

Summary
The adaptor protein missing-in-metastasis (MIM) contains independent F- and G-actin-binding domains, consisting respectively of an N-terminal 250-aa IRSp53/MIM homology domain (IMD) and a C-terminal WASP-homology domain 2 (WH2). We determined the crystal structures of MIM’s IMD and that of its WH2 bound to actin. The IMD forms a dimer, with each subunit folded as an antiparallel three-helix bundle. This fold is related to that of the BAR domain. Like the BAR domain, the IMD has been implicated in membrane binding. Comparison of the structures reveals that the putative membrane-binding surfaces of the two domains have opposite curvatures, which could be directly linked to the type of curvature of the interacting membrane. The WH2 of MIM is longer than the prototypical WH2, interacting with all four subdomains of actin. We characterize a similar WH2 at the C-terminus of IRSp53 and propose that in these two proteins WH2 performs a scaffolding function.
Introduction
Missing-in-metastasis (MIM) and insulin receptor tyrosine kinase substrate p53 (IRSp53) form part of a new family of actin cytoskeleton adaptor proteins (Bompard et al., 2005; Funato et al., 2004; Miki et al., 2000; Woodings et al., 2003). Like most actin-binding proteins, MIM and IRSp53 are multidomain proteins, containing protein-protein interaction modules, involved in signaling and localization, and structurally conserved actin-binding motifs.
A gene coding for a 356-aa C-terminal fragment of MIM was originally isolated using mRNA differential display, and identified as a protein whose expression appeared to be down-regulated in certain bladder cancer cell lines (Lee et al., 2002). Full-length MIM was subsequently cloned and shown to contain 759-aa (Woodings et al., 2003). Although it was initially proposed that MIM might function as a metastasis suppressor protein (Lee et al., 2002), this role has not been confirmed (Bompard et al., 2005; Nixdorf et al., 2004). Instead, MIM seems to play a role in cytoskeleton remodeling (Lin et al., 2005; Mattila et al., 2003; Yamagishi et al., 2004), possibly downstream of tyrosine kinase signaling (Gonzalez-Quevedo et al., 2005; Woodings et al., 2003) and Rho-family GTPases (Bompard et al., 2005). MIM localizes to areas of dynamic actin assembly and its overexpression induces the formation of actin-rich protrusions resembling surface ruffles and microspikes (Woodings et al., 2003). MIM has also been identified as a sonic hedgehog inducible protein that potentiates Gli transcription (Callahan et al., 2004).
MIM is a modular protein (Figure 1A). Its actin-binding function can be attributed to two spatially separated actin-binding domains: an N-terminal 250-aa IRSp53/MIM homology domain (IMD) (Yamagishi et al., 2004) and a C-terminal 30-aa WASP-homology domain 2 (WH2) (Mattila et al., 2003). The 475-aa central region sandwiched in between these two actin-binding domains is rich in Pro, Ser and Thr residues. This region appears to play regulatory/scaffolding roles; it binds receptor protein tyrosine phosphatase δ (RPTP δ) (Gonzalez-Quevedo et al., 2005; Woodings et al., 2003), the transcription factor Gli and the tumor suppressor Sufu (Callahan et al., 2004), and the SH3 domain of cortactin (Lin et al., 2005), a protein implicated in the nucleation and stabilization of Arp2/3-mediated filament branches (Uruno et al., 2001; Weaver et al., 2001).
Figure 1.
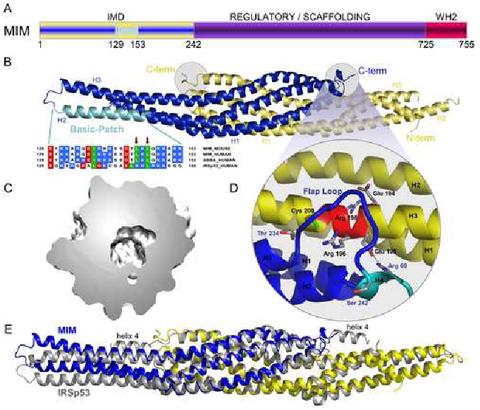
Crystal structure of the IMD of mouse MIM. (A) Schematic representation of MIM (yellow/blue, IMD; purple, middle regulatory/scaffolding region; red, WH2). (B) Ribbon representation of the structure of the IMD dimer (figure made with the program PyMOL, http://www.pymol.org). The two subunit of the dimer are colored blue and yellow. Helices 1 to 3 of each subunit are labeled H1, H2 and H3. Also shown is a sequence alignment corresponding to the conserved basic cluster at the symmetric ends of the IMD dimer (highlighted cyan in one of the subunits of the structure). In this alignment, red, blue, and green represent negatively charged, positively charged, and hydrophobic conserved amino acids, respectively. Accession numbers are: MIM_MOUSE, Q8R1S4; MIM_HUMAN, O43312; ABBA_HUMAN, Q765P7; IRSp53_HUMAN, Q9UQB8. Red arrows point to amino acids Leu 145 and Leu 147, which were mutated in this study. (C) Slice cut through the middle of the molecular surface of the IMD dimer revealing the interior cavity. (D) Close-up view of the “flap” loop between helices 3 and 4 that covers the “signature sequence” (Yamagishi et al., 2004) of the IMD, which is a charged and conserved sequence that is buried in the structure (red colored area of helix 3). (E) Superimposition of the structures of the IMDs of MIM and IRSp53. The two structures were superimposed based on the best overlapping central region (amino acids 26-68, 72-110 and 24-66, 69-107 of both chains of MIM and IRSp53, respectively). The view is as in part B and figure 3. This orientation highlights the differences between the A chains of the two proteins (blue). Although not well seen from this angle, similar differences occur between the B chains (yellow). Notice that helix 4 of the IMD of IRSp53 is missing in MIM.
The relationship between MIM and IRSp53 first emerged from the discovery that the two proteins share similar N-terminal IMDs, an actin-binding domain that has also been implicated in actin bundling (Yamagishi et al., 2004). Like MIM, IRSp53 is an adaptor protein that plays a role in actin cytoskeleton remodeling by linking Rho-family GTPases, such as Rac and Cdc42, to effector proteins, such as Mena (Krugmann et al., 2001) and the Arp2/3-complex activator protein WAVE (Miki et al., 2000). The crystal structure of the IMD of IRSp53 has been determined, consisting of a dimer, with each subunit forming an extended four-helix bundle (Millard et al., 2005).
Here we describe the crystal structures of the IMD of MIM and that of its WH2 bound to actin. Despite low sequence similarity, the IMDs of MIM and IRSp53 are structurally similar, and therefore may bind actin and Rac in a similar fashion. The structure of the IMD is generally related to that of the BAR (Bin-Amphiphysin-Rvs) domain, a fold involved in membrane binding (Peter et al., 2004). However, the overall shape of the two domains is markedly different, which probably explains their different roles in membrane curvature sensing. The WH2 of MIM is unusual, both because of its localization in the protein and the way in which it interacts with actin. We characterize a similar WH2 in IRSp53, further expanding the relationship between these two adaptor proteins.
