

Abstract
Background
Formation of long-term memories is critically dependent on extracellular-regulated kinase (ERK) signaling. Activation of the ERK pathway by the sequential recruitment of mitogen-activated protein kinases is well understood. In contrast, the proteins that inactivate this pathway are not as well characterized.
Methods
Here we tested the hypothesis that the brain-specific protein tyrosine phosphatase STEP plays a key role in neuroplasticity and fear memory formation by its ability to regulate ERK1/2 activation.
Results
STEP co-localizes with the ERKs within neurons of the lateral amygdala. A substrate-trapping STEP protein (TAT-STEP) binds to the ERKs and prevents their nuclear translocation after glutamate stimulation in primary cell cultures. Administration of TAT-STEP into the lateral amygdala disrupts long-term potentiation (LTP) and selectively disrupts fear memory consolidation. Fear conditioning induces a bi-phasic activation of ERK1/2 in the lateral amygdala (LA) with an initial activation within 5 minutes of training, a return to baseline levels by 15 minutes, and an increase again at 1 hour. In addition, fear conditioning results in the de novo translation of STEP. Inhibitors of ERK1/2 activation or of protein translation block the synthesis of STEP within the LA after fear conditioning.
Conclusions
Together, these data imply a role for STEP in experience-dependent plasticity, and suggest that STEP modulates the activation of ERK1/2 during amygdala–dependent memory formation. The regulation of emotional memory by modulating STEP activity may represent a target for the treatment of psychiatric disorders such as PTSD, panic, and anxiety disorders.
Keywords: STEP, striatal-enriched tyrosine phosphatase, fear conditioning, amygdala, protein tyrosine phosphatase, local protein synthesis, signal transduction
Introduction
The involvement of the extracellular-regulated kinase (ERK1/2) pathway in the formation of long-term memories has added to our understanding of the development of synaptic plasticity (Sweatt, 2004). The requirement for ERK1/2 signaling in the consolidation of persistent memories has been demonstrated across numerous paradigms, such as conditioned taste aversion in the insular cortex (Berman et al., 1998) and spatial memory in the hippocampus (Blum et al., 1999). This is also true for Pavlovian auditory fear conditioning in the amygdala where converging stimulation of auditory thalamic inputs related to the conditioning stimulus (tone) and somatosensory thalamic inputs linked to the unconditioned stimulus (electrical foot shock) during training results in activation of ERK1/2 in neurons within the lateral amygdala (LA). Both inputs are required for the establishment of LTP in the LA and for the acquisition and consolidation of auditory fear conditioning (Schafe and LeDoux, 2000; Blair et al., 2001), and blockade of ERK activation impairs the consolidation of fear memory (Schafe et al., 2000; Rodriques et al., 2004).
Although the involvement of ERK1/2 in memory consolidation is well established, the mechanisms that regulate ERK1/2 signaling are not known. Previous studies have shown that the protein tyrosine phosphatase (PTP) STEP modulates ERK signaling pathways by dephosphorylating a regulatory Tyr204 within the activation domain of ERK1/2 and thereby inactivating these proteins (Paul et al., 2003; Valjent et al., 2005). Processes that increase the availability of active STEP should attenuate ERK1/2 activity, while mechanisms that prevent STEP from interacting with the ERKs should prolong ERK1/2 signaling. The effects of STEP on ERK1/2 signaling would be predicted to influence memory formation, but a direct role for STEP in memory processes has yet to be demonstrated.
STEP is localized to brain regions involved in learning and memory, including the striatum, hippocampus, amygdala, nucleus accumbens, and cortex (Lombroso et al., 1993; Boulanger et al., 1995). Alternative splicing of a single STEP gene produces a 46 kDa cytosolic member (STEP46) and a 61 kDa membrane-associated member (STEP61) (Bult et al., 1996). Lower molecular weight isoforms (STEP38 and STEP20) are also present in striatal tissue (Lombroso et al., 1993; Sharma et al., 1995).
STEP61 contains an additional 172 amino acids at its N-terminus that include a number of motifs not found in STEP46. These include two transmembrane domains, two polyproline domains, and two PEST sequences. Electron microscopy and biochemical analyses have localized STEP61 to the endoplasmic reticulum and postsynaptic terminal, where it is associated both with the postsynaptic density (Bult et al., 1996; Boulanger et al., 1995; Oyama et al., 1995) and the N-methyl-D-aspartate (NMDA) receptor protein complex (Pelkey et al., 2002). STEP-family proteins contain an ERK binding domain, the kinase interacting motif (KIM), which is required for the association of STEP with the ERKs (Paul et al., 2003; Pulido et al., 1998).
The present study tested the hypothesis that STEP regulates the cellular processes underlying memory formation. We constructed a TAT-STEP fusion protein with a point mutation in the catalytic domain that renders it inactive. The inactive STEP protein acts as a substrate-trapping protein: it binds to ERK1/2, prevents their translocation into the nucleus, and thereby disrupts ERK1/2 signaling. Infusion of this substrate-trapping construct into the lateral amygdala led to a selective impairment in memory consolidation, while the acquisition of fear learning remained intact. Parallel manipulations blocked the induction of LTP in amygdala slices after stimulation of thalamic afferent pathways. Pavlovian auditory fear conditioning was rapidly followed by the induction of STEP proteins in the lateral amygdala where, prior to training, only STEP61 is normally found. This de novo STEP expression was prevented by systemic pretreatment with the specific MEK inhibitor SL327 or the protein synthesis inhibitor cycloheximide. Two important conclusions are that STEP activity can be modulated by experience, as well as that STEP regulates neuroplasticity underlying long-term memory consolidation.
MATERIALS AND METHODS
Reagents
Cycloheximide, SL327, and glutamate were from Calbiochem (La Jolla, CA). The anti-ERK2 polyclonal antibody that only recognizes ERK2 (C-14) and the anti-myc monoclonal antibody (sc-40) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p44/42 ERK antibodies that detect ERK1/2 when dually phosphorylated at Thr-202 and Tyr-204 (TPEYP) were obtained from Cell Signaling Technology (Beverly, MA). Bicuculline was obtained from Tocris Cookson (Ballwin, MO). All other biochemicals were obtained from Sigma (St. Louis, MO) and media was purchased from Invitrogen-Gibco-BRL (Gaithersburg, MD).
Purification of TAT-STEP
A point mutant within the catalytic domain of STEP46 (C300S) was made by polymerase chain reaction using site-directed mutagenesis and verified by nucleotide sequencing. This mutation in the catalytic domain renders STEP enzymatically inactive. It still binds to its substrate ERK1/2, but is unable to dephosphorylates them. It thus acts as a substrate-trapping mutant that continues to bind to ERK proteins and not release them. We inserted the TAT nucleotide sequence (TAC-GGT-CGT-AAA-AAA-CGT-CGT-CAG-CGT-CGT-CGT) at the N-terminal of the STEP46 cDNA, subcloned it in pTrcHis-TOPO expression vector and transformed into E. Coli, BL21 (Invitrogen, Carlsbad, CA). Six histidines and a myc tag were added to the C-terminus to purify the fusion protein and to track it in vivo, respectively. Fusion proteins were induced with IPTG and affinity purified, and single bands on westerns blotted with myc- and STEP-antibodies were used as an indication of purity. A TAT-myc peptide was synthesized by the core facility at Yale University.
Cell culture
E16-17 day old rat embryos from Sprague-Dawley rats (Charles River Laboratory, Wilmington, MA) were used to obtain primary neuronal cultures. Pregnant dams were euthanized with CO2 and embryos removed through Caesarean section. Cerebrum was dissected under a microscope and tissue was mechanically dissociated. Cells were re-suspended in DMEM/F-12 (1:1)-containing 5% fetal calf serum. Poly-L-lysine-coated 100 mm tissue culture dishes were used and cells plated at a density of 3 x 106 per dish. For immunocytochemistry, cells were grown on poly-D-lysine coated 2-well culture slides (Becton Dickinson). Cells were grown for 10–12 days at 37°C in a humidified atmosphere of 5% CO2 and 95% air. 10 μM of cytosine D-arabinofuranoside was added to the cultures 72 hrs after plating to prevent proliferation of non-neuronal cells. For some experiments, neurons were incubated with TAT-STEP (4 μM) for 1 hr prior to treatment. For glutamate stimulation, cells were washed twice with MEM followed by the addition of glutamate (100 μM) for 5 min at 37°C, in the presence or absence of TAT-STEP. For immunoprecipitation, cells were first washed with PBS and then lysed in a buffer containing (in mM): 50 Tris-HCl, pH 7.4, 150 NaCl, 50 NaF, 10 Na4P2O7, 1 Na3VO4, 0.5% NP-40 and a cocktail of protease inhibitors. Lysates were centrifuged for 10 min at 14,000 rpm to remove insoluble material, and then pre-cleared with protein G sepharose. For immunoprecipitation of either TAT-STEP or TAT-myc, samples were incubated for 1–2 hr with the anti-myc antibody. The immune complexes were incubated with 50 μl of protein G-sepharose for 2–12 hr at 4°C. Controls included adding protein G-sepharose only. Beads were collected by centrifugation at 1000 rpm for 2 min, washed five times with NP-40-containing lysis buffer, and proteins were eluted using SDS-sample buffer.
