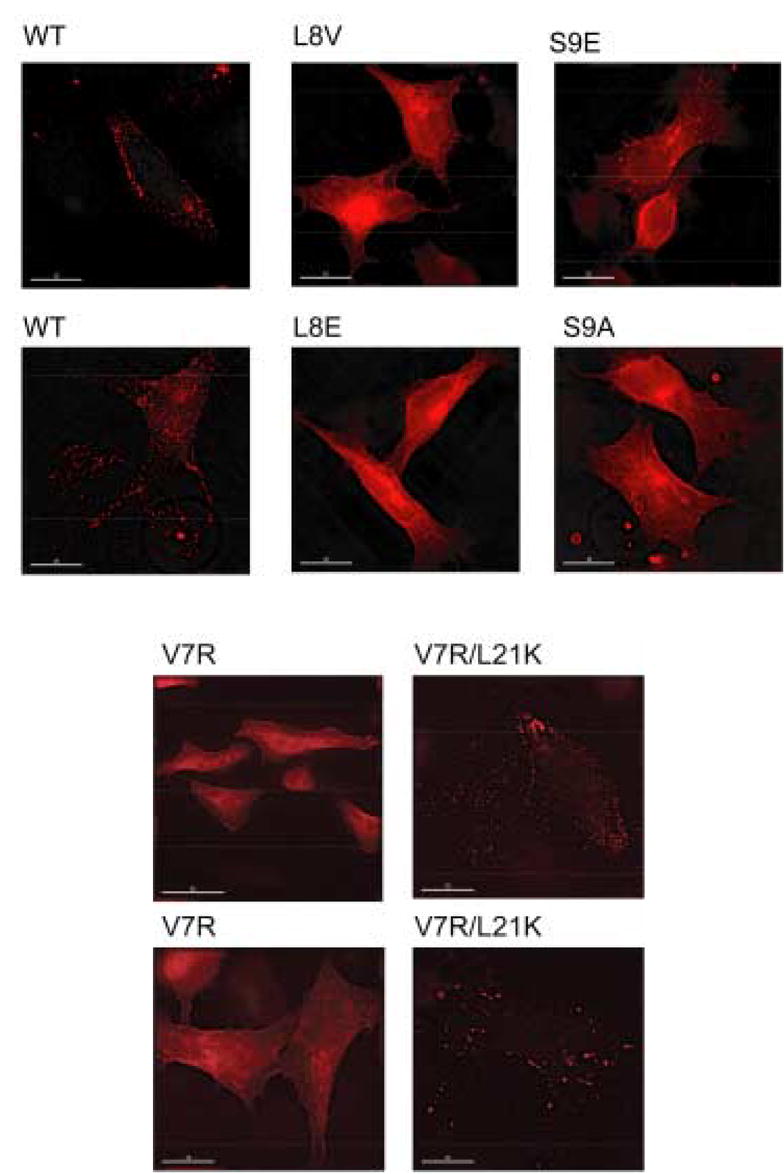
Subcellular localization of MA mutants. HeLa cells were transfected with WT pNL4-3 or derivatives encoding the indicated MA mutants. Cells were fixed, permeabilized, and stained with anti-p17 monoclonal antibody and Texas red-conjugated anti-mouse IgG. Cells were mounted and examined using a Delta Vision RT microscope and the images were deconvolved. Two representative fields for WT, V7R, and V7R/L21K are shown.
Discussion
Previous in vivo mutagenesis studies have provided important insights into the mechanism of retrovirus assembly. Mutations of the first five residues of the HIV-1 MA domain that inhibit myristoylation lead to the accumulation of Gag in the cytosol, indicating that myristoylation is required for efficient membrane binding. Substitution of basic residues between Lys-18 and Lys-32, which form a basic patch on the surface of MA, retargets Gag to a cytoplasmic compartment 5,12,19,35, and in vitro membrane binding studies support the hypothesis that the myristyl group and basic patch cooperatively facilitate membrane binding 5,12,19,35. Mutations at positions 7 and 8 of the MA domain (V7R, L8A, and L8I), which do not inhibit myristoylation, have also been shown to inhibit membrane binding in cells, suggesting that the mutations might inhibit myristate exposure. 19,24–26 The findings presented here demonstrate that these mutations inhibit PM localization in vivo and turn off the myristyl switch. The myristyl switch equilibrium is sensitive to subtle molecular changes, and even the conservative substitution of Leu-8 by Ile is sufficient to block myristate exposure under conditions in which exposure is normally highly favored. These studies further demonstrate that the mutations lead to only minor structural changes that are proximal to the mutation sites.