Results
Structure of the IMD of MIM
The crystal structure of the IMD of MIM (N-terminal 250 amino acids) was determined to 1.85 Å resolution, using the single anomalous dispersion method and X-ray data collected from a Se-Met-substituted crystal (Experimental Procedures and Table 1). The IMD forms a dimer (Figure 1B). The structure is well defined in the electron density map, except for three areas, which are disordered: the last six amino acids of chain A, the last eight amino acids of chain B, and amino acids Asp 155 to Ser 168 of chain B. The electron density map also reveals three amino acids from the expression vector (Ala-Gly-His) at the N-terminal ends of both chains.
Table 1.
Crystallographic Data And Refinement Statistics
| WH2 | IMD | ||
|---|---|---|---|
| High resolution | Se-Peak | ||
| Diffraction statistics | |||
| Space group | P 21 21 21 | P 21 | P 21 |
| Cell parameters | |||
| a, b, c, Å | 42.1, 75.5, 229.0 | 53.5, 37.3, 129.0 | 53.5, 37.3, 128.9 |
| α, β, γ, ° | 90.0, 90.0, 90.0 | 90.0, 94.07, 90.0 | 90.0, 94.08, 90.0 |
| Resolution | |||
| Total, Å | 41.4-2.5 | 48-1.85 | 48-2.1 |
| Highest shell, Å | 2.59-2.5 | 1.92-1.85 | 2.17-2.1 |
| Completeness, % | 88.7 (80.1) | 98.7 (86.7) | 99.4 (99.1) |
| Redundancy | 8.9 (5.1) | 10.7 (4.5) | 6.8 (6.5) |
| Unique reflections | 23589 (2069) | 43479 (3873) | 30686 (3335) |
| R-merge1, % | 7.2 (23.2) | 5.7 (38) | 8.6 (33.3) |
| Average I/σ | 25.8 (7.2) | 30.4 (3.6) | 11.8 (3.5) |
| Refinement statistics | |||
| R-factor2, % | 21.7 | 18.4 | |
| R-free3, % | 28.4 | 22.9 | |
| Rms deviations | |||
| Bond length, Å | 0.012 | 0.014 | |
| Bond angles, ° | 1.38 | 1.13 | |
| Average B-factor | |||
| Protein atoms, Å2 | 59.1 | 33.2 | |
| Solvent atoms, Å2 | 43.7 | 39.9 | |
| PDB code | 2D1K | 2D1L | |
R-merge=∑(/-</>)/∑/; / and </> are the intensity and the mean value of all the measurements of an individual reflection
R-factor=∑|Fo - Fc|/∑ |Fo|; Fo and Fc are the observed and calculated structure factors
R-free; R-factor calculated for a randomly selected subset of the reflections (5%) that were omitted during the refinement Values in parentheses correspond to highest resolution shell
Each chain is folded as an extended (∼135 Å) antiparallel three-helix bundle (Figure 1B). The two subunits in the dimer are oriented opposite to one another and interact extensively. Thus, the contact area between subunits is 2941 Å2 (calculated with CCP4 program AreaIMOL, using a 1.4 Å probe). The six α-helices that comprise the IMD dimer form a twisted ellipsoid ∼183 Å in length and ∼30 Å in diameter (at the widest point). Despite extensive contacts between adjacent α-helices, the IMD cannot be classified as a coiled-coil structure. Indeed, an analysis of the structure using the program Socket (Walshaw and Woolfson, 2001) reveals that there exist short, scattered regions of coiled-coil between pairs of α-helices, but not a single region of the six-helix bundle displays the classical knobs-into-holes layer extending through all the α-helices.
The dimer features a sizable 1396 Å3 (calculated with the program Swiss-PDB using a 1.4 Å probe) cavity in the middle (Figure 1C). This cavity contains a number of water molecules. Although the side chains that are directed toward the cavity are predominantly hydrophobic, a number of polar amino acids, including Thr 47, His 86, Glu 213 and Glu 195, also point toward this cavity. These amino acids are involved in interactions among them. Thus, the O δ of Thr 47 of one chain is hydrogen bonded to the same atom from the other chain. Similarly, Glu 213 and Glu 195 of one chain form salt bridges with His 86 and Arg 69 from the other chain.
The structure of the IMD of IRSp53 has also been determined (Figure 1E) (Millard et al., 2005). The fact that this structure could not be used as a molecular replacement model to determine the current structure suggested from the beginning that important differences were to be expected. Indeed, an alignment of the sequences based on a superimposition of the structures, reveals that the IMDs of MIM and IRSp53 share only ∼19.3 % sequence identity and although generally similar, the structures superimpose with a relatively large RMS deviation of 2.8 Å. The differences remain important within the core (or middle) region (RMS deviation 1.64 Å), defined as the region where the two subunits of the dimer overlap (MIM residues 22-119 and 192-235). However, the two structures differ more significantly toward the N- and C-termini and the distal ends of the ellipsoid (corresponding to the loop between helices 2 and 3). These differences may be ascribed mainly to increased flexibility in these regions, since the identical molecules that form the IMD dimers also display large RMS deviations (2.0 Å for MIM and 1.5 Å for IRSp53).
The IMD of IRSp53 presents a short α-helix at the C-terminus (helix 4). In the structure of MIM’s IMD, only one turn of this α-helix is observed for chain A, whereas the helix is fully missing in chain B, possibly due to local disorder in the structure (Figure 1B). In contrast, the loop Thr 234-Ser 242 preceding helix 4 (Figure 1D) is well defined in the electron density map for both chains. The conformation of this loop is very similar between the two chains of MIM, as well as between the two chains of IRSp53. However, the conformation of the loop differs quite significantly between the two proteins, which may have functional implications. This loop forms a “flap” that covers the so-called signature sequence of the IMD (Yamagishi et al., 2004) of the other molecule in the dimer (Figure 1D). The signature sequence, EER[R/G]R (MIM residues Glu 194-Arg198), is located within α-helix 3. MIM presents a Gly at the fourth position of this motif, whereas a bulkier residue (Arg 192) at this position in IRSp53 is directed toward the flap loop and affects its conformation. Another important difference is that a putative disulfide bond in IRSp53 (Millard et al., 2005), between Cys 195 of the signature sequence and Cys 230 of the flap loop, is missing in MIM which lacks the latter Cys (corresponding to Thr 234 in MIM).
Because the signature sequence forms part of an α-helix, some of the charged amino acids in this sequence are directed inward, while others are covered by the flap loop (Figure 1D). As a result, MIM amino acids Glu 195, Arg 196 and Arg 198 are all buried in the structure and make electrostatic contacts with main chain atoms, as well as a salt bridge between Glu 195 of one chain and Arg 69 of the other chain. Charged amino acids are rarely buried and their occurrence typically points to important regions of the structure. The occurrence of buried and charged side chains within the signature sequence and the interaction with the “flap” loop, which connects the IMD to other domains of the protein, suggest an important role for this region of the IMD, possibly in the control or protein-protein interactions involving the IMD.