Amygdala slice preparation
All procedures were carried out in compliance with the guidelines of the Canadian Council for Animal Care. Sprague-Dawley rats (21–42 days old) were deeply anesthetized with urethane (1.5 g/kg, i.p.) and decapitated. The brain was removed and transferred into ice-cold oxygenated (95% O2-5% CO2) artificial cerebrospinal fluid (aCSF) containing (in mM): 125 NaCl, 2.5 KCl, 2 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, and 25 D-glucose, pH 7.4. Coronal slices (400 μm thick; 3–4 slices) containing the LA were cut using a vibratome (Leica VT1000 S, Leica Microsystems Richmond Hill, ON, Canada) in ice-cold oxygenated aCSF as described by Weisskopf and colleagues (1999). Slices were incubated at 36°C for 1 hr and then stored at room temperature (22–24°C).
Recording and stimulation
Amygdala slices from 150 g male Sprague Dawley rats were superfused (flow rate 2–4 μl/min) at room temperature (22–24°C) with oxygenated (95% O2-5% CO2) aCSF. Tat-myc or TAT-STEP proteins were dissolved in aCSF (final concentration 300 nM) and applied by superfusion. For recording field excitatory postsynaptic potentials (fEPSPs) 5–10 μM bicuculline was included to block inhibitory GABAergic synaptic inputs. To record NMDAR-mediated fEPSPs, CNQX (10 μM) was added to the aCSF. The LA was identifiable as a triangular-shaped region bounded laterally by the external capsule and medially by the lateral association bundle. fEPSPs (AMPAR- and NMDAR-mediated) were recorded with a patch pipette pulled from thin-walled borosilicate glass (WPI, Sarasota, FL) filled with aCSF. To record pharmacologically-isolated NMDAR-mediated fEPSPs, CNQX (10 μM) was added to the aCSF and continuously superfused. At the conclusion of the experiment, the NMDAR-antagonist AP5 (50μM) was superfused onto the slice to confirm the fEPSP was mediated by NMDARs. Bipolar stainless steel stimulating electrodes (Frederick Haer, Bowhoiden, ME) were positioned in the ventral striatum, medial to the LA. A portion of these fibers is thought to originate in the auditory thalamus (Weisskopf et al., 1999). Synaptic responses were evoked at a frequency of 0.05–0.1 Hz (stimulus duration 150 μsec). After 20 min of baseline recording LTP was induced by tetanic stimulation (3 trains; 100 Hz; 1 sec duration; 90 sec inter-train interval). Responses were recorded for 60 min following tetanus.
Electrophysiology data analysis
Data were acquired and analyzed using pCLAMP 8.0 (Axon Instruments, Inc.). fEPSP amplitude was defined as the maximum DC voltage of a vertical line running tangent to the points of fEPSP onset and offset. In some recordings, fEPSP slope (10–60%) was also analyzed. However, the slope measurements were more variable and sensitive to noise, making them difficult to analyze reliably. Therefore, we chose response amplitude for the analysis of extracellular recordings. Responses were averaged and normalized to baseline, which was defined as the mean value obtained in the 10 min prior to tetanus or the 10 min prior to drug application. The amount of potentiation was analyzed by comparing pre-tetanus values (10 min before tetanus) with those collected 50 min after LTP induction. Significance of potentiation relative to baseline was tested with a paired Student's t-test. Differences were considered significant if p < 0.05. Statistical comparisons were made using GraphPad Instat 3.05 (GraphPad Software Inc., San Diego, CA) or Microsoft Excel. Figures were produced with Origin 7.0 (OriginLab Corp., Northhampton, MA) or Microsoft Excel (Microsoft, Redmond, WA).
Behavioral procedures
Behavioral studies were conducted as described previously (Schafe et al., 2000). Rats were anesthetized with Equithesin (4.32 ml/kg i.p.). The guide cannulae (22 gauge) fitted with dummy cannulae were implanted just above LA using the following coordinates from Paxinos and Watson (1986): 2.8 mm posterior to bregma, 5.3 mm lateral to midline, and 8.0 mm ventral to the skull surface. The guide cannulae were anchored to the skull by stainless steel screws in the skull and dental cement. All surgical procedures were conducted in accordance to the NIH Guide for the Care and Use of Experimental Animals and were approved by the Yale University Animal Care and Use Committee. Rats were allowed at least 5 days to recover before any experiment was initiated.
On the day before conditioning (day 1), rats were habituated to the training chambers (dark operant boxes with bar floors wiped with 70% ethanol) for 15 min. On the day of conditioning (day 2), animals were randomly assigned to two groups and infused with 1 μl of either TAT-STEP (4 μM) or TAT-myc peptide (control) bilaterally into the amygdala at a rate of 0.25 μl/min, either 1 hr before or immediately after training. A control experiment examined the potential non-specific effects of TAT-STEP on expression of fear and used infusions made 24 hours after training.
In all experiments, injector cannulae were left in place for an additional 2 min to allow diffusion of the solution from the cannula tip before replacing it with the dummy cannulae. For training sessions, rats were presented with the two pairings of a 30 sec tone CS (4.5 kHz, 75 dB) that co-terminated with a foot shock US (1 sec, 1.5 mA). The intertrial interval was 2 min. Testing for conditioned fear responses (freezing) was conducted at 1 hour (short term memory, STM) or 24 hours (long term memory, LTM) after conditioning. Animals assigned to the expression test control group were tested 24 hours after infusion, i.e., 48 hours after conditioning.
During the test session, animals were placed in a novel testing chamber (illuminated 5-hole boxes with grid floors wiped with Nolvasan) and allowed to explore for 5 min. The rats were then exposed to one (STM) or three (LTM) subsequent CS presentations, 30 sec each with a 2 min interval in between. The testing session was videotaped and behavior subsequently scored by an investigator blind to the experimental treatments. The total duration of freezing (in seconds) during the CS presentations was scored for each rat, and this number was expressed as a percentage of the total CS presentation time. All data were analyzed with ANOVA. Differences were considered significant if p<0.05. At the end of each behavioral experiment, rats were anesthetized with Equithesin and perfused with 4% buffered paraformaldehyde. Nissl staining and light microscopy was used to verify the location of the cannula tips within the amygdala. Animals with cannula tips beyond 0.5 mm of the boundaries of the LA were excluded from further analyses and were not included in any of the figures. Anti-myc antibody was used in some experiments (1:250) to determine the extent and time course of TAT-STEP diffusion.
Immunohistochemistry
To study the time kinetics of infusion of TAT-STEP in vivo, rats were implanted bilaterally with cannulae aimed at the lateral amygdala followed by infusion of TAT-STEP, as described above. At the appropriate time points (30, 60, 120, 180, and 360 min) after infusion, rats were rapidly anesthetized with Equithesin (4.32 ml/kg i.p.) and perfused intracardially with ice-cold 4% paraformaldehyde in 0.1M phosphate buffered saline (PBS). Brains were removed and post-fixed in the same fixative solution for 4 hours and cryoprotected in 15% and 30% sucrose in 0.1M PBS and then frozen in O.C.T. compound. Immunohistochemistry was then performed on 10 μm sections with anti-myc antibody. Briefly, sections were blocked with 10% normal goat serum, 3% bovine serum albumin (BSA) in PBS-T (0.1M PBS and 0.2% triton X-100) incubated at 4ºC overnight in anti-myc antibody. After extensive washes in PBS-T, tissue sections were incubated in Alexa Fluor 488 goat anti-mouse antibody (1:250, Molecular Probes, Eugene, OR) for 1 hr at room temperature. Sections were extensively rinsed in PBS-T and cover-slipped for viewing using a fluorescent microscope (Zeiss Axiovert 2000). In some experiments, 300 μm thick amygdala slices were incubated with TAT-STEP (2 μM) at 30°C for 5 and 10 min, fixed overnight with 4% paraformaldehyde in PBS, and cryoprotected serially in 10, 20 and 30% sucrose in PBS. 12 μm thick cryostat sections of these slices were mounted onto slides and processed for immunohistochemistry using the anti-myc antibody as described above.
