

Abstract
Nucleotide variation at the genomic region encompassing the odorant-binding protein genes OS-E and OS-F (OS region) was surveyed in two populations of Drosophila simulans, one from Europe and the other from Africa. We found that the European population shows an atypical and large haplotype structure, which extends throughout the ∼5-kb surveyed genomic region. This structure is depicted by two major haplotype groups segregating at intermediate frequency in the sample, one haplogroup with nearly no variation, and the other at levels more typical for this species. This pattern of variation was incompatible with neutral predictions for a population at a stationary equilibrium. Nevertheless, neutrality tests contrasting polymorphism and divergence data fail to detect any departure from the standard neutral model in this species, whereas they confirm the non-neutral behavior previously observed at the OS-E gene in D. melanogaster. Although positive Darwinian selection may have been responsible for the observed unusual nucleotide variation structure, coalescent simulation results do not allow rejecting the hypothesis that the pattern was generated by a recent bottleneck in the history of European populations of D. simulans.
SMELL is one of the oldest and most important senses of animals. Olfaction allows for the recognition and discrimination of the chemical signals that provide animals the essential information needed to detect and assess food, to identify mating partners and predators, and to adopt individual and collective behavior. Positive natural selection can therefore play a major role in the evolution of olfactory system genes; indeed, human olfaction-involved genes are among the most fast-evolving genes (e.g., Clark et al. 2003; Gilad et al. 2003; Nielsen et al. 2005; see also Gimelbrant et al. 2004). Furthermore, Darwinian positive selection has been proposed to be driving the evolution of some olfactory system genes in rodents (Emes et al. 2004), channel catfish (Ngai et al. 1993), salamander (Watts et al. 2004; Palmer et al. 2005), and insects (Willett 2000; Krieger and Ross 2002, 2005).
The olfactory system constitutes the principal sensory modality of invertebrates, showing a high specificity and sensitivity. Odorant receptors (OR) are located in the external membrane of specialized sensory neurons, which extend their dendrites into an aqueous fluid. Hydrophobic odors traverse the fluid space bound to odorant-binding proteins (OBPs), which deliver them close to receptors. The OBP multigene family includes two different subfamilies of proteins: the general odorant-binding proteins, which bind and transport general odorants, and the pheromone-binding proteins, which specialize in pheromone perception (Vogt and Riddiford 1981; Pelosi and Maida 1995). Phylogenetic analysis indicates that these OBPs show a monophyletic origin (Vogt et al. 1999; Hekmat-Scafe et al. 2000). There is little knowledge about the evolution of the Drosophila OBP multigene family. This family contains 51 putative members located in clusters and scattered across the genome (Hekmat-Scafe et al. 2002). Surprisingly, this number is very close to the actual number of OR genes (∼61 members; Vosshall 2000). This fact, together with the different odorant-binding specificities and gene expression patterns (e.g., Vogt et al. 1991, 1999; Galindo and Smith 2001; Vogt et al. 2002), suggests that OBPs not only are odorant carriers, but also have an important role in the olfactory coding.
Recently, we studied the molecular evolution (at intraspecific and interspecific levels) of two members of the OBP gene family, the OS-E and OS-F genes, in different Drosophila species (Sánchez-Gracia et al. 2003; see also Hekmat-Scafe et al. 2000). These genes most likely originated from an old gene duplication event (>40 MYA) and still maintain a high degree of conservation at the gene structure, amino acid, and nucleotide levels. In D. melanogaster, we detected a significant gradient of silent nucleotide polymorphism along the OS region (the ∼5-kb genomic region including the OS-E and the OS-F genes along with their intergenic region), and an excess of amino acid replacements fixed at the OS-E gene in this species. Although the results are unlikely for a neutral evolving region, we could not discriminate among the different selection scenarios that might accommodate the data.