Actin-binding and bundling activities of the IMD of MIM
The IMD was originally described as an actin-binding and bundling domain (Yamagishi et al., 2004). However, the ability of the IMD of human MIM to bundle actin in vitro has lead to conflicting results, ranging from significant bundling (Bompard et al., 2005) to weak (Yamagishi et al., 2004) or no bundling activity (Gonzalez-Quevedo et al., 2005). We decided to test the actin-binding and bundling activities of the IMD of mouse MIM, which presents a four amino acid insert (154 VDAQ157) (Mattila et al., 2003) near what has been described as the actin-binding site (Bompard et al., 2005; Millard et al., 2005). F-actin-binding by the IMD of mouse MIM was confirmed using a high-speed co-sedimentation experiment (Figure 2A and Experimental Procedures). A similar experiment, carried out at varying IMD concentrations, resulted in a Kd estimate of ∼17 μM (Figure 2B). This value is similar to that obtained for IRSp53 (5 μM) (Millard et al., 2005).
Figure 2.
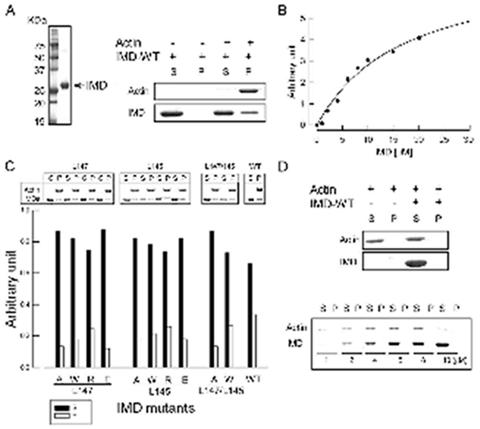
Testing the actin binding and bundling activities of the IMD of MIM. (A) Purified IMD (left panel) and high-speed F-actin binding assay (right panel). 10 μM aliquots of IMD were ultracentrifuged at 400,000 X g, in the presence (+) and the absence (-) of 5 μM F-actin. Equal aliquots of supernatant (S) and pellet (P) were analyzed on a SDS-PAGE gel. (B) Quantification of the F-actin binding affinity of the IMD. 2 μM F-actin aliquots were incubated with increasing amounts of IMD (0, 1, 2, 4, 6, 8, 10, 15 and 20 μM) and analyzed on gel as in part 1A. Each point corresponds to the average densitometric reading of three independent experiments. The line represents the best fit of the data to the Michaelis-Menten equation. (C) High-speed co-sedimentation analysis of IMD mutants. 10 μM aliquots of each mutant were incubated with 5 μM F-actin and analyzed on gel (upper panel). The percentage of F-actin-bound IMD was calculated as the IMD fraction in pellet. Before each experiment the mutants were ultracentrifuged at 400,000 X g to eliminate potential aggregates. The lower panel illustrates the percentage of bound (empty bars) vs. unbound (black-filled bars) mutants from three independent experiments. (D) Low-speed analysis of the F-actin bundling activity of the IMD. 5 μM F-actin was centrifuged at 10,000 X g in the absence (-) and the presence (+) of 10 μM wild-type IMD, and the supernatant (S) and pellet (P) were analyzed on gel (upper panel). F-actin stays in the supernatant, indicating the lack of bundling activity. This result was confirmed by experiments carried out at increasing ratios IMD to F-actin (1, 2, 4, 6, 8, 10) (lower panel).
A cluster of basic amino acids at the distal ends of the IMD dimer has been implicated in actin binding in IRSp53 (Millard et al., 2005). Although mutations of individual amino acids in this cluster had no effect in actin binding, a construct where Lys residues 142, 143, 146, and 147 were simultaneously mutated to Glu showed somewhat reduced actin binding (10 μM vs 5 μM for the wild type IMD) (Millard et al., 2005). This result was interpreted as evidence that the basic cluster is involved in actin binding. However, the change in binding affinity appears minor, in particular considering the effect that a substitution of four positive charges by negative charges could have on the general stability and electrostatic properties of the IMD dimer. The IMD of MIM also presents positively charged clusters at the extremes of the dimer (Figure 3A), featuring a total of 10 positively charged side chains in the region between Lys 131 and Lys 153 (Figures 1B). The same group reported a similar reduction in the actin-binding affinity of MIM’s IMD when Lys residues 149, 150, 152, 153 in this cluster were simultaneously replaced by Asp (Bompard et al., 2005).
Figure 3.
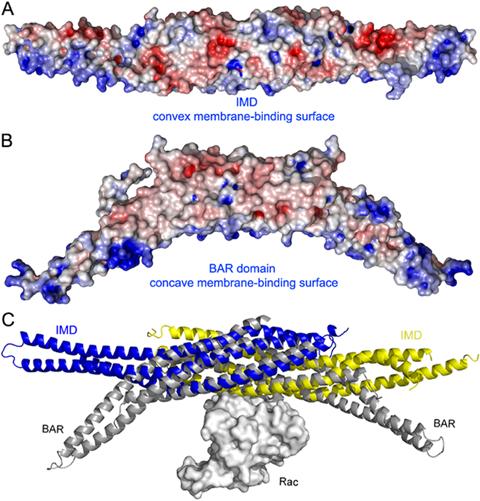
Structural and functional relationship between the IMD and BAR domains. (A) Electrostatic surface representation of the IMD dimer calculated with the program APBS (Baker et al., 2001) and displayed with the program PyMOL (http://www.pymol.org). Red and blue indicate negatively and positively charged regions, respectively (red, -6 kTe-1, blue +6 kTe-1). Note the positively charged and slightly convex surface, thought to mediate the interactions with membranes of the IMD (Suetsugu et al., 2006). (B) Similar electrostatic representation of the BAR domain of amphiphysin (Peter et al., 2004). The orientation is the same as in part A. Note that the shape of the positively charged membrane binding surface of the BAR domain is concave. (C) Superimposition of the structures of the IMD of MIM (blue, yellow) with that of the BAR domain of arfaptin complexed with Rac (Tarricone et al., 2001) (gray). The orientation is the same as in parts A and B. The two folds have different curvatures, but superimpose well in the middle section where the dimers overlap, suggesting that this region may also mediate the binding of Rac in MIM and IRSp53.