To determine the time kinetics of ERK1/2 phosphorylation in the amygdala following fear conditioning, rats were anesthetized at 0, 5, 10, 30, 60, 120 and 360 minutes after training, perfused, and brains were post-fixed as described above. Cryostat cut 30 μm free-floating sections were obtained, and every third section was processed for pERK immunoreactivity. Endogenous peroxidase activity was eliminated by incubation with 0.3% H2O2 in PBS followed by blocking with 10% normal goat serum and 3% BSA in PBS-T. Sections were incubated at 4ºC overnight with anti-phospho-p44/42 MAPK antibody (mouse monoclonal antibody, 1:200) in PBS-T containing 1% normal goat serum and 3% BSA. After extensive washes in PBS-T, tissue sections were incubated in biotinylated goat anti-mouse antibody (1:200, Vector Laboratories, Burlingame, CA) for 1 hr at room temperature. Tissue was again rinsed in PBS-T followed by incubation in avidin-biotin-peroxidase complex solution (final dilution 1:50, Vector Laboratories, Burlingame, CA) for 1 hr at room temperature, then rinsed in PBS, and developed in DAB-nickel chloride (Vector Laboratories, Burlingame, CA). Sections were mounted on SuperFrost Plus slides (VWR International, Bridgeport, NJ), counterstained with Nuclear Fast Red (Vector Laboratories), dehydrated and cover-slipped for light microscopic examination. The number of pERK1/2 labeled cells was quantified using ImageJ (NIH) and counts were normalized to the total number of cells as detected by the Nuclear Fast Red staining. Briefly, the lateral amygdala was marked using the polygonal tool, the total number of cells and the number of pERK1/2 positive cells were obtained using the analyze particles tool. The number of pERK1/2 positive cells per 100 cells from 5 sections for each time point was averaged and a paired Student’s t-test was performed against the control values.
Immunoblotting of amygdala punches
For immunoblotting studies, rats were decapitated at 5, 10, 15, 20, and 60 minutes after training without anesthesia, and the brains were frozen on dry ice and stored at −80°C until processed. Punches from the lateral amygdala were taken using a 0.5 mm tissue corer (Fine Science Tools, Foster City, CA) from 400 μM thick cryostat sections. The tissue were briefly sonicated in 150 μl of Laemmli sample buffer, boiled at 100°C for 10 min and processed for SDS-polyacrylamide gel electrophoresis and immunoblotting with anti-pERK and anti-ERK antibodies.
RESULTS
STEP is present in the amygdala and co-localizes with ERK2
We first investigated whether STEP is co-expressed with ERK2 in the LA neurons relevant to fear conditioning by using immunofluorescent staining of rat brain coronal sections. For these experiments, we used an antibody specific to ERK2. STEP and ERK2 co-localized in most cells of the dorso-lateral and ventro-lateral amygdala (LAd and LAv) (Fig. 1A). To determine which STEP isoforms were expressed in LA neurons, we performed western blot analyses on tissue punches and compared levels of STEP within the LA, the central nucleus of the amygdala (CeN), and the striatum (Fig. 1B). STEP61 is the only isoform expressed in the LA, while the alternatively spliced variants STEP61, STEP46, and STEP38 are expressed in the CeN and the striatum (Bult et al., 1997). These data demonstrate that STEP is expressed in LA neurons that co-express ERK2, and that the major isoform of STEP expressed during basal conditions is STEP61.
Figure 1. STEP and ERK co-localize in the lateral amygdala.
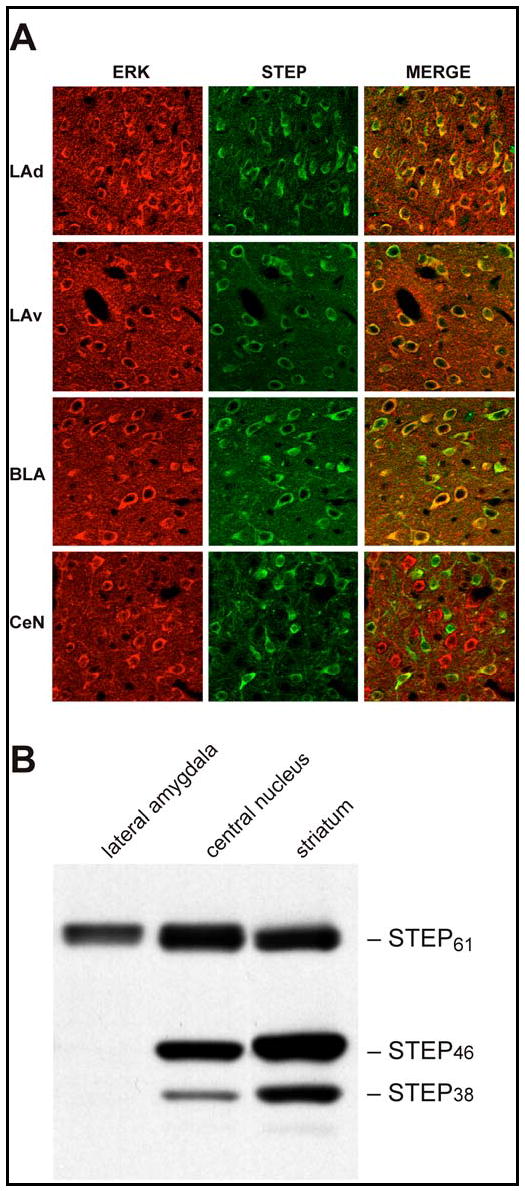
(A) Immunohistochemical analysis of ERK and STEP in the dorso-lateral amygdala (LAd), ventro-lateral amygdala (LAv), baso-lateral amygdala (BLA) and the central nucleus (CeN). Rat brain coronal sections were double-labeled with anti-ERK2 and anti-STEP antibodies. The ‘Merge’ column illustrates the co-localization of the two proteins in the same cell. (B) Tissue punches from the LA, CeN and the striatum were analyzed by SDS-polyacrylamide gel electrophoresis and western blots were probed with anti-STEP antibody to show the distribution of STEP isoforms in these regions. Note that STEP61 is the only isoform expressed in the LA.
TAT-STEP blocks nuclear translocation of ERK2 in neurons
To investigate the functional role of STEP, we used a catalytically inactive mutant of STEP46 (STEP46 C-S), in which the essential cysteine in the catalytic domain was converted to a serine (Pelkey et al, 2002). This mutation inactivates STEP, allowing it to bind constitutively to substrates but not to dephosphorylate them. STEP46 C-S thus acts as a substrate-trapping protein (Flint et al., 1997). We rendered STEP46 C-S cell-permeable by fusing it to the protein transduction domain of the human immunodeficiency virus-type 1, TAT (Frankel and Pabo, 1988; Green and Loewenstein, 1988; Aarts et al., 2002). In addition, we added a myc epitope tag at the carboxyl terminal to distinguish this construct from endogenous STEP and to permit immunoprecipitation (Fig. 2A). This construct is hereafter referred to as TAT-STEP.
Figure 2. Functional characterization of TAT-STEP in primary neuronal cultures.
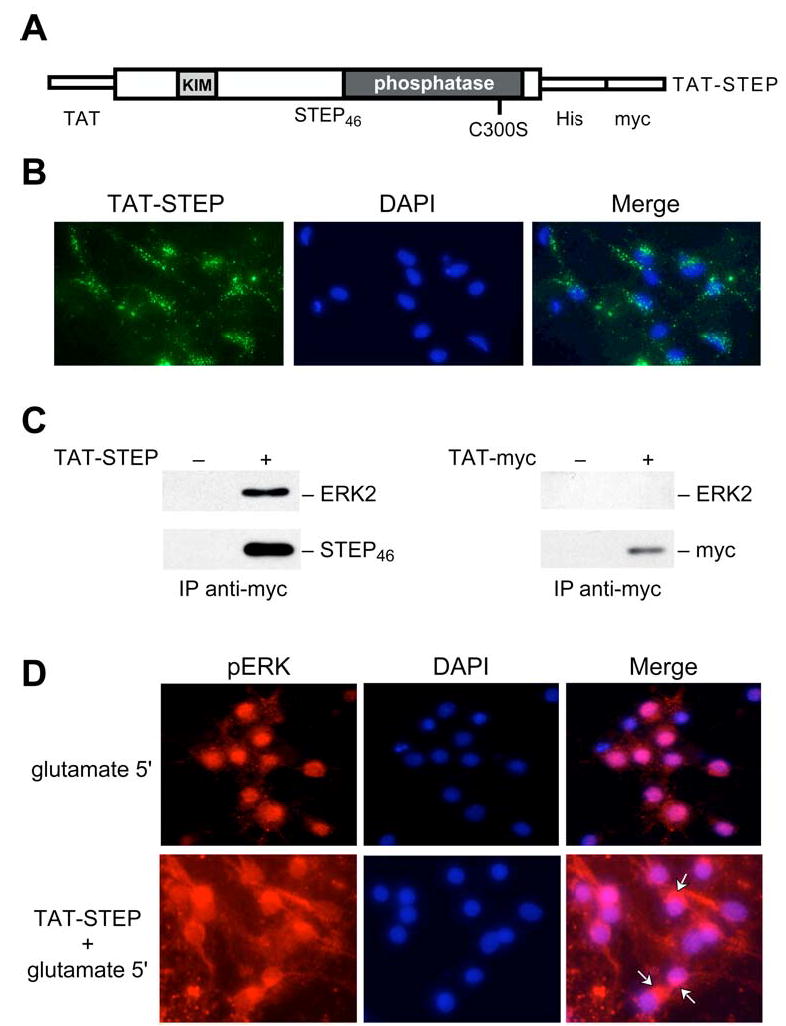
(A) Schematic diagram of TAT-STEP46 C-S indicating the positions of the TAT-peptide, six-histidine (His) and myc tags, the KIM domain, and the C-S mutation site within the phosphatase domain. (B) Immunocytochemical staining with anti-myc antibody and DAPI to characterize the transduction of TAT-STEP. (C) Neuron cultures were incubated with either TAT-myc or TAT-STEP, which were immunoprecipitated with anti-myc antibody, analyzed by SDS-PAGE, and probed with anti-ERK2 antibody (upper panels). The blots were then reprobed with anti-myc antibody (lower panels). (D) Immunocytochemical analysis using an antibody that recognizes phospho-ERK1/2 illustrates the cytoplasmic and nuclear distribution of pERK1/2 following glutamate treatment for 5 min, in the absence or presence of TAT-STEP. Arrows point to perinuclear pERK1/2.