Recent studies indicate that PI(4,5)P2, a member of a family of differentially phosphorylated phosphatidylinositides that serve as membrane markers for specific cellular proteins 28–30, plays a critical role in the ultimate localization of Gag at the PM.32 PI(4,5)P2 is normally associated with the inner leaflet of the plasma membrane 29, and depletion of PI(4,5)P2 leads to accumulation of Gag at the membranes of late endosomes and MVBs 32. In addition, induction of PI(4,5)P2-enriched endosomes retargets Gag to endosomes and induces intra-vesicle budding 32. In both cases, virus production is severely attenuated32. PI(4,5)P2 binds MA in an “extended lipid” conformation in which the inositol head group and the 2’-fatty acid chain bind to a hydrophobic cleft formed by helices II and V, and a meandering β-hairpin (β-II–V), and the remaining 1’-fatty acid and exposed myristyl group bracket a conserved basic surface patch previously implicated in membrane binding36. These findings suggested to us that the position-7 and –8 mutations, and the revertant mutants, might affect PI(4,5)P2 binding or the sensitivity of PI(4,5)P2 binding on the myristyl switch. The present studies demonstrate that PI(4,5)P2 binds to V7R, L8A, and L8I with affinities similar to that observed for the native protein. Furthermore, the NMR chemical shift and NOE data indicate these mutants bind PI(4,5)P2 in a manner that is essentially identical to that observed for the wild-type protein. However, PI(4,5)P2 binding does not trigger myristate exposure in the V7R, L8A and L8I mutants, indicating that the coupling between the PI(4,5)P2 binding site and the pocket that sequesters the myristyl group is weak. The fact that PI(4,5)P2 binding is not attenuated by these mutations suggests that membrane binding in vivo may be mediated by a tripartite mechanism, in which the exposed myristyl group, the extended PI(4,5)P2 bridge, and the basic patch collectively contribute to binding.
The defects in virus assembly and membrane binding imposed by amino acid substitutions at positions 7 and 8 can be reversed by intragenic second-site changes in the core of the protein24–26. In particular, L21K and K98E reversed the phenotype imposed by V7R, which led to the suggestion that these second mutations likely trigger myristate exposure to facilitate membrane binding. The present studies unexpectedly reveal that substitution of Leu-21 by Lys does not promote myristate exposure in either the L21K or L21K/V7R MA proteins. Instead, these mutants bind PI(4,5)P2 with ~2-fold greater affinity than either the WT or V7R, L8A, and L8I mutants of the MA protein. It thus appears that this rather modest increase in binding affinity is sufficient to compensate for the inability of the protein to expose its myristyl group.
In related studies, the substitution of Val-7 and Leu-8 by glutamine (V7Q and L8Q) in the simian immunodeficiency virus (SIV) MA protein was shown to dramatically inhibit virus production and membrane association without affecting myristoylation 37. HIV-1 and SIV MA proteins are highly homologous (~50% sequence identity and ~80% homologous), and the three-dimensional structures of the unmyristoylated MA proteins solved by NMR and X-ray techniques are very similar 36,38,39. It is thus likely that the mechanism used by SIV to regulate membrane targeting and binding is similar to the mechanism employed by HIV-1.
In summary, we have demonstrated that mutations near the amino-terminus of the HIV-1 MA protein that inhibit membrane binding in vitro also lead to cytosolic localization of Gag in vivo. This loss of membrane association is due to the inability of the mutant MA proteins to expose their myristyl group. The myristyl switch equilibrium is highly sensitive to mutations near the N-terminus of the protein, and even the conservative substitution of Leu 8 by Ile is sufficient to “lock off” the myristyl switch. The myristyl switch unexpectedly remains locked off in the revertant mutant V7R/L21K, which assembles at the PM and exhibits a wild-type phenotype. This behavior appears to result from enhanced interactions with PI(4,5)P2, which binds L21K- and V7R/L21K-MA with affinities that are about two-fold greater than that of the native MA protein. However, we cannot rule out the possibility that the increased charge and location of the L21K substitution might also directly influence membrane binding, and future studies of the interactions between these proteins with membranes, including those containing PI(4,5)P2, are warranted.