Amino acids Ile 137, Leu 145 and Leu 147 (equivalent to IRSp53 Leu 134, Lys 142 and Leu 144) are the only hydrophobic amino acids within the basic cluster of MIM (Figure 1B). While Ile 137 is buried in the structure, Leu 147 is partially exposed and Leu 145 is fully exposed. Given the general importance of hydrophobic amino acids in protein-protein recognition (Jones and Thornton, 1996) we decided to mutate these two Leu residues. Ten mutations were generated, with both leucine residues being replaced individually to Ala, Trp, Arg and Glu, and simultaneously to Ala or Trp. These mutations were designed to change the electrostatic character of the basic patch, as well as to increase or reduce the size of the two exposed hydrophobic side chains within this area. In addition, we expected these mutations to alter the local conformation of the basic patch, and thereby actin binding. To our surprise, none of the mutations affected actin binding significantly (Figure 2C). More importantly, the double mutations did not contribute additively to a lesser actin-binding efficiency. These results, which are quantitatively similar to those obtained for IRSp53 (Millard et al., 2005), suggest that the basic cluster is unlikely to form a major (or a single) actin-binding site. Thus, although the actin-binding interface appears to involve the region around the basic cluster, other parts of the IMD are most likely involved. Note further that the structurally related BAR domain (see below) presents a similar cluster of basic amino acids at the distal ends of the dimer (Peter et al., 2004), and yet the BAR domain is not typically involved in actin binding. Instead, the basic cluster of the BAR domain binds negatively charged phospholipid membranes (Peter et al., 2004).
We further tested the ability of the IMD of MIM to bundle actin at physiological salt concentration. First, the quality of F-actin for this experiment was checked using low-speed sedimentation (10,000 X g) and rotary-shadowing electron microscopy, to guarantee that no bundles were formed in the absence of the IMD. In contrast with two previous reports (Bompard et al., 2005; Yamagishi et al., 2004), we found that the IMD of MIM did not bundle F-actin under any of the conditions tested (Figure 2D, and Experimental Procedures). This result is in agreement, however, with another report that a slightly longer MIM construct (amino acids 1-277) showed markedly reduced bundling activity compared to the full-length protein (Gonzalez-Quevedo et al., 2005). The disagreement between different laboratories concerning the bundling activity of the IMD may have resulted from nonspecific aggregation of the IMD at low ionic strengths, or the use of F-actin preparations that appear to sediment even in the absence of the IMD construct.
However, it could be also questioned whether the IMD construct studied here is dimeric in solution. To answer this question, we determined the molecular mass of the IMD in solution using Multi-Angle Light Scattering combined with particle separation by Asymmetric Field Flow Fractionation (Figure S1 and legend; see the Supplementary Data available with this article online). The molar mass of the elution peak determined by this method was 56.7 ± 3.4 kDa, which is in excellent agreement with the expected theoretical mass of the dimer (56.3 kDa). Combined, the lack of bundling activity and the finding that the IMD is a dimer in solution would suggest that the actin-binding surface of the IMD spans over the two subunits of the dimer.
Structure of the WH2 of MIM complexed with actin
MIM presents a second actin-binding site at the C-terminus, consisting of a WH2 domain (Mattila et al., 2003). We made a synthetic peptide corresponding to this WH2, comprising amino acids Asp 724 to the C-terminus (Ser 755) of human MIM. With the exception of the first two amino acids, which display conservative mutations, this sequence is identical to that of mouse MIM (Figure 4A). The crystal structure of this WH2 (Figure 4B) was determined as a ternary complex with actin-DNase I. DNase I, which was necessary in order to prevent actin polymerization during crystallization, does not appear to have a significant effect on the actin-binding affinity of WH2 (Chereau et al., 2005), and makes no contacts with the WH2 peptide in the current structure. However, the last two amino acids of the peptide (Phe 754 and Ser 755) are disordered in the structure, and we cannot distinguish whether this is due to a local effect of DNase I, or that these amino acids do not normally interact with actin and therefore become disordered. Also disordered in the structure is the first amino acid of the WH2 peptide, which does not appear to be important for actin binding, since the interactions with actin start after residue Gly 728 of the WH2 peptide.
Figure 4.
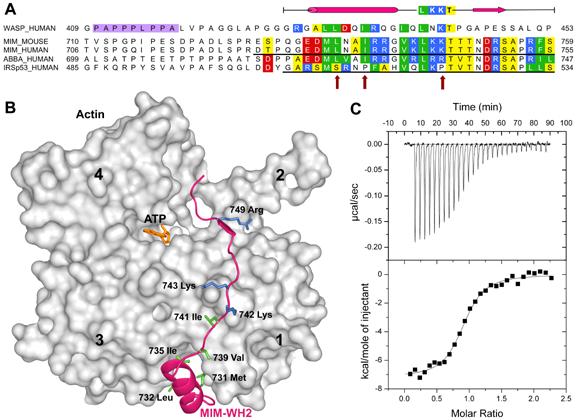
The WASP-homology domain 2 (WH2) of MIM and IRSp53. (A), Comparison of a classical WH2 (represented by WASP, Wiskott-Aldrich Syndrome Protein) with the WH2s of MIM, ABBA and IRSp53. Red, blue, green and yellow correspond to negatively charged, positively charged, hydrophobic and small (Thr, Val, Ser, Ala) conserved amino acids, respectively. The diagram above the sequences represents a secondary structure assignment based on the structure determined here (cylinder, α-helix; arrow, β-strand). Accession numbers are as in figure 1, and WASP_HUMAN, P42768. Red arrows point to non-canonical amino acids present in the WH2 of IRSp53. (B) Structure of the WH2 of MIM (red ribbon) bound to actin (gray surface). Numbers 1-4 indicate actin’s four subdomains. The side chains of some of the amino acids involved in interactions with actin are shown (green, hydrophobic; blue, positively charged). (C) Binding of the WH2 of IRSp53 to actin measured by ITC. The upper graph corresponds to the heat evolved upon repeated 10 μL injections of a 100 μM solution of the WH2 peptide into a 10 μM solution of actin in G-buffer. The lower graph shows the binding isotherm produced by integration of the heat for each injection. The line represents a nonlinear least squares fit to the data using a single-site binding model. The following thermodynamic parameters were determined from the fitting: dissociation constant Kd =0.28 ± 0.04 μM; molar enthalpy ΔH = -7.2 ± 0.1 kcal.mol-1; and stoichiometry n = 0.9.