The initial characterization of TAT-STEP was in 10–12 day old primary neuronal cultures. Immunocytochemistry with anti-myc antibody demonstrated intracellular uptake of the fusion protein in most neurons (Fig. 2B). TAT-STEP was detectable within twenty minutes of transduction and was still present 6 hours later (data not shown). To determine whether transduced TAT-STEP was bound to ERK2, we performed immunoprecipitation with anti-myc antibody 1 hour after transduction, followed by immunoblotting with anti-ERK2 antibody. The results show that TAT-STEP forms a stable complex with ERK2. A control peptide consisting of the TAT transduction sequence and the myc epitope did not interact with ERK2 (Fig. 2C).
We also investigated whether the formation of the TAT-STEP/ERK complex could affect nuclear translocation of phospho-ERK1/2 (pERK1/2) following glutamate application. Glutamate normally leads to the rapid activation of pERK1/2. Activated ERKs translocate into nuclei where they directly or indirectly lead to the phosphorylation of transcription factors required for transcriptional activation. Neuronal cultures exposed to glutamate displayed the expected rapid phosphorylation and translocation of pERK1/2 into the nucleus (Fig. 2D, top panels). In contrast, cultures exposed to the substrate trapping TAT-STEP and then stimulated with glutamate showed diminished nuclear pERK1/2 (Fig. 2D, bottom panel, arrows). Quantification of the results showed pERK1/2 translocation in 88% of nuclei after 5 minutes treatment with glutamate alone, while pERK1/2 was detected in 29% of nuclei in the presence of TAT-STEP and glutamate (glutamate alone, 88 ± 3%; TAT-STEP + glutamate, 29 ± 8%; p<0.002; counts from 4 separate slides per condition). The control experiment with TAT-myc showed little difference in the number of pERK1/2 nuclear translocations compared to the glutamate alone condition (TAT-myc + glutamate, 94± 3%). Taken together, these results demonstrate that TAT-STEP transduces into neurons, binds to, and blocks nuclear translocation of pERK1/2.
TAT-STEP disrupts LTP in slices from the lateral amygdala
We next examined the effect of TAT-STEP on synaptic transmission and plasticity in the LA in vitro. We first established that TAT-STEP was able to enter cells in slices and found that TAT-STEP was detected within 5 min of addition to the artificial CSF (aCSF) (Fig. 3A). We recorded extracellularly from acute brain slices containing the LA from adult rats. Orthodromic stimulation of the internal capsule evoked monosynaptic field excitatory postsynaptic potentials (fEPSP) that were observed to be stable over periods of 80 min (Fig. 3B, left panel) or longer (not illustrated). We found that superfusion with aCSF containing the TAT-myc control peptide (Fig. 3B, left panel) had no effect on fEPSP amplitude over a period of 60 min. Likewise, fEPSP amplitudes were unaffected by bath application of TAT-STEP. Bath application of TAT-STEP also had no effect on pharmacologically-isolated NMDAR-mediated fEPSP amplitude (Fig. 3B, right panel). Furthermore TAT-STEP had no effect on paired-pulse facilitation of the fEPSP (data not shown). Thus, TAT-STEP had no effect on basal synaptic responses in the LA.
Figure 3. TAT-STEP blocks induction of LTP in LA without affect basal synaptic transmission.
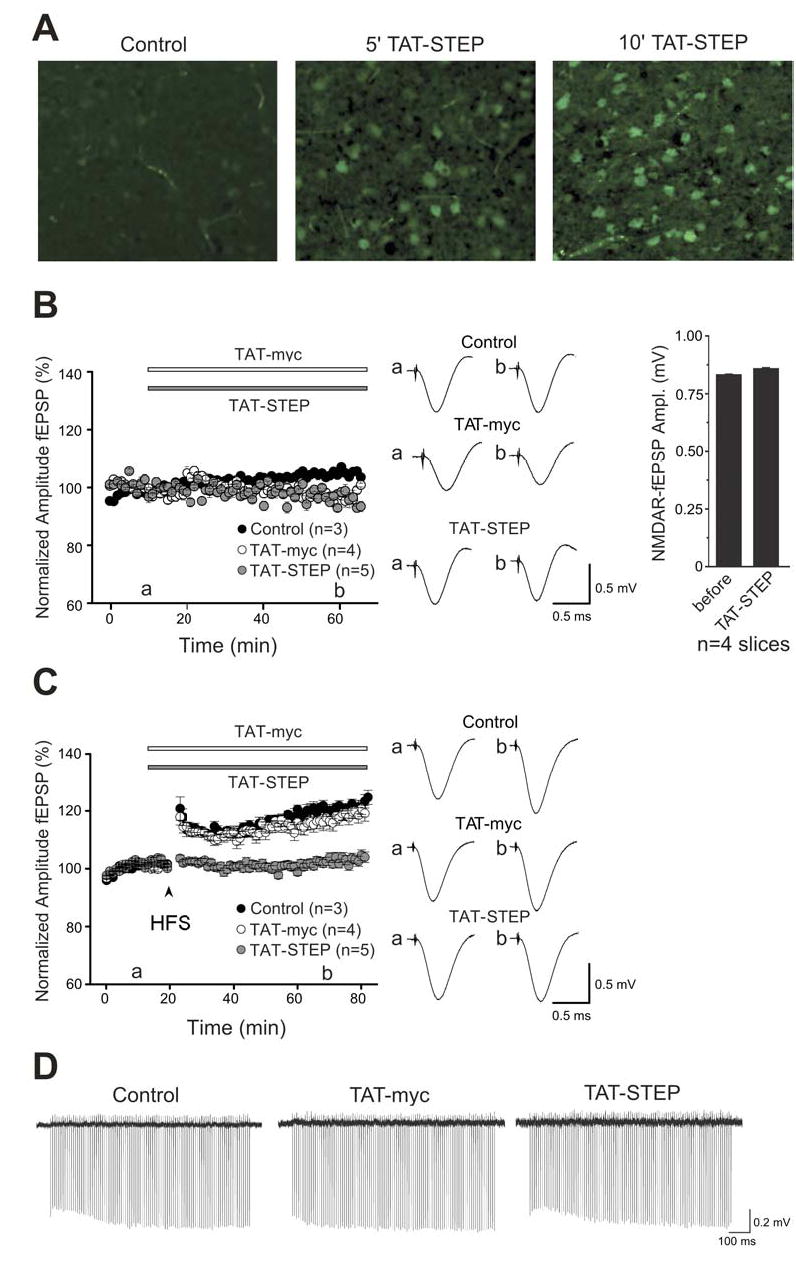
(A) aCSF (control) or TAT-STEP (2 μM) was added to 300 μm amygdala slices and tissue processed with anti-myc antibody at 5’ and 10’ after start of incubation. (B) Left panel. Field EPSP (fEPSP) amplitude (mean ± SEM) is plotted every 1 min for control slices (n = 3 slices, closed circles), for a slices treated with 300 nM TAT-myc (n = 4 slices, open circles) and for slices treated with 300 nM TAT-STEP (n = 5, gray circles) that was applied during the period indicated by the bar. In the middle are averaged traces from control slices (upper panel) or before (a) application of either TAT-myc or TAT-STEP and (b) 50 min after application of TAT-myc (middle panel) or TAT-STEP (lower panel). On the right, histogram showing average NMDAR mediated fEPSPs before or during application of TAT-STEP (20 min after start of application). (C) Left panel. Field EPSP (fEPSP) amplitude (mean ± SEM) is plotted every 1 min for control slices (n = 6 slices, closed circles), for a slices treated with 300 nM TAT-myc (n = 6 slices, open circles) and for slices treated with 300 nM TAT-STEP (n = 7, gray circles) that was applied during the period indicated by the bar. Tetanus (HFS; three trains of 100 Hz stimuli, 1 sec duration, intertrain interval of 90 sec) is indicated by the arrowhead. On the right are averaged traces taken from control slices (upper panel) or before (a) application of either TAT-myc (middle panel) or TAT-STEP (lower panel) and (b) 50 min after tetanus. Six sweeps were averaged to produce the responses shown. Scale bar, 0.5 msec, 0.5 mV for B and C. (D) Representative, continuous field recordings illustrating responses during HFS trains in control, TAT-myc- or TAT-STEP-treated slices. Scale bar, 100 msec, 0.2 mV.