Here, we analyze levels and patterns of DNA polymorphism and divergence along the OS genomic region in two populations of D. simulans to provide new insights into the evolution of these olfactory genes and, particularly, to examine if the evolutionary pattern observed in D. melanogaster is a species-specific feature or if it is instead shared with other Drosophila species. We found that both levels of silent variation and estimates of recombination rates are considerably higher in D. simulans than in D. melanogaster, whereas the non-neutral behavior previously detected at the OS-E gene in the D. melanogaster lineage might have been caused by a relaxation of functional constraints in this species. Surprisingly, we detected that the European population of D. simulans is highly structured, with a very unusual haplotype configuration depicted by two clearly different haplogroups segregating at intermediate frequencies in the sample, one almost invariant (only one segregating site) and the other with a high level of nucleotide polymorphism (83 segregating sites and 10 indel polymorphisms). We discuss these findings along with their implications for the molecular population genetics of D. simulans.
MATERIALS AND METHODS
Drosophila strains:
Twenty-two highly inbred D. simulans lines (obtained after 10 generations of sib mating) randomly sampled from two natural populations were surveyed: 11 lines from a European population (Montblanc, Spain; S lines) and 11 from an African sample (Maputo, Mozambique; MZ lines) (Rozas et al. 2001). This survey also includes the 14 lines of D. melanogaster (Córdoba, Spain; M lines) and the lines of D. mauritiana and D. erecta reported in Sánchez-Gracia et al. (2003).
DNA extraction, PCR amplification, and DNA sequencing:
Genomic DNA of D. simulans was extracted using a modification of protocol 48 from Ashburner (1989). For the European sample, an ∼5-kb fragment (referred to as fragment 1), including the OS-E and OS-F genes along with their intergenic region, was amplified by PCR (Saiki et al. 1988), while in the African lines, the amplified fragment (an ∼2-kb fragment referred to as fragment 2) included only the transcribed OS-E region, the intergenic region, and the first untranslated exon of the OS-F gene (Figure 1). PCR products were cycle sequenced and separated on a Perkin-Elmer (Norwalk, CT) ABI PRISM 377 automated DNA sequencer, following the manufacturer's instructions. For each line, the DNA was sequenced on both strands. The new reported nucleotide sequences have been deposited in the EMBL nucleotide sequence database under accession nos. AM490947–AM490968.
Figure 1.—

Structure of the OSregion in D. melanogaster. Solid and shaded boxes indicate the coding regions of the OS-E and OS-F genes, respectively. Introns are indicated by a “V.” The two amplified fragments are also indicated.
Data analysis:
Nucleotide sequences were assembled using the SeqMan version 5.53 software (DNASTAR, Madison, WI), multiple aligned with ClustalX (Thompson et al. 1997), and edited in MacClade 3.06 (Maddison and Maddison 1992). Phylogenetic analysis was performed using the neighbor-joining algorithm (Saitou and Nei 1987) implemented in MEGA3 (Kumar et al. 2004). Clade support measures were based on 1000 bootstrap replicates. We estimated the number of synonymous and nonsynonymous substitutions in each branch of the tree by using the codeml program from the PAML 3.14 package (Yang 1997). DnaSP 4.0 (Rozas et al. 2003) was used for most intraspecific and some interspecific analyses. The level of DNA polymorphism was estimated as the per-site nucleotide diversity (π; Nei 1987), Watterson's parameter (θ; Watterson 1975), and haplotype diversity (h; Nei 1987). Nucleotide divergence between species was estimated as K, and the number of substitutions per site was corrected according to Jukes and Cantor (1969).
The Tajima (1989), Fu and Li (1993), Fu (1997), and Wall (1999) tests were conducted to examine whether the DNA polymorphism pattern conformed to the neutral expectations. Fay and Wu's (2000) test was used to assess the presence of high-frequency-derived nucleotide variants in the sample. The correlation between polymorphism and divergence expected under the neutral model was tested using the HKA test (Hudson et al. 1987). We used the McDonald–Kreitman (MK) test (McDonald and Kreitman 1991) to test for the expected relationship between the ratio of replacement-to-synonymous fixed differences between species and the ratio of replacement-to-synonymous polymorphisms within species. The putative genetic differentiation between populations was determined by a permutation test (1000 replicates) using the Snn statistic of Hudson (2000). The confidence intervals and P-values of the neutrality tests were obtained by Monte Carlo simulations based on the neutral coalescent process assuming the infinite-sites model in a large constant-size population (Hudson 1990). Coalescent simulations were performed either assuming no intragenic recombination or with variable levels of recombination (10,000 replicates). Simulations were carried out fixing the value of θ (θ = 4Neυ, where Ne is the effective population size and υ is the per-gene mutation rate) or fixing the number of segregating sites; since both methods yielded similar results, we will show only the results based on the later method.