The structure of the WH2 of MIM can be conceptually subdivided into two parts, an N-terminal amphiphilic α-helix, comprising amino acids Gly 728 to Gly 738, and a C-terminal extended region from Val 739 to Arg 753 (Figure 4B). As previously shown (Chereau et al., 2005), the most important contribution to the interaction with actin comes from the N-terminal α-helix that binds in the hydrophobic cleft between actin subdomains 1 and 3. In the current structure, amino acids Met 731, Leu 732, and Ile 735, on the hydrophobic side of this α-helix, are embedded within the hydrophobic cleft in actin (Figure 4B). The extended portion of the WH2 of MIM follows a path alongside the actin surface, climbing to the top of actin subdomains 2 and 4. Amino acids Val 739 and Leu 741 within this region bind in a hydrophobic pocket on the actin surface formed by amino acids Ile 341, Ile 345 and Leu 349. Leu 741 of the WH2 peptide forms part of the canonical LKKT sequence, found in other actin-binding proteins, such as thymosin β4 (Paunola et al., 2002) and the linker region between gelsolin domains 1 and 2 (Irobi et al., 2003). Interestingly, the two Lys residues of this canonical sequence bind atop actin residues Asp 24 and Asp 25, but interact with actin only via main chain atoms. Thus, the main chain nitrogen and oxygen atoms of Lys 742 are hydrogen bonded to the oxygenand nitrogen atoms of actin residues Gly 23 and Asp 25, respectively. The other important element of the interaction for this part of the WH2 peptide involves the segment Thr 746 to Arg 749, which is incorporated as an additional β-strand into a β-sheet in actin subdomain 1 (running parallel to actin β-strand Arg 28 to Phe 31). Finally, Arg 749 of the WH2 peptide forms a salt bridge with actin residue Glu 93. The remaining portion of WH2 extends across the cleft between actin subdomains 2 and 4, and appears to interact only weakly with actin, which is consistent with the limited contribution of this portion of WH2 to the actin-binding affinity (Chereau et al., 2005).
An alternatively spliced WH2 at the C-terminus of IRSp53
IRSp53 is another actin cytoskeleton adaptor protein, which like MIM presents an N-terminal IMD. Six isoforms of IRSp53 have been identified. In addition to the IMD, all six isoforms present identical CRIB, SH3, and WW protein-protein interaction modules. The differences between isoforms occur at the C-termini. Two of the isoforms present a C-terminal extension consisting of a PDZ-binding sequence (Soltau et al., 2004), another two present WH2-related extensions, and the remaining two appear to have no functionally identifiable extensions. The WH2 of IRSp53 is unusual (Figure 4A). While the C-terminal portion of this WH2 is nearly identical to that of MIM, the N-terminal α-helix, known to play a critical role in actin binding (Chereau et al., 2005), has a non-canonical sequence. Indeed, the periodicity of hydrophobic amino acids in the segment corresponding to the α-helix is altered, and there are conserved Pro and Gly residues in this region, which could prevent the formation of an α-helix. In addition, one of the Lys residues in the canonical LKKT sequence is replaced by Pro in IRSp53 (Figure 4A). Taken together these observations raised doubts about the capacity of this WH2 to bind actin. We decided to study the binding of this WH2 to actin using ITC (Figure 4C). A peptide corresponding to the WH2 of human IRSp53 isoform 2, amino acids Gly 506 to Ser 534, was synthesized (Figure 4A). The actin-binding affinity of this peptide (Kd =0.28 μM) was found to be surprisingly similar to that measured previously by us under identical conditions for the WH2 of MIM (Kd = 0.23 μM) (Chereau et al., 2005). Therefore, we conclude that, like MIM, certain isoforms of IRSp53 present two independent actin-binding domains at the N- and C-terminal ends, which in both proteins enclose a large central region featuring various protein-protein interaction modules.
Discussion
The IMD of MIM is an all α-helical structure, which dimerizes to form a twisted ellipsoid ∼183 Å in length, with a large cavity in the middle (Figure 1). Despite low sequence similarity the structures of the IMDs of MIM and IRSp53 (Millard et al., 2005) are generally similar. The loop following α-helix 3 of MIM’s IMD forms a “flap” that covers the so-called “signature sequence” of the IMD, a conserved and charged sequence that is conspicuously buried in the structure (Figure 1D).
While we were able to confirm that the IMD binds F-actin with ∼17 μM affinity (Figure 2A and B), we found that the symmetric patches of basic amino acids at the distal ends of the dimer (Figure 3A) play only a limited role in this interaction (Figure 2C). Furthermore, we did not observe any bundling activity for the IMD (Figure 2D), which would have been consistent with the symmetric ends of the dimer being involved in actin binding. If as previously suggested (Yamagishi et al., 2004) MIM is an actin bundling protein, this function may require other parts of the molecule that lie outside the IMD. Gonzalez-Quevedo et al. (Gonzalez-Quevedo et al., 2005) reached a similar conclusion by studying various fragments of MIM. They showed that most of the bundling activity could be restored by a construct comprising amino acids 1 to 408 of MIM. Another possibility is that bundling is regulated (or potentiated) in vivo by still unknown factors.
The IMD of IRSp53 interacts with Rac, possibly functioning as an intermediate for the activation of WAVE, which is recruited by the SH3 domain of IRSp53 (Miki et al., 2000). Similarly, the IMD of MIM has been shown to bind and activate Rac, suggesting that MIM could link Rac to effector proteins involved in lamellipodia formation, such as WAVE (Bompard et al., 2005). The structural basis for the Rac-IMD interaction is unknown. Interestingly, the structure of the IMD resembles that of the BAR domain, which also binds small GTPases (Habermann, 2004). The crystal structures of various BAR-domain proteins, including arfaptin (Tarricone et al., 2001), amphiphysin (Peter et al., 2004) and endophilin (Weissenhorn, 2005) have been determined. Although the BAR domain is curved and the IMD is relatively straight, the two folds superimpose remarkably well in the middle section, where the two subunits that conform these two domains overlap (Figure 3C). It is via this well-overlapping middle section that the binding of small GTPases appears to take place. Indeed, the structure of arfaptin was also determined bound to Rac (Tarricone et al., 2001). One molecule of Rac sits at the midpoint of the arfaptin BAR dimer. It is likely that the IMDs of MIM and IRSp53 bind Rac in a similar fashion, as illustrated by a superimposition of the structures of MIM and arfaptin-Rac (Figure 3C). Note, however, that this superimposition does not represent an accurate model of the interaction, since there is no obvious sequence similarity between the IMD and BAR domains and local changes are likely.
The binding of Rac and actin by the IMD of MIM appear to be mutually exclusive (Bompard et al., 2005). Although this study did not determine the total extent of the actin-binding interface, the lack of bundling activity (Figure 2D) and the fact that the distal ends of the IMD dimer do not constitute a major actin-binding site (Figure 2C) would suggest that the middle section of the IMD dimer also participates in actin binding. As suggested by the analogy with the BAR domain, the binding of Rac may also involve the middle section of the IMD dimer (Figure 3C), possibly explaining why actin and Rac bind in a mutually exclusive manner.