We then determined the effect of TAT-STEP on LTP in the LA (Fig. 3C). Tetanic stimulation (three 1 sec, 100 Hz trains, 90 sec inter-train interval) to the internal capsule in control slices produced a sustained increase in fEPSP amplitude. fEPSP was 124 ± 2.0% (mean ± SEM; n = 6 slices; p <0.0001, paired t-test) of the baseline level 60 min after tetanus. In slices treated with the TAT-myc peptide, the fEPSP amplitude was 119 ± 2.7% (mean ± SEM; n = 6 slices; p >0.1 compared with control slices) of the baseline level 60 min after tetanus. On the other hand, during application of TAT-STEP, the fEPSP amplitude was 103 ± 1.8% (mean ± SEM; n = 7 slices; p < 0.01 compared with aCSF control) of the baseline level 60 min after tetanus. We found that TAT-STEP did not affect the progressive, short-lasting enhancement of synaptic responses that occurred during the tetanus (Fig. 3D). Comparing the amplitude of the first response during the tetanus with that of the last response showed that synaptic responses increased to 113 ± 4.2% in control slices (n = 6), 122 ± 7.2% in TAT-myc treated slices (n = 6) and 111 ± 4.6% in TAT-STEP-treated slices (n = 7; p=0.293; one-way ANOVA). Thus, TAT-STEP but not the TAT-myc control peptide, prevented induction of LTP in the LA but did not affect the short-term facilitation during high-frequency stimulation.
TAT-STEP blocks Pavlovian fear conditioning
Based on these electrophysiological results, we investigated whether TAT-STEP infused in vivo blocks the consolidation of fear conditioning, as this depends upon synaptic plasticity in the LA (Schafe et al., 2000). For this purpose, rats were implanted with guide cannulae aimed at the LA. Following one week of recovery, animals received bilateral infusions of TAT-STEP. We initially determined whether TAT-STEP or TAT-myc were able to get into cells and the extent of the diffusion over time. Coronal brain sections of the amygdala were taken at different time points after infusion and sections examined immunohistochemically with anti-myc antibody. No myc staining was observed in uninfused control animals, whereas myc staining was detectable in TAT-STEP and TAT-myc infused animals up to 6 hours, the longest period examined, and its distribution was restricted to the amygdala (data not shown).
In behavioral experiments, rats received bilateral intra-amygdala infusions of either TAT-STEP or TAT-myc control peptide 1 hour prior to training in an auditory fear conditioning paradigm. The training consisted of two trials in which a 30 second tone was paired with electrical shock (1.5 mA of 1 sec). The expression of fear memory was examined at 1 and 24 hours after training. TAT-STEP infusion had no effect on short-term memory (STM) formation as indicated by a similar degree of freezing in animals infused with TAT-STEP or TAT-myc controls during CS presentation (F1,11=0.61; p>0.45; Fig. 4A). Freezing prior to CS presentation was low, demonstrating little generalization between the training and testing contexts, and there were no significant differences between the experimental groups (F1,11=0.01; p>0.91).
Figure 4. Effect of intra-amygdala infusion of TAT-STEP prior to fear conditioning.
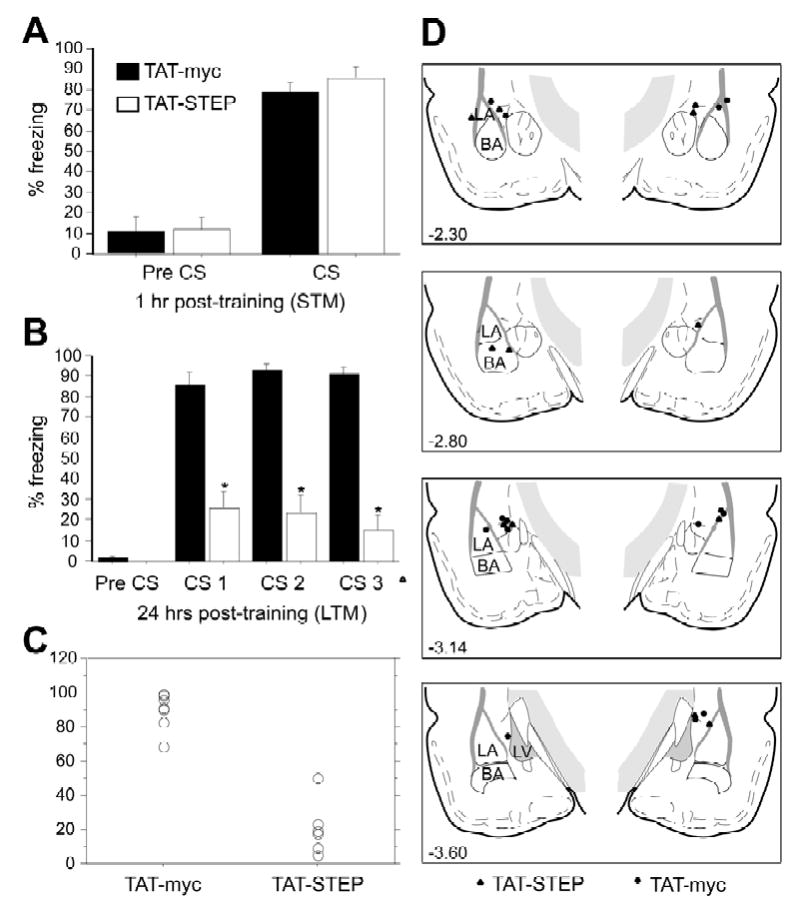
Rats were infused with TAT-STEP (n=6) or TAT-myc (control; n=7) 1 hr prior to training on Pavlovian fear conditioning (two pairings of 30 sec tone and 1 sec 1.5 mA shock). They were then assessed for fear memory formation at 1 hr (A, short term memory, STM) or 24 hr (B, long-term memory, LTM) after training. (A & B) Mean ± S.E. freezing in rats 30 sec prior to CS presentation (pre CS) and during the three tone presentations (CS). *p < 0.0001 relative to the TAT-myc control. (C) Scattergram demonstrating the distribution of freezing in TAT-myc and TAT-TAT-STEP in the LTM test. (D) Histological verification of cannula tip placements in rats infused with either TAT-STEP (▴) or TAT-myc (•).
TAT-STEP significantly reduced long-term memory (LTM) assessed 24 hours after training (Fig. 4B). A repeated measures ANOVA using TAT-STEP treatment as the independent variable and freezing behavior during the three CS presentations as the repeated measure demonstrated a significant main effect of the treatment (F1,11=80.89; p<0.0001). Post-hoc analysis confirmed significantly less freezing in animals receiving infusion of TAT-STEP prior to training compared to those receiving TAT-myc infusions (p<0.0001). Both groups showed comparable freezing during the pre-CS period (F1,11=0.95; p=0.35). Figure 4C represents a scattergram demonstrating the distribution of freezing in TAT-myc and TAT-STEP infused animals in the LTM test. Histological examination of all bilateral cannulae placements within the amygdala are shown in Figure 4D. Only rats with cannula tips in, or immediately adjacent to, the LA were included in statistical analyses: out of 16 animals, three rats were excluded on the basis of these criteria (final group sizes; TAT-STEP, n=6; controls, n=7).
To directly examine the effect of STEP on fear memory consolidation, a second series of experiments was performed using post-session infusion of TAT-STEP. Post-session infusions can be used to dissociate the effects of an experimental manipulation on memory consolidation from those on learning, as well as non-specific performance effects. Rats were infused with TAT-STEP or TAT-myc control peptides immediately after training. Post-session TAT-STEP infusion into the amygdala blocked memory consolidation. Repeated measures ANOVA showed a significant main effect of the treatment on freezing behavior in the LTM test (F1,12=9.23; p<0.01; Fig. 5B). Post-hoc analysis confirmed that TAT-STEP elicited significantly less freezing behavior than TAT-myc infused control animals in this test (p≤0.01). ANOVA demonstrated no effect of TAT-STEP treatment on freezing behavior prior to (F1,12=1.49; p=0.25; pre-CS; Fig 5A) or during CS presentation (F1,12=1.63; p=0.23; fig 5A) in the STM test. Moreover, there was no difference between the groups on freezing in response to exposure to the novel test chambers in the LTM test (F1,12=0.706; p=0.42; Fig 5B).
Figure 5. Effect of post-session intra-amygdala infusion of TAT-STEP.