Materials and Methods
Sample preparation
HIV-1 myrMA constructs containing amino acid changes V7R, L8I, L8A, L21K and V7R/L21K were made by using QuickChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) and sequenced at the DNA sequencing facility, UMBC. Protein samples were prepared as described,27,38 except that a modified protocol was applied to prepare 15N-, and 13C-,15N-labeled samples.40 Cells were first grown in 4L LB media at 37°C until O.D.600 reached ~0.6 – 0.7. Next, cells were spun down and washed with 1X M9 salt before transferring them to 1L M9 minimal media containing 15NH4Cl as the sole nitrogen source to produce 15N-labeled protein. At this stage, cells were supplemented with myristic acid (10 mg/liter; Sigma) then were grown for 1h before induction with isopropyl -D-thiogalactoside (1 mM). Cells were grown at 37°C for ~12 – 14h, lysed using microfluidizer, and the proteins purified by cobalt affinity chromatography (Clontech, Mountain View, CA) and ion exchange column chromatography (SP column). Traces of unmyristoylated species (for L21K) were separated from myristoylated sample by butyl-Sepharose hydrophobicity chromatography (GE Healthcare, Pittsburg, PA). We obtained a high yield (~25 mg/L) of 93 – 97% isotopically 15N- and 15N/13C-enriched proteins. Molecular weights and efficiency of myristylation were confirmed by electrospray ionization mass spectrometry. Phosphoinositide di-C4-PI(4,5)P2 (Echelon, Salt Lake City, UT) was obtained commercially and used without further purification. Samples for all NMR experiments were prepared in 50 mM sodium phosphate at pH 5.5, 100 mM NaCl and 5 mM DTT. NaCl was excluded from the NMR buffer for di-C4-PI(4,5)P2 titration experiments.
NMR spectroscopy
NMR data were collected with Bruker DRX (800 MHz 1H) and DMX (600 MHz 1H) spectrometers equipped with cryoprobes, processed with NMRPIPE41 and analyzed with NMRVIEW42. A combination of two-, three- and four-dimensional NOESY data were obtained for combinations of natural abundance, 15N- and 15N-,13C-labeled protein samples (35 °C). Protein backbone signals were assigned using standard triple resonance methods (HNCA and HNCOCA), and side chain signals were assigned from 3D and 4D 15N-, 13C-, and 15N/13C-edited NOESY data. Intermolecular 1H-1H NOEs between 15N-,13C-labeled protein and unlabeled myristate group were assigned from 2D (1H-1H) and 3D (13C- and 13C-edited/12C-double-half-filtered) NOESY data (see references 43–45 and citations there-in). 1H-15N residual dipolar couplings were measured using a modified in-phase/anti-phase (IPAP) HSQC experiment 46 for V7R and L8A samples aligned in polyacrylamide gel 47. Binding isotherms from 1H-15N NMR HSQC titration experiments were calculated with ORIGIN 7.0 software (MicoCal, Northampton, MA).
Analytical ultracentrifugation
Sedimentation equilibrium measurements were collected on a Beckman XL-I Optima system equipped with a four-hole An-60 rotor (Beckman Coulter) and cells were equipped with double-sectored centerpieces of path length 12 or 3 mm and quartz windows. Protein samples were prepared in 50 mM sodium phosphate buffer at pH 5.5 containing 100 mM NaCl and 2 mM Tris (2-carboxy-ethyl)-phosphine-HCl. Loading concentrations varied from 50 to 150 μM for all samples. Rotor speeds were at 22,000 and 26,000 rpm, and temperature was 20 °C. Scans were obtained by using the absorption optics system with 0.002 cm step size and four averages per point, and acquired at wavelengths of 280 and 295 nm. Partial specific volumes (v-bar) and molar extinction coefficients were calculated by using the program SEDNTERP (www.jphilo.mail-way.com) and buffer densities were measured pycnometrically. Data analysis was performed by using NONLIN.48 Equilibrium association constants were determined by global analysis of data acquired from samples prepared at two loading concentrations and centrifuged at two rotor speeds.