We have stressed here the striking resemblance between the IMD and BAR folds, including their shared ability to bind small GTPases. In addition, both domains present similar clusters of positively charged amino acids (Figures 3A and 3B), which in the BAR domain coincide with the concave surface of the dimer and are involved in phospholipid membrane binding (Peter et al., 2004). The most noticeable difference between the two folds is that the IMD forms relatively straight dimers (Millard et al., 2005), whereas the BAR domain forms curved “banana-shaped” dimers (Peter et al., 2004; Tarricone et al., 2001; Weissenhorn, 2005). However, the curvature of the BAR domain varies from protein to protein (arfaptin > amphiphysin > endophilin), which may facilitate the binding to membranes with different curvatures. The IMD was discovered independently and due to the lack of sequence similarity was not originally considered a member of the BAR domain family (Yamagishi et al., 2004). A comparison of the structures of the IMD and BAR domains would now suggest that the two domains are not only structurally but also functionally related to each other (Figures 3). Indeed, it was recently reported that like the BAR domain the IMD also binds membranes and that this function is mediated by the clusters of basic amino acids at the distal ends of the dimer (Suetsugu et al., 2006). Interestingly, the directionality of membrane deformation by the IMD (outward) was found to be opposite to that produced by the BAR domain (inward). The structures may provide an explanation for this observation since the concave and positively charged surface implicated in membrane binding in the BAR domain adopts a somewhat convex shape in the IMD (Figure 3). Therefore, the evidence to date suggests that the IMD is a multifunctional module, linking the actin cytoskeleton to the formation of membrane protrusions by direct interactions with both F-actin and membranes, all under the control of the small GTPase Rac.
The WH2 of MIM interacts with all four subdomains of actin (Figure 4B). It consists of an N-terminal amphiphilic α-helix that binds in the cleft between actin subdomains 1 and 3 and a C-terminal extended region that binds along the actin surface and the nucleotide cleft reaching the top of actin subdomains 2 and 4. Note that the end of this WH2 coincides with the C-terminus of the MIM protein. The prototypical WH2 found among WASP-family proteins tends to be shorter (Figure 4A), and presents little or no interactions with actin after the LKKT sequence (Chereau et al., 2005).
We demonstrated here that certain isoforms of IRSp53 present a C-terminal WH2 that binds actin with similar affinity to that of MIM’s WH2, further extending the relationship between these two actin-cytoskeleton scaffolding proteins. WH2 is the smallest actin-binding motif known. Based on their sequences and structures, we have identified two types of WH2s; long and short (Chereau et al., 2005). Short WH2s consist solely of the N-terminal α-helix and the LKKT-related sequence (for example WASP’s WH2, Figure 4A). Long WH2s present an additional ∼10 amino acids at the C-terminus. The extra amino acids of long WH2s share sequence similarity with T β4 and make similar contacts with actin (Irobi et al., 2004), supporting a previously proposed relationship between the WH2 and T β families (Paunola et al., 2002). However, it remains unclear whether the extra amino acids of long WH2s play any specific role, since they don’t seem to contribute significantly to the actin binding affinity nor the nucleotide exchange inhibition by actin (Chereau et al., 2005).
What is the role of WH2 in MIM and IRSp53? WH2 could serve two possible functions: recruit actin monomers, or recruit a protein to a specific actin cytoskeletal network. Actin filament nucleation and elongation factors, including WASP, Ena/VASP and spire, form the main group of WH2-containing proteins. These proteins present short WH2s, typically positioned C-terminal to Pro-rich sequences (Figure 4A). In WASP WH2 is followed by the central (or C) region that binds one of the subunits of Arp2/3 complex, whereas in VASP WH2 is known as the G-actin-binding domain (GAB) and is followed by the F-actin-binding domain (FAB). The C region of WASP and the FAB domain of VASP are related to each other, and both constitute specialized forms of WH2 (Chereau and Dominguez, 2006). Spire, on the other hand, contains four WH2s in tandem (Quinlan et al., 2005). We have proposed that in these proteins WH2 becomes involved in nucleation and elongation by forming nuclei for actin assembly and by mediating the incorporation of profilin-actin at the barbed end of growing filaments (Chereau and Dominguez, 2006; Chereau et al., 2005). So far, we have identified long WH2s in actobindin, WIP, MIM (Chereau et al., 2005), and now in IRSp53. It appears that in MIM and IRSp53 WH2 occurs within a different domain organization than in most cytoskeletal proteins (Figure 4A). Thus, in MIM and IRSp53 WH2 is found in isolation at the C-terminal end; i.e. not immediately preceded by Pro-rich sequences nor followed by other WH2s (or WH2-related sequences). Unlike the actin monomer-trapping molecule T β4 and the nucleation-elongation factors described above, MIM and IRSp53 function as scaffolding proteins. It is therefore likely that WH2 helps recruit MIM and IRSp53, as well as their multiple binding partners, to specific cytoskeletal networks. Consistent with this idea, images of cells overexpressing full-length MIM show a significant loss of stress fibers (Gonzalez-Quevedo et al., 2005; Mattila et al., 2003; Woodings et al., 2003), but this effect appears diminished for MIM constructs lacking the WH2 region (Bompard et al., 2005; Gonzalez-Quevedo et al., 2005).
What is the spatial relationship between the IMD and WH2 domains? Hydrophobic cluster analysis (Callebaut et al., 1997) suggests that the region sandwiched in between the IMD and WH2 of MIM is mostly unstructured, with only two segments with predicted globular or inducible folding (Figure S2). Given these characteristics and the antiparallel organization of the IMD dimer, the two WH2s could be located far apart from each other in the protein, which would imply a lack of communication between them. More likely, however, the various domains of MIM and IRSp53 fold back into a more compact structure, possibly mediated by auto-regulatory interactions involving the IMD and other parts of the molecule.
Experimental procedures
Preparation of proteins and peptides
The cDNA encoding for full-length mouse MIM was purchased from ATCC. Amino acids 1-250, corresponding to MIM’s IMD, were amplified by PCR and inserted into vector pTYB12 (New England Biolabs). This vector comprises a chitin affinity purification tag and an intein self-cleavage domain. IMD mutants (Leu 145 to Ala, Trp, Asp, Arg; Leu 147 to Ala, Trp, Asp, Arg and the double mutants Leu-145 and Leu 147 to Ala; Leu 145 and Leu 147 to Trp) were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
BL21(DE3) cells (Invitrogen) were transformed with the various IMD constructs and grown in LB medium at 37°C until the OD at 600 nm reached a value of 0.8. Expression was induced by addition of 1 mM isopropylthio-_-D-galactoside (IPTG) and carried out overnight at 20°C. Cells were harvested by centrifugation and resuspended in chitin-affinity-column equilibration buffer (20 mM Tris pH 7.5, 500 mM NaCl, 1 mM EDTA, 100 μM PMSF), followed by standard purification on a chitin affinity column at 4°C(New England Biolabs manual). The proteins were eluted from this column following DTT-induced self-cleavage of the intein. The proteins were then dialyzed against 20 mM Tris pH 7.5, 50 mM NaCl and 1 mM DTT, and purified to homogeneity on a MonoQ column (Pharmacia). A Se-Met substituted IMD protein was obtained following a similar procedure by growing cells in M9 media, supplemented with 70 mg/ml Se-Met. Actin was prepared from rabbit muscle as described (Graceffa and Dominguez, 2003). Ultra pure grade bovine pancreatic DNAse I was purchased from BioWorld. WH2 domains, corresponding to human MIM724-755 and human IRSp53506-534, were synthesized on an ABI431 peptide synthesizer and purified by HPLC. The concentrations of the peptides were determined by amino acid analysis (Dana-Farber Cancer Institute, Boston, MA).