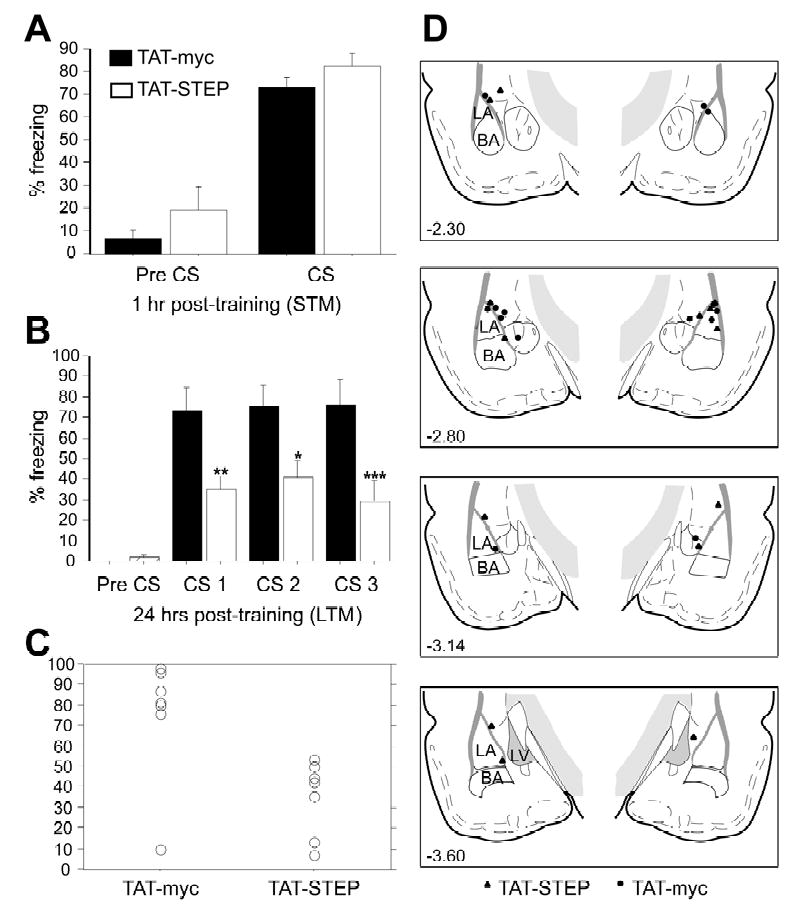
Rats were infused with TAT-STEP (n=7) or TAT-myc (control; n=7) immediately following training on Pavlovian fear conditioning (two pairings of 30 sec tone and 1 sec 1.5 mA shock). They were then assessed for fear memory formation at 1 hr (A, short term memory, STM) or 24 hr (B, long-term memory, LTM) after training. (A & B) (A & B) Mean ± S.E. freezing in rats prior to the tone (pre CS) and the three tone presentations (CS). *p < 0.05, **p < 0.01, ***p < 0.001 relative to the TAT-myc control. (C) Scattergram demonstrating the distribution of freezing in TAT-myc and TAT-STEP in the LTM test. (D) Histological verification of cannula tip placements in rats infused with either TAT-STEP (▴) or TAT-myc (•).
Importantly, the administration of TAT-STEP after the fear memory was established 24 hours after conditioning did not influence the fear response. Mice infused with either TAT-STEP or the TAT-myc control showed no significant differences when tested 24 hours after the infusion (F1,10=0.830; p=0.38). Thus, non-specific effects of the TAT-STEP infusion on expression of freezing can not account for the impairment observed in the LTM test. Similarly, increases in locomotor activity as a result of the TAT-STEP infusion are unlikely to explain the data, since 30 min prior to the LTM test there was no effect of infusion on baseline locomotor activity (F1,12=0.95; p=0.35; data not shown). Figure 5C shows a scattergram representing the distribution of freezing in the LTM test in TAT-myc and TAT-STEP animals. Histological examination of all bilateral cannulae placements within the amygdala are shown in Figure 5D. Out of 17 animals, three rats were excluded from this figure as well as from the statistical analysis due to either misplaced cannulae or, in one case, extensive damage to the LA (final group sizes; TAT-STEP, n=7; controls, n=7). Post-training infusion of TAT-STEP thus selectively impaired auditory fear memory consolidation, indicating that STEP function is required for long-term memory.
Fear conditioning is associated with ERK activation and de novo translation of STEP in the lateral amygdala
We next determined the time course of pERK1/2 activation following auditory fear conditioning. Immunohistochemical data showed that prior to conditioning, ERK1/2 is primarily in its dephosphorylated form in the LA (Fig. 6A). However, pERK1/2 immunoreactive cells were present throughout this region 5 minutes after fear conditioning. The number of pERK1/2 positive cells at 5 minutes following fear conditioning was significantly higher than what was detected in the untrained control group (Fig. 6B, p< 0.001). Western blots confirmed these findings with an increase in pERK1/2 detected by 5 minutes, followed by a decrease at 15 minutes, and a return to higher pERK1/2 levels at 1 hour post-training (Fig. 6C and D). We also determined whether tetanic stimulation would induce rapid activation of ERK proteins in LA slices. Punches of LA were processed by immunoblotting and the results indicate an increase in pERK1/2 in stimulated slices (n=3) to 168% of control slices (n=3) by 5 min (p<0.008) with a return to baseline levels by 10 min (p>0.88).
Figure 6. Time kinetics of ERK1/2 activation in the lateral amygdala.
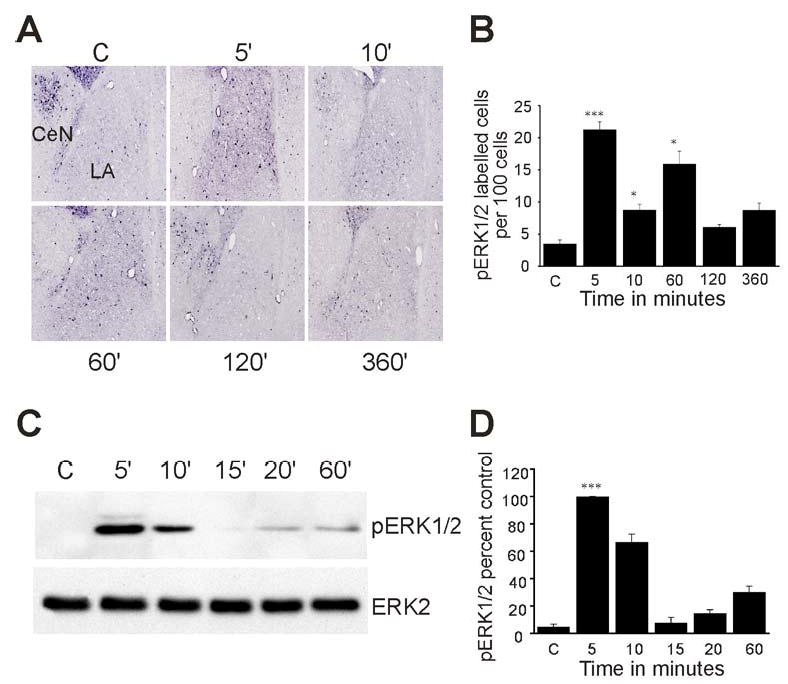
Rats were presented with two pairs of tone and shock and processed for immunohistochemistry or immunoblotting. (A) Representative immunohistochemical staining of pERK1/2 in the amygdala at 0, 5, 10, 60, 120 and 360 min after fear conditioning. (B) Bar diagram showing pERK1/2 immunoreactive cells per 100 cells in serial sections through the LA (mean ± S.E., n = 5 sections per time point). *p < 0.05, **p < 0.01, ***p < 0.001 relative to control. (C) pERK1/2 immunoblot of tissue punches taken from the LA at 5, 10, 15, 20 and 60 min after fear conditioning training (upper panel). The blots were reprobed with total ERK2 antibody (lower panel). (D) Quantification of pERK1/2 levels from 3 separate experiments (***p < 0.001).
To further characterize the role of STEP in fear conditioning, we determined the profile of STEP expression in the LA at different time points following titanic stimulation as well as fear training (Fig. 7A). Punches from high-frequency stimulated slices were processed at 5 and 10 minutes, and no significant changes were detected in STEP levels (data not shown). However, the situation was quite different from punches obtained in vivo. In the absence of training, only the STEP61 isoform was detected (lane C). Unexpectedly, within ten minutes of fear conditioning, the cytosolic variant, STEP46, appeared and remained present for the time course of the study (Fig. 7A). An increase in STEP61 was also observed (approximately 20% in Fig. 7B, lane 3; p<0.05). Importantly, there was no detectable increase in STEP expression in animals exposed to either shock or tone alone (Fig. 7B). The levels of pERK1/2 were again examined and the same biphasic activation was clearly visible. The decrease in pERK1/2 correlated with the appearance of STEP46 (compare Fig. 7A and 7C, 5 and 10 minute time points).
Figure 7. De novo synthesis of STEP in the lateral amygdala following fear conditioning.