The composite-likelihood method of Kim and Stephan (2002) was used to determine the compatibility of the data with a selective-sweep model. This method relies on the detection of the local skew in the frequency spectrum of mutations caused by a hitchhiking event. The statistical test is based on the likelihood ratio of the neutral and selective-sweep models, which is a function of θ, the recombination rate, and the strength and location of the selected site. The null distribution of the likelihood ratio is obtained from coalescent simulations under the standard neutral model with recombination. The modified version of Meiklejohn et al. (2004) was applied to test for a partial-sweep hypothesis. We estimated the age of the putative selective sweep, assuming that all mutations detected in haplogroup H1 were new mutations originated after the complete reduction of variation caused by the hitchhiking effect (Rozas et al. 2001).
Recombination:
The recombination parameter C (in Drosophila, C = 2Ner, where r is the per-generation recombination rate for the studied region) was estimated using three different methods. The Hudson (1987) method estimates C (CH) from the variance of the average number of nucleotide differences. The Hudson and Kaplan (1985) method estimates C (CR) from the minimum number of recombination events in the sample (RM) by using coalescent simulations. Estimates of C based on the D. simulans recombination map (CM) (Andolfatto and Przeworski 2000) were obtained assuming that r = 1.04 × 10−8 (i.e., assuming that the OS and Gld regions, which are located in chromosomal bands 83CD and 84D, respectively, have the same recombination rate) and that Ne is 2 × 106. We also used computer simulations to estimate the CL value (Rozas et al. 2001), that is, the minimum value of C compatible at 5% with the observed RM value. The effect of intragenic recombination on nucleotide variation was also analyzed using the ZZ statistic (Rozas et al. 2001), which compares the average pairwise linkage disequilibrium between all sites to that between adjacent sites.
Demographic scenario:
We performed coalescent simulations (10,000 replicates) to investigate the compatibility of the data under specific population bottleneck scenarios. We considered a bottleneck model where a panmictic population of effective size NA was instantaneously reduced in size to bNA, Tb generations ago, and persisted for Td generations with this population size until time T0, when it instantly recovered the current population size, fNA (Figure 2). Fay and Wu (1999) have shown that bottleneck severity (Sb) is approximately proportional to the product of the duration of the bottleneck (Td/NA) to its reduction in population size (NA/bNA). Therefore, Sb and Tb are the key parameters determining patterns of nucleotide variation after a reduction of population size (Galtier et al. 2000). We analyzed an exhaustive number of combinations of Tb and Sb to explore a significant part of the parameter space of this model. Bottleneck times ranging from 0.005 to 0.05 and severities ranging from 0.05 to 2 were considered (times are scaled in units of 4Ne generations). We considered two different values of f: f = 1, i.e., equal sizes for the ancestral and present populations, and f = 0.5. Since coalescent simulations fixing S are not accurate for models different from the standard neutral model (S. Ramos-Onsins, personal communication) and the uncertainty in the estimates of the true population mutation parameter θ, we conducted the simulations using the rejection algorithm of Tavaré et al. (1997). For the analysis, we chose values of the θ extracted from a uniform distribution (ranging from 0.0005 to 0.06/bp); the candidate replicate was accepted if the value of θ was compatible with the number of segregating sites observed in the data. Simulations were performed using different values of the population recombination parameter. We computed two summary statistics: the observed number of identical lines (Hudson et al. 1994; Rozas et al. 2001), allowing for just one segregating site, l1, and the Q statistic (Wall 1999), which measures the association among segregating sites. For the l1 statistic, we used a one-tailed test since the explored population bottleneck scenarios likely increase the number of identical lines. For the Q statistic, however, there is no prior hypothesis, and a two-tailed test was applied. Given that the distribution of the Q statistic presents a small number of points, we calculated the two-tailed probability as follows: let p1 and p2 be the probabilities of obtaining a value greater and lower, respectively, than that observed in the data sample; the two-tailed probability, P, was computed as
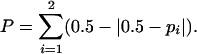 |
Figure 2.—

Graphic representation of the bottleneck scenario.