F-actin-binding assay
For this experiment actin underwent additional purification through a gel filtration Sephacryl S300HR column (Pharmacia). Actin in G-buffer (2 mM Tris pH 7.4, 0.2 mM CaCl2, 0.2 mM ATP, 1 mM DTT, 1 mM NaN3) was polymerized by addition of 100 mM KCl, 2 mM MgCl2, and 2 mM EGTA (F-buffer). IMD and IMD mutants were dialyzed against the same F-buffer, and centrifuged at 400,000 X g for 30 min before the experiments, to remove potential aggregates. 10 μM IMD (or IMD mutants) were mixed with 5 μM F-actin on ice, and incubated for 30 min. The protein mixtures were then centrifuged at 400,000 X g for 30 min. Equal volumes of supernatant and pellet were analyzed on a 15% SDS-PAGE gel. A quantification of the F-actin binding affinity was obtained by adding increasing amounts of IMD (0, 1, 2, 4, 6, 8, 10, 15, 20 μM) to 2 μM F-actin and analyzed as described above. The Coomassie Blue-stained gels were then scanned at high resolution and the intensities of the bands were quantified using the program ImageJ version 1.34S (http://rsb.info.nih.gov/).
F-actin-bundling assay
Actin was purified and polymerized as described above. Before each bundling experiment, both F-actin and the IMD were centrifuged for 30 min at 10,000 and 400,000 X g, respectively. Note that this step is important in order to remove potential high molecular weight aggregates. F-actin samples were then visualized on a Philips EM 300 electron microscope to ensure that actin bundles were not present prior to the addition of the IMD. 10 μM IMD were then mixed with 5 μM F-actin and incubated for 1 h at room temperature. Next, the mixture was centrifuged at low speed (10,000 X g) for 30 min at room temperature. Supernatant and pellet were analyzed by SDS-PAGE gel as described above. A quantitative bundling assay was done in a similar way by adding increasing amounts of IMD (1, 2, 4, 6, 8, 10 μM) to 1 μM F-actin.
Crystallization, data collection and structure determination
Actin-DNase I complex at a 1:1 molar ratio was mixed with MIM’s WH2 domain peptide at 1.5 molar excess. The ternary complex was then dialyzed against G-buffer (2 mM Tris pH 7.5, 0.2 mM CaCl2, 0.2 mM ATP, 1 mM NaN3), and concentrated to ∼10 mg/ml using a Centricon device (Millipore). The complex was crystallized at 20°C, using the hanging drop vapor diffusion method. The 2 μL hanging drops consisted of a 1:1 (v/v) mixture of protein solution and a well solution containing 13-14 % PEG 3350, 50 mM Na cacodylate pH 6.8-7.2, and 100 mM Na-formate. Before crystallization MIM’s IMD was dialyzed against 20 mM Tris pH 7.4, 50 mM NaCl, and 5 mM DTT and concentrated to ∼20 mg/ml. Crystals were obtained at 4°C in 0.1 M Tris pH 7.4, 0.2 M LiCl and 16% PEG 2000 MME. The crystals were flash-frozen in propane, using 25% glycerol as cryoprotectant. X-ray datasets were collected at the BioCARS beamlines 14-BM-D (IMD) and 14-BM-C (actin-WH2) at the Advance Photon Source (Argonne, IL). The datasets were indexed and scaled with program HKL-2000 (HKL Research, Inc.). The structure of the actin-WH2 complex was determined by molecular replacement using CCP4 program AMoRe and the structure of WIP-actin-DNase I as a search model (Chereau et al., 2005). Model building and refinement were done with the program Coot (Emsley and Cowtan, 2004), and CCP4 program Refmac (Table 1).
The structure of the IMD was determined from the anomalous signal of a Se-Met substituted crystal, using the single anomalous dispersion (SAD) method and data collected to 2.1 Å resolution at the absorption peak wavelength of the Se atoms (Table 1). The positions of 11 out of 18 Se atoms in the structure were found with the program SnB (Weeks and Miller, 1999). These positions were then refined and phases were calculated with the program Solve (Terwilliger and Berendzen, 1999). About 70% of the model was built automatically with the program ARP/wARP (Morris et al., 2003), and using a 1.85 Å resolution X-ray dataset collected from the same Se-Met substituted crystal. Further model building and refinement were carried out with the program Coot (Emsley and Cowtan, 2004), and CCP4 program Refmac. Data collection and refinement statistics are given in Table 1.
Isothermal titration calorimetry
These measurements were done using a VP-ITC (MicroCal, Northampton, MA). To determine βH and Ka of WH2-actin association, the WH2 peptide of IRSp53 at a concentration of 100 μM was titrated, in 10 μL injections, into 1.44 mL of 10 μM actin in G-buffer at 25°C. The duration of each injection was 10 s, with an interval of 3 min between injections. The heat of binding was corrected for the small exothermic heat of injection, determined by injecting WH2 peptides into buffer. Data were analyzed using MicroCal’s Origin program.
Supplementary Material
Acknowledgments
Supported by NIH grant GM073791. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. W-31-109-Eng-38. Use of the BioCARS facilities was supported by NIH grant RR07707. The authors thank Andrei Lupas for important comments about coiled-coil structures and Pekka Lappalainen for helpful discussions.