Structure calculations
Structures were calculated in torsion angle space with CYANA (http://www.las.jp/index_eg.html) starting from random initial angles. Upper interproton distance bounds of 2.7, 3.3 and 5.0 Å (with appropriate corrections for pseudoatoms) were employed for NOE cross peaks of strong, medium, and weak intensity respectively, which were qualitatively determined following intensity normalization of the different NOE data sets. No backbone hydrogen bond or chemical shift-based torsion angle restraints were employed. Statistical information is provided in Table 1. Structure figures were generated with PyMOL (http://pymol.sourceforge.net).
Fluorescence microscopy
Construction of derivatives of the HIV-1 pNL4-3 clone containing MA mutations V7R, L8V, L8E, S9A, S9E and V7R/L21K has been described elsewhere.19,24,25 The numbering scheme of these mutations has been changed to be consistent with the scheme we used in earlier structural studies. HeLa cells cultured in chamber slides (Nunc) were transfected with either wild type pNL4-3 or the indicated MA mutants in the pNL4-3 backbone using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). Cells were fixed 24–48 hr posttransfection with 3.7% formaldehyde in 100 mM sodium phosphate buffer (pH 7.2) for 20 min, and permeabilized with phosphate-buffered saline (PBS) containing 0.1% Triton X-100. Gag protein in transfected cells was visualized by incubating with anti-p17 monoclonal antibody (Advanced Biotechnologioes Inc, Columbia, MD) diluted in PBS containing 3% BSA followed by Texas red-conjugated anti-mouse IgG. Cells were mounted with Aqua Poly Mount (Polysciences Inc, Warrington, PA) and examined using a Delta Vision RT microscope.
Atomic coordinates
The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org, and are available under the accession codes 2JMG and 2NV3 for V7R and L8A, respectively.
Supplementary Material
Acknowledgments
This work was supported by NIH grant AI30917. We thank the HHMI staff and colleagues at UMBC, Dr. Dorothy Beckett (University of Maryland, College Park), and David King (HHMI, U.C. Berkeley) for technical support. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (EOF). P.J. and M.L. are Meyerhoff undergraduate scholars, and E.L. and J.T. are UMBC Presidential and MARC undergraduate scholars, respectively.
Abbreviations used
- MA
myristoylated HIV-1 matrix protein
- Gag
myristoylated HIV-1 Gag polyprotein
- myr(-)
unmyristoylated
- myr(s)
myristate-sequestered state
- myr(e)
myristate-exposed state
- PI(4,5)P2
phosphatidylinositol-(4,5)-bisphosphate
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single quantum coherence
- AU
analytical ultracentrifugation
- RDC
residual dipolar coupling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coffin JM, Hughes SH, Varmus HE. Retroviruses. Cold Spring Harbor Laboratory Press; Plainview, N.Y.: 1997. [PubMed] [Google Scholar]
- 2.Morita E, Sundquist WI. Retrovirus Budding. Annual Review of Cell and Develpomental Biology. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 3.von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, Scott A, Krausslich HG, Kaplan J, Morham SG, Sundquist WI. The protein network of HIV budding. Cell. 2003;114:701–713. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 4.Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, Peters TR, Dermody TS, Woodruff E, Wang JJ, Spearman P. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell. 2005;120:663–674. doi: 10.1016/j.cell.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 5.Hermida-Matsumoto L, Resh MD. Localization of Human Immunodeficiency virus Type 1 Gag and env at the Plasma Membrane by Confocal Imagine. Journal of Virology. 2000;74:8670–8679. doi: 10.1128/jvi.74.18.8670-8679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen DG, Booth A, Gould SJ, Hildreth JE. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J Biol Chem. 2003;278:52347–52354. doi: 10.1074/jbc.M309009200. [DOI] [PubMed] [Google Scholar]