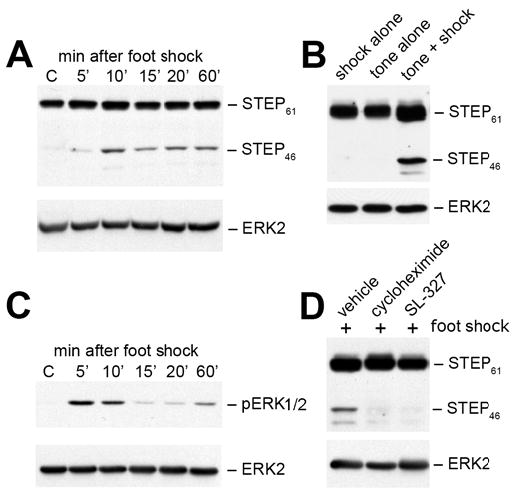
(A) Tissue punches taken from LA at 5, 10, 15, 20 and 60 min after fear conditioning or from control animals (no tone or shock, lane C) were analyzed by SDS-PAGE and processed for immunoblotting with anti-STEP (upper panel). (B) Rats were exposed to either shock or tone alone and punches were taken from the LA at 10 minutes, when STEP levels are at their maximum after fear conditioning (n = 3 independent trials with 3 rats per treatment). Note the increase in both STEP isoforms after fear conditioning. (C) pERK1/2 levels were determined after fear conditioning (upper panel). (D) Rats were injected intraperitoneally with DMSO (vehicle), cycloheximide (protein synthesis inhibitor) or SL327 (MEK inhibitor) 1 hr prior to fear conditioning. Tissue punches were again processed at 10 minutes for immunoblotting with anti-STEP antibody (upper panel). These experiments were repeated in triplicate with p<0.02 for both inhibitors. All blots were reprobed with anti-ERK2 antibody to allow for comparisons of loading levels (A, B, C & D, all lower panels).
To determine whether the rapid expression of STEP resulted from de novo protein synthesis, we tested the effects of cycloheximide, an inhibitor of protein translation. Rats were injected with cycloheximide (200 mg/kg i.p.) or vehicle 1 hour prior to training and sacrificed 10 minutes following training. Tissue punches from the LA were processed for immunoblotting. Control rats injected with vehicle again demonstrated the rapid expression of STEP after training (Fig. 7D, lane 1). In contrast, rats injected with cycloheximide prior to fear conditioning did not show increased levels of STEP (Fig. 7D, lane 2; p<0.02). These results suggest that STEP isoforms are translated in the LA within minutes of fear conditioning.
We noted that the appearance of STEP immediately follows activation of the ERK pathways, raising the possibility that ERK1/2 might regulate the expression of STEP. We therefore tested this potential regulatory mechanism by injecting rats prior to training with SL327 (50 mg/kg i.p.), a MEK inhibitor that has previously been shown to disrupt consolidation of fear conditioning (Selcher et al., 1999). In the presence of the MEK inhibitor, no increases in STEP levels were detected after fear conditioning (Fig. 7D, lane 3; p<0.02). Taken together, these results indicate that fear conditioning results in the activation of ERK1/2 in the LA within 5 minutes of training, that ERK1/2 activation is required for the rapid translation of STEP46 by 10 minutes, and that there is a subsequent decrease in pERK1/2 levels.
DISCUSSION
Here we report the ability of the protein tyrosine phosphatase STEP to interfere with memory consolidation in a Pavlovian fear conditioning paradigm and does so, in part, through its ability to regulate the ERK signaling pathway. In the lateral amygdala, activation of the ERK pathway results in long-term changes in synaptic activity that are thought to underlie the consolidation of a Pavlovian auditory fear memory (Schafe et al., 2000; Rodrigues et al., 2004; Schafe and LeDoux, 2000). Activated ERK1/2 is required for the induction of LTP in the hippocampus (Wu et al., 1999; English and Sweatt, 2002). Once activated, ERK1/2 translocate into the nucleus where they induce cAMP response element-containing genes (Impey et al., 1998a, 1998b; Davis et al., 2000).
However, the ERK cascades have additional functions beyond their effects on gene transcription. An emerging model suggests that activated ERK1/2 participate in several pathways that work together to induce synaptic plasticity (English and Sweatt, 1997; Lamprecht and LeDoux, 2004). Thus, ERK1/2 modulate back-propagating action potentials by phosphorylating dendritic A-type K+ channels (Kv4.2 subunits) (Watanabe et al., 2002; Yuan et al., 2002; Morozov et al., 2003), and is one of several signaling cascades shown to initiate local protein synthesis after synaptic activation through phosphorylation of translation factors (eIF4E) and ribosomal protein S6 (Kelleher et al., 2004a, 2004b).
The present data suggest that STEP might act as an inhibitory constraint on the development of synaptic plasticity through its ability to regulate the ERK pathway. It is now established that a subfamily of PTPs exists that are the only proteins found to date to contain a MAP kinase binding site termed the KIM domain. These PTPs inactivate MAP kinase family members by dephosphorylation of the regulatory tyrosine residue in the activation domain (Paul et al., 2003; Saxena et al., 2000; Blanco-Aparicio et al., 1999; Pulido et al., 1998). Taken together with our present findings, a parsimonious explanation for the results is that wildtype STEP regulates the ERK pathway, and that the TAT-STEP construct used here disrupts ERK1/2 signaling. Although we have demonstrated TAT-STEP’s ability to block nuclear translocation of ERK1/2, future studies are needed to determine whether TAT-STEP also blocks additional functions such as the generation of back-propagating action potentials or the initiation of local protein synthesis.
The observed disruption of nuclear translocation of pERK1/2 stimulated a second series of experiments examining the ability of the TAT-STEP to influence memory formation. We first demonstrate that TAT-STEP has a robust effect on the induction of LTP in the LA: superfusion of TAT-STEP blocks tetanus-induced LTP, whereas LTP is unimpaired in the presence of the TAT-myc peptide or aCSF. Tetanus-induced LTP in the LA is dependent upon NMDA receptor activation (Bauer et al, 2002) but we found no depression of NMDAR synaptic responses by TAT-STEP. Thus, the suppression of LTP induction was not caused by blockade of NMDARs. Furthermore, we observed that neither the ability of the synapses to respond at high-frequency nor the short-term facilitation during the tetanic stimulation was affected by TAT-STEP. Thus, the synaptic transmission responsible for initiating LTP was not perturbed by TAT-STEP, indicating that its site of action is downstream in the LTP-inducing signaling cascade.
In the CA1 region of the hippocampus, STEP acts as a brake on the induction of LTP by tonically suppressing NMDARs, and inhibiting STEP causes potentiation that occludes LTP (Pelkey et al., 2002). However, in LA slices treated with TAT-STEP we did not observe any change in fEPSP amplitude, or in pharmacologically isolated NMDAR-mediated synaptic responses. Thus, in the LA, in contrast to hippocampal CA1, there appears to be no tonic, ongoing regulation of NMDARs. Since we did not observe increases in fEPSP amplitude following application of TAT-STEP, but TAT-STEP did block induction of LTP, this suggests that STEP is critical for LTP induction and its mechanism is other than through regulation of glutamatergic receptors within neurons of the lateral amygdala.
The ability of TAT-STEP to block the induction of LTP within the lateral amygdala suggested that it might also block memory formation in the Pavlovian fear conditioning paradigm. This form of learning is believed to depend on LTP and neural plasticity in the LA (Nader et al., 2000; Schafe et al., 2000; Schafe and LeDoux, 2000; Blair et al., 2001; Rodrigues et al., 2004). Our data indicate that TAT-STEP impairs the consolidation of long-term memory without affecting the acquisition of memories. These observations and the ability of TAT-STEP to disrupt ERK signaling are consistent with a role for the ERK cascade in the formation of fear memories (Schafe et al., 2000). Moreover, the data suggest for the first time a potential role for STEP in memory consolidation.
ERK1/2, however, are not the only substrate of STEP, and it is possible that the disruption of memory consolidation and LTP induction occurs through the ability of STEP to dephosphorylate additional signaling proteins. While the focus of the present study is on the ERK pathway, previous data have identified two additional substrates for STEP: the NMDAR subunit NR2B and the tyrosine kinase Fyn. STEP isoforms are a component of the NMDAR protein complex, depress NMDAR activity (Pelkey et al., 2002), and participate in NMDAR internalization in hippocampal cultures and tissue (Snyder et al., 2005). However, the lack of effect of TAT-STEP on NMDAR synaptic responses found in the present study suggest that this receptor may not be a relevant of target of STEP in LA neurons. STEP also binds to Fyn, dephosphorylates one of its two regulatory sites (Tyr420), and thereby inactivates Fyn (Nguyen et al., 2002). Whether regulation of Fyn by STEP contributes to synaptic plasticity and memory formation in the LA remains to be investigated.
A striking observation is the rapid activation of the ERK signaling cascade after fear conditioning. Phosphorylated ERK appears within 5 minutes of training. The earlier time points, reported here for the first time, extend previous findings that demonstrated the ERK pathway is activated 1 hour post-training (Schafe et al., 2000). The reason for the difference is that the earlier paper did not investigate time points prior to 15 minutes, by which time our data indicate pERK1/2 had returned to baseline levels. The rapid activation shown here is consistent with reports indicating activation of the ERK pathways within 5 minutes of behavioral testing for habituation (Murphy et al., 2005) or after stimulation with BDNF (Takei et al., 2001). We also observed a biphasic activation, with pERK1/2 levels decreasing to baseline levels by 15 minutes before increasing again at one hour. Biphasic regulation of the ERK pathway has been reported for novel taste learning, although the cycling occurred at later time points (Sweatt and Swank, 2001). The role for a biphasic activation of the ERK pathway remains unclear, although it may function as a positive feed-back signal for the development of synaptic plasticity (Sweatt and Swank, 2001).