Abbreviations
- MIM
Missing-in-metastasis
- IRSp53
insulin receptor tyrosine kinase substrate p53
- IMD
IRSp53/MIM homology domain
- WASP
Wiskott-Aldrich Syndrome Protein
- WH2
WASP-homology domain 2
- BAR
Bin-Amphiphysin-Rvs
- ABBA
actin-bundling protein with BAIAP2 homology
- ITC
isothermal titration calorimetry
Footnotes
Atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2D1L and 2D1K).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompard G, Sharp SJ, Freiss G, Machesky LM. Involvement of Rac in actin cytoskeleton rearrangements induced by MIM-B. J Cell Sci. 2005;118:5393–5403. doi: 10.1242/jcs.02640. [DOI] [PubMed] [Google Scholar]
- Callahan CA, Ofstad T, Horng L, Wang JK, Zhen HH, Coulombe PA, Oro AE. MIM/BEG4, a Sonic hedgehog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 2004;18:2724–2729. doi: 10.1101/gad.1221804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callebaut I, Labesse G, Durand P, Poupon A, Canard L, Chomilier J, Henrissat B, Mornon JP. Deciphering protein sequence information through hydrophobic cluster analysis (HCA): current status and perspectives. Cell Mol Life Sci. 1997;53:621–645. doi: 10.1007/s000180050082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereau D, Dominguez R. Understanding the role of the G-actin-binding domain of Ena/VASP in actin assembly. J Struct Biol. 2006 doi: 10.1016/j.jsb.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Chereau D, Kerff F, Graceffa P, Grabarek Z, Langsetmo K, Dominguez R. Actin-bound structures of Wiskott-Aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. Proc Natl Acad Sci U S A. 2005;102:16644–16649. doi: 10.1073/pnas.0507021102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Funato Y, Terabayashi T, Suenaga N, Seiki M, Takenawa T, Miki H. IRSp53/Eps8 complex is important for positive regulation of Rac and cancer cell motility/invasiveness. Cancer Res. 2004;64:5237–5244. doi: 10.1158/0008-5472.CAN-04-0327. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Quevedo R, Shoffer M, Horng L, Oro AE. Receptor tyrosine phosphatase-dependent cytoskeletal remodeling by the hedgehog-responsive gene MIM/BEG4. J Cell Biol. 2005;168:453–463. doi: 10.1083/jcb.200409078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graceffa P, Dominguez R. Crystal Structure of Monomeric Actin in the ATP State: Structural Basis of Nucleotide-Dependent Actin Dynamics. J Biol Chem. 2003;278:34172–34180. doi: 10.1074/jbc.M303689200. [DOI] [PubMed] [Google Scholar]
- Habermann B. The BAR-domain family of proteins: a case of bending and binding? EMBO Rep. 2004;5:250–255. doi: 10.1038/sj.embor.7400105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irobi E, Aguda AH, Larsson M, Guerin C, Yin HL, Burtnick LD, Blanchoin L, Robinson RC. Structural basis of actin sequestration by thymosin-beta4: implications for WH2 proteins. Embo J. 2004;23:3599–3608. doi: 10.1038/sj.emboj.7600372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irobi E, Burtnick LD, Urosev D, Narayan K, Robinson RC. From the first to the second domain of gelsolin: a common path on the surface of actin? FEBS Lett. 2003;552:86–90. doi: 10.1016/s0014-5793(03)00934-7. [DOI] [PubMed] [Google Scholar]
- Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci U S A. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krugmann S, Jordens I, Gevaert K, Driessens M, Vandekerckhove J, Hall A. Cdc42 induces filopodia by promoting the formation of an IRSp53:Mena complex. Curr Biol. 2001;11:1645–1655. doi: 10.1016/s0960-9822(01)00506-1. [DOI] [PubMed] [Google Scholar]
- Lee YG, Macoska JA, Korenchuk S, Pienta KJ. MIM, a potential metastasis suppressor gene in bladder cancer. Neoplasia. 2002;4:291–294. doi: 10.1038/sj.neo.7900231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Liu J, Wang Y, Zhu J, Zhou K, Smith N, Zhan X. Differential regulation of cortactin and N-WASP-mediated actin polymerization by missing in metastasis (MIM) protein. Oncogene. 2005;24:2059–2066. doi: 10.1038/sj.onc.1208412. [DOI] [PubMed] [Google Scholar]
- Mattila PK, Salminen M, Yamashiro T, Lappalainen P. Mouse MIM, a tissue-specific regulator of cytoskeletal dynamics, interacts with ATP-actin monomers through its C-terminal WH2 domain. J Biol Chem. 2003;278:8452–8459. doi: 10.1074/jbc.M212113200. [DOI] [PubMed] [Google Scholar]
- Miki H, Yamaguchi H, Suetsugu S, Takenawa T. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature. 2000;408:732–735. doi: 10.1038/35047107. [DOI] [PubMed] [Google Scholar]
- Millard TH, Bompard G, Heung MY, Dafforn TR, Scott DJ, Machesky LM, Futterer K. Structural basis of filopodia formation induced by the IRSp53/MIM homology domain of human IRSp53. Embo J. 2005;24:240–250. doi: 10.1038/sj.emboj.7600535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RJ, Perrakis A, Lamzin VS. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 2003;374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- Nixdorf S, Grimm MO, Loberg R, Marreiros A, Russell PJ, Pienta KJ, Jackson P. Expression and regulation of MIM (Missing In Metastasis), a novel putative metastasis suppressor gene, and MIM-B, in bladder cancer cell lines. Cancer Lett. 2004;215:209–220. doi: 10.1016/j.canlet.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Paunola E, Mattila PK, Lappalainen P. WH2 domain: a small, versatile adapter for actin monomers. FEBS Lett. 2002;513:92–97. doi: 10.1016/s0014-5793(01)03242-2. [DOI] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Quinlan ME, Heuser JE, Kerkhoff E, Mullins RD. Drosophila Spire is an actin nucleation factor. Nature. 2005;433:382–388. doi: 10.1038/nature03241. [DOI] [PubMed] [Google Scholar]
- Soltau M, Berhorster K, Kindler S, Buck F, Richter D, Kreienkamp HJ. Insulin receptor substrate of 53 kDa links postsynaptic shank to PSD-95. J Neurochem. 2004;90:659–665. doi: 10.1111/j.1471-4159.2004.02523.x. [DOI] [PubMed] [Google Scholar]
- Suetsugu S, Murayama K, Sakamoto A, Hanawa-Suetsugu K, Seto A, Oikawa T, Mishima C, Shirouzu M, Takenawa T, Yokoyama S. The RAC-binding domain/IRSP53-MIM homology domain of IRSP53 induces RAC-dependent membrane deformation. J Biol Chem. 2006 doi: 10.1074/jbc.M606814200. [DOI] [PubMed] [Google Scholar]
- Tarricone C, Xiao B, Justin N, Walker PA, Rittinger K, Gamblin SJ, Smerdon SJ. The structural basis of Arfaptin-mediated cross-talk between Rac and Arf signalling pathways. Nature. 2001;411:215–219. doi: 10.1038/35075620. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uruno T, Liu J, Zhang P, Fan Y, Egile C, Li R, Mueller SC, Zhan X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat Cell Biol. 2001;3:259–266. doi: 10.1038/35060051. [DOI] [PubMed] [Google Scholar]
- Walshaw J, Woolfson DN. Socket: a program for identifying and analysing coiled-coil motifs within protein structures. J Mol Biol. 2001;307:1427–1450. doi: 10.1006/jmbi.2001.4545. [DOI] [PubMed] [Google Scholar]
- Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, Cooper JA. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001;11:370–374. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- Weeks CM, Miller R. The design and implementation of SnB v2.0. J Appl Cryst. 1999;32:120–124. [Google Scholar]
- Weissenhorn W. Crystal structure of the endophilin-A1 BAR domain. J Mol Biol. 2005;351:653–661. doi: 10.1016/j.jmb.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Woodings JA, Sharp SJ, Machesky LM. MIM-B, a putative metastasis suppressor protein, binds to actin and to protein tyrosine phosphatase delta. Biochem J. 2003;371:463–471. doi: 10.1042/BJ20021962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi A, Masuda M, Ohki T, Onishi H, Mochizuki N. A novel actin bundling/filopodium-forming domain conserved in insulin receptor tyrosine kinase substrate p53 and missing in metastasis protein. J Biol Chem. 2004;279:14929–14936. doi: 10.1074/jbc.M309408200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.