- 7.Ono A, Freed EO. Cell-Type-Dependent Tageting of Human Immunodeficiency Virus Type 1 Assembly to the Plasma Membrane and the Multivesicular body. Journal of Virology. 2004;78:1552–1563. doi: 10.1128/JVI.78.3.1552-1563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raposo G, Moore M, Innes D, Leijendekker R, Leigh-Brown A, Benaroch P, Geuze H. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic. 2002;3:718–729. doi: 10.1034/j.1600-0854.2002.31004.x. [DOI] [PubMed] [Google Scholar]
- 9.Jouvenet N, Neil SJD, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. PLoS Biology. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci USA. 2001;98:13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ono A, Freed EO. Role of lipid rafts in virus replication. Adv Virus Res. 2005;64:311–358. doi: 10.1016/S0065-3527(05)64010-9. [DOI] [PubMed] [Google Scholar]
- 12.Yuan X, Yu X, Lee TH, Essex M. Mutations in the N-Terminal Region of Human Immunodeficiency Virus Type I Matrix Protein Block Intracellular Transport of the Gag Precursor. Journal of Virology. 1993;67:6387–6394. doi: 10.1128/jvi.67.11.6387-6394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou W, Parent LJ, Wills JW, Resh MD. Identification of a Membrane-Binding Domain Within the Amino-Terminal Region of Human Immunodeficiency Virus Type 1 Gag Protein Which Interacts with Acidic Phospholipids. Journal of Virology. 1994;68:2556–2569. doi: 10.1128/jvi.68.4.2556-2569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bryant M, Ratner L. Myristoylation-Dependent Replication and Assembly of Human Immunodeficiency Virus 1. Proceedings of the National Academy of Sciences USA. 1990;87:523–527. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Copeland NG, Jenkins NA, Nexo B, Schultz AM, Rein A, Mikkelsen T, Jorgensen P. Poorly expressed endogenous ecotropic provirus of DBA/2 mice encodes a mutant Pr65gag protein that is not myristylated. J Virol. 1988;62:479–487. doi: 10.1128/jvi.62.2.479-487.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Göttlinger HG, Sodroski JG, Haseltine WA. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proceedings of the National Academy of Sciences USA. 1989;86 :5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spearman P, Horton R, Ratner L, Kuli-Zade I. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J Virol. 1997;71:6582–6592. doi: 10.1128/jvi.71.9.6582-6592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spearman P, Wang JJ, Vander Heyden N, Ratner L. Identification of Human Immunodeficiency Virus Type 1 Gag Protein Domains Essential to Membrane Binding and Particle Assembly. Journal of Virology. 1994;68:3232–3242. doi: 10.1128/jvi.68.5.3232-3242.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freed EO, Orenstein JM, Buckler-White AJ, Martin MA. Single Amino Acid Changes in the Human Immunodeficiency Virus Type 1 Matrix Protein Block Virus Particle Production. Journal of Virology. 1994;68:5311–5320. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ono A, Orenstein JM, Freed EO. Role of the Gag Matrix Domain in Targeting Human Immunodeficiency Virus Type 1 assembly. Journal of Virology. 2000;74:2855–2866. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouamr F, Scarlata S, Carter CA. Role of myristylation in HIV-1 Gag assembly. Biochemistry. 2003;42:6408–6417. doi: 10.1021/bi020692z. [DOI] [PubMed] [Google Scholar]
- 22.Zhou W, Resh MD. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J Virol. 1996;70:8540–8548. doi: 10.1128/jvi.70.12.8540-8548.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindwasser OW, Resh MD. Myristoylation as a target for inhibiting HIV assembly: Unsaturated fatty acids block viral budding. Proc Natl Acad Sci USA. 2002;99:13037–13042. doi: 10.1073/pnas.212409999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ono A, Freed EO. Binding of Human Immunodeficiency Virus Type 1 gag to membrane: Role of the matrix amino terminus. J Virol. 1999;73:4136–4144. doi: 10.1128/jvi.73.5.4136-4144.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ono A, Huang M, Freed EO. Characterization of human immunodeficiency virus type 1 matrix revertants: effects on virus assembly, Gag processing, and Env incorporation into virions. J Virol. 1997;71:4409–4418. doi: 10.1128/jvi.71.6.4409-4418.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paillart JC, Gottlinger HG. Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of Gag membrane targeting. J Virol. 1999;73:2604–2612. doi: 10.1128/jvi.73.4.2604-2612.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci USA. 2004;101:517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin TFJ. PI(4,5)P2 regulation of surface membrane traffic. Curr Opin Cell Biol. 2001;13:493–499. doi: 10.1016/s0955-0674(00)00241-6. [DOI] [PubMed] [Google Scholar]