The rapid activation of the ERK pathway was replicated in punches processed from LA slices within 5 minutes of HFS, with a return to baseline levels by 10 minutes. We did not observe an increase in STEP translation under these conditions. A possible explanation for differences between the in vivo and slice work is the absence of neuromodulatory signals in the slices. NMDA-receptor-dependent synaptic plasticity often requires additional signals through neuromodulatory receptors such as the Gs-coupled β1 adrenergic receptor (Thomas et al., 1996; Winder et al., 1999). In fact, preliminary data with β1 adrenergic stimulation of slices have demonstrated a rapid translation of STEP within 10 minutes (Hu and Lombroso, unpublished data).
The finding that STEP is rapidly translated in response to fear conditioning is the first indication that experience-dependent plasticity regulates STEP activity in addition to previously described phosphorylation mechanisms (Paul et al., 2000, 2003). Within minutes of the observed activation of ERK1/2, both STEP46 and STEP61 levels are increased, and the upregulation of STEP is followed by ERK1/2 dephosphorylation. The rapid appearance of STEP argues against nuclear transcription, with subsequent translation and transport of STEP. Moreover, the ability of a MEK inhibitor to block STEP translation suggests that the synthesis of STEP is itself regulated by ERK1/2 activation. Thus, activation of ERK1/2 leads to the rapid synthesis of STEP that in turn may decrease the ERKs activity and contribute to the observed bi-phasic pattern of ERK signaling. Taken together, the data suggest that STEP may provide a feedback mechanism to limit the duration that the ERKs are active. Our study does not clarify whether STEP interacts primarily with ERK1 or ERK2. Preliminary experiments performed using primary neuronal cultures suggest that TAT-STEP specifically immunoprecipitates pERK2 and not pERK1. Further experiments are needed to address the differences in STEP’s interaction with ERK1 and/or ERK2.
The results indicate a potential for STEP to dynamically regulate fear conditioning within the amygdala and oppose the development of synaptic plasticity. These findings have broader implications for ERK-dependent synaptic plasticity outside the amygdala as STEP co-localizes with ERK1/2 in numerous brain regions involved in long-term memory formation. Whether STEP modulates synaptic plasticity in other brain regions is the subject of on-going investigations. The current findings also point to STEP as a potential target for therapeutic interventions, as dampening STEP activity would lead to enhancement of ERK activity and promote cognitive function (Braithwaite et al., 2006).
Acknowledgments
The National Association of Research on Schizophrenia and Depression (NARSAD, SP and PJL), and the National Institutes of Health grants MH01527(PJL), MH52711 (PJL) and DA15222 (JRT), supported this work. We thank Drs. Wenya Linda Bi, Janice Naegele, Angus Nairn, Marilee Ogren, and Glenn Schafe for their helpful comments on the manuscript.
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;29:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neuroscience. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DE, Hazvi S, Rosenblum K, Seger R, Dudai Y. Specific and differential activation of mitogen-activated protein kinase cascades by unfamiliar taste in the insular cortex of the behaving rat. J Neurosci. 1998;18:10037–10044. doi: 10.1523/JNEUROSCI.18-23-10037.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem. 2001;8:229–242. doi: 10.1101/lm.30901. [DOI] [PubMed] [Google Scholar]
- Blanco-Aparicio C, Torres J, Pulido R. A novel regulatory mechanism of MAP kinases activation and nuclear translocation mediated by PKA and the PTP-SL tyrosine phosphatase. J Cell Biol. 1999;147:1129–1136. doi: 10.1083/jcb.147.6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum S, Moore AN, Adams F, Dash PK. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, Naegele JR. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J Neurosci. 1995;15:1532–1544. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Paul S, Nairn AC, Lombroso PJ. Synaptic plasticity: One STEP at a time. TINS. 2006 doi: 10.1016/j.tins.2006.06.007. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R, Sharma E, Lukacsi E, Solimena M, Naegele JR, Lombroso PJ. STEP61: A new member of a family of brain-enriched PTPs is localized to the ER. J Neurosci. 1996;16:7821–7831. doi: 10.1523/JNEUROSCI.16-24-07821.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R, Raghunathan A, Solimena M, Lombroso PJ. STEP: A family of brain enriched PTPs: Alternative splicing produces transmembrane, cytosolic and truncated isoforms. European Journal of Cell Biology. 1997;72:337–344. [PubMed] [Google Scholar]
- Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Flint A, Tganis T, Barford D, Tonks N. Development of substrate trapping mutants to identify physiological substrates of PTPs. PNAS. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998a;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong S, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998b;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004a;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004b;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- Lombroso PJ, Naegele JR, Sharma E, Lerner M. A protein tyrosine phosphatase expressed within dopaminoceptive neurons of the basal ganglia and related structures. J Neurosci. 1993;13:3064–3074. doi: 10.1523/JNEUROSCI.13-07-03064.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Armentia M, Sah P. Development and subunit composition of synaptic NMDA receptors in the amygdala: NR2B synapses in the adult central amygdala. J Neurosci. 2003;23:6876–6883. doi: 10.1523/JNEUROSCI.23-17-06876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron. 2003;39:309–325. doi: 10.1016/s0896-6273(03)00404-5. [DOI] [PubMed] [Google Scholar]
- Murphy ES, Harding JW, Muhunthan K, Holtfreter KL, Wright JW. Role of mitogen-activated protein kinases during recovery from head-shake response habituation in rats. Brain Res. 2005;1050:170–179. doi: 10.1016/j.brainres.2005.05.040. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Nguyen TH, Liu J, Lombroso PJ. Striatal enriched phosphatase 61 (STEP61) dephosphorylates Fyn at phosphotyrosine 420. J Biol Chem. 2002;277:24274–24279. doi: 10.1074/jbc.M111683200. [DOI] [PubMed] [Google Scholar]
- Oyama T, Goto S, Nishi T, Sato K, Yamada K, Yoshikawa M, Ushio Y. Immunocytochemical localization of the striatal enriched protein tyrosine phosphatase in the rat striatum: EM study. Neuroscience. 1995;69:869–880. doi: 10.1016/0306-4522(95)00278-q. [DOI] [PubMed] [Google Scholar]
- Paul S, Hisayuki Y, Snider G, Picciotto M, Nairn A, Lombroso PJ. Dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase, STEP, through a PKA-mediated pathway. 5630–5638. J Neurosci. 2000;20:5630–5638. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Nairn A, Wang P, Lombroso PJ. NMDA-mediated activation of the protein tyrosine phosphatase, STEP, regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1986. [Google Scholar]
- Pelkey K, Askalan R, Paul S, Kalia LV, Nguyen TH, Pitcher GM, Salter MW, Lombroso PJ. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34:127–138. doi: 10.1016/s0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Pulido R, Zuniga A, Ulrich A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 1998;17:7337–7350. doi: 10.1093/emboj/17.24.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44:75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Sah P, Lopez De Armentia M. Excitatory synaptic transmission in the lateral and central amygdala. Ann NY Acad Sci. 2003;985:67–77. doi: 10.1111/j.1749-6632.2003.tb07072.x. [DOI] [PubMed] [Google Scholar]
- Saxena M, Williams S, Brockdorff J, Gilman J, Mustelin T. Inhibition of T cell signaling by mitogen-activated protein kinase-targeted hematopoietic tyrosine phosphatase (HePTP) J Biol Chem. 2000;274:11693–11700. doi: 10.1074/jbc.274.17.11693. [DOI] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory consolidation of auditory Pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20:RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of Pavlovian fear conditioning. J Neurosci. 2000;2:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma E, Zhao F, Bult A, Lombroso PJ. Identification of two alternatively spliced transcripts of STEP: a subfamily of brain-enriched protein tyrosine phosphatases. Molecular Brain Research. 1995;32:87–93. doi: 10.1016/0169-328x(95)00066-2. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Swank MW, Sweatt JD. Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning. J Neurosci. 2001;21:3383–3391. doi: 10.1523/JNEUROSCI.21-10-03383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Cur Opin Neurobio. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Szinyei C, Stork O, Pape HC. Contribution of NR2B subunits to synaptic transmission in amygdaloid interneurons. J Neurosci. 2003;23:2549–2556. doi: 10.1523/JNEUROSCI.23-07-02549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Kawamura M, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor enhances neuronal translation by activating multiple initiation processes: comparison with the effects of insulin. J Biol Chem. 2001;276:42818–42825. doi: 10.1074/jbc.M103237200. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol J-C, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Hervé D, Girault J-A. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. PNAS. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoose AM, Winder DG. NMDA and beta1-adrenergic receptors differentially signal phosphorylation of glutamate receptor type 1 in area CA1 of hippocampus. J Neurosci. 2003;23:5827–5834. doi: 10.1523/JNEUROSCI.23-13-05827.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. PNAS. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci. 1999;19:10512–10519. doi: 10.1523/JNEUROSCI.19-23-10512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- Wu SP, Lu KT, Chang WC, Gean PW. Involvement of mitogen activated protein kinase in hippocampal long-term potentiation. J Biomed Sci. 1999;6:409–417. doi: 10.1007/BF02253672. [DOI] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]