- 29.Behnia R, Munro S. Organelle identity and the signposts for membrane traffic. Nature. 2005;438:597–604. doi: 10.1038/nature04397. [DOI] [PubMed] [Google Scholar]
- 30.McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 31.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci USA. 2006;103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proceedings of the National Academy of Sciences. 2004;101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joshi A, Nagashima K, Freed EO. Mutation of dileucine-like motifs in the human immunodeficiency virus type 1 capsid disrupts virus assembly, gag-gag interactions, gag-membrane binding, and virion maturation. Journal of Virology. 2006;80:7939–7951. doi: 10.1128/JVI.00355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ono A, Demirov D, Freed EO. Relationship between human immunodeficiency virus Type-1 Gag multimerization and membrane binding. J Virol. 2000;74:5142–5150. doi: 10.1128/jvi.74.11.5142-5150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freed EO, Englund G, Martin AM. Role of the Basic Domain of Human Immondeficiency Virus Type 1 Matrix in Macrophage Infection. Journal of Virology. 1995;69:3949–3954. doi: 10.1128/jvi.69.6.3949-3954.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. Crystal Structures of the Trimeric HIV-1 Matrix Protein: Implications for Membrane Association. Proceedings of the National Academy of Sciences USA. 1996;93:3099–3104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzalez SA, Affranchino JL. Substitution of Leucine 8 in Simian Immunodeficiency Virus Matrix Protein Impairs Particle Formation Without Affecting N-Myristylation of the Gag Precursor. Virology. 1998;240:27–35. doi: 10.1006/viro.1997.8919. [DOI] [PubMed] [Google Scholar]
- 38.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. Three dimensional structure of the human immunodeficiency virus type 1 matrix protein. J Mol Biol. 1994;244:198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 39.Rao Z, Belyaev AS, Fry E, Roy P, Jones IM, Stuart DI. Crystal structure of SIV matrix antigen and implications for virus assembly. Nature. 1995;378:743–747. doi: 10.1038/378743a0. [DOI] [PubMed] [Google Scholar]
- 40.Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. Journal of Biomolecular NMR. 2001;20:71–75. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- 41.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 42.Johnson BA, Blevins RA. NMRview: a Computer Program for the Visualization and Analysis of NMR Data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 43.Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons; New York: 1986. [Google Scholar]
- 44.Kay LE, Clore GM, Bax A, Gronenborn AM. Four-dimensional heteronuclear triple-resonance NMR spectroscopy of interleukin-1β in solution. Science. 1990;249:411–414. doi: 10.1126/science.2377896. [DOI] [PubMed] [Google Scholar]
- 45.Folkers PJM, Folmer RHA, Konings RNH, Hilbers CW. Overcoming the ambiguity problem encountered in the analysis of nuclear Overhauser magnetic resonance spectra of symmetric dimer proteins. J Am Chem Soc. 1993;115:3798–3799. [Google Scholar]
- 46.Ishii Y, Markus MA, Tycko R. Controlling residual dipolar couplings in high-resolution NMR of proteins by strain induced alignment in a gel. J Biomol NMR. 2001;21:141–151. doi: 10.1023/a:1012417721455. [DOI] [PubMed] [Google Scholar]
- 47.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating streched polyacrylamide gels, yeilding uniform alignment of proteins and detergent micelles. J Biomol NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 48.Johnson ML, Faunt LM. Parameter estimation by least-squares methods. Methods Enzymol. 1992;210:1–37. doi: 10.1016/0076-6879(92)10003-v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.