The combined probability of the two statistics, Λ, was obtained using the formula (see Voight et al. 2005)
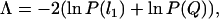 |
where P(l1) and P(Q) denote the individual P-values for l1 and Q statistics, respectively. We used coalescent simulations (10,000 replicates) to obtain the empirical distribution of Λ. In a particular replicate, we calculated the two-tailed P-values associated with each summary statistic from the empirical distribution of the remaining 9999 replicates (i.e., considering the value of this particular replicate as the “observed” value; see also Voight et al. 2005). These probabilities were used to obtain the Λ value associated with each simulation replicate and, thus, to construct its empirical distribution. We conducted a one-tailed test to obtain the probability of observing an equal or higher value of Λ.
RESULTS
European sample:
DNA sequence variation:
We initially surveyed a genomic region that included the OS-E and OS-F genes with their intergenic region (4896 bp, fragment 1; Figure 1) in 11 European lines of D. simulans. A total of 96 nucleotide polymorphic sites (consisting of a minimum of 99 mutation events) and 12 insertion/deletion polymorphisms (ranging from 1 to 60 bp in length) were detected. All nucleotide substitution polymorphisms were silent: 10 were synonymous (6 and 4 in the OS-E and OS-F coding regions, respectively), while the rest were in noncoding regions (Figure 3). All length polymorphisms were in noncoding regions. Table 1 shows estimates of nucleotide variation for the different OS region functional parts. As in previous reports, levels of synonymous variation (at the coding region) were slightly higher than those present at noncoding fragments. Estimates of the silent nucleotide diversity (πSIL = 0.0088) were similar to those obtained in other surveys of the same European population (Cirera and Aguadé 1997; Aguadé 1998, 1999; Rozas et al. 2001) or in other European samples (Baudry et al. 2006) of this species.
Figure 3.—

Nucleotide polymorphisms found at the complete OS region of the Montblanc population. Dots represent nucleotide variants identical to the first sequence. For length polymorphisms, the position indicates the first affected site. Site 1 corresponds to the first position of the OS-E translation start codon. Coding positions are shaded. The last two rows indicate the nucleotide information present in D. mauritiana (D.mau) and D. melanogaster (D.mel) for each polymorphic site detected in D. simulans. Dashes indicate the absence of the corresponding variant. H#1, haplogroup 1; H#2, haplogroup 2; E, OS-E coding region; F, OS-F coding region; d, deletion; i, insertion; Rec, putative recombinant sequences.
TABLE 1.
Summary of the nucleotide variation at the OS-region in the Montblanc population of D. simulans
| 5′ | OS-E | Intergenic | OS-F | Total | |
|---|---|---|---|---|---|
| Silent | |||||
| No. of sitesa | 75 | 308.5 | 934 | 2609.3 | 3926.8 |
| S | 2 | 12 | 18 | 64 | 96 |
| π | 0.0087 | 0.0144 | 0.0070 | 0.0088 | 0.0088 |
| θ | 0.0091 | 0.0133 | 0.0066 | 0.0088 | 0.0086 |
| K | 0.0194 | 0.1076 | 0.0522 | 0.0462 | 0.0519 |
| Synonymous | |||||
| No. of sitesa | 88.5 | 101.3 | 189.8 | ||
| S | 6 | 4 | 10 | ||
| π | 0.0275 | 0.0111 | 0.0188 | ||
| θ | 0.0232 | 0.0135 | 0.0180 | ||
| K | 0.1260 | 0.0578 | 0.0887 | ||
| Noncoding | |||||
| No. of sitesa | 75 | 220 | 934 | 2508 | 3737 |
| S | 2 | 6 | 18 | 60 | 86 |
| π | 0.0087 | 0.0091 | 0.0070 | 0.0087 | 0.0083 |
| θ | 0.0091 | 0.0093 | 0.0066 | 0.0086 | 0.0081 |
| K | 0.0194 | 0.1000 | 0.0522 | 0.0457 | 0.0499 |
S, number of segregating sites; K, nucleotide divergence between D. simulans and D. melanogaster.
Number of sites in the polymorphism data set.
Current silent nucleotide variation levels in D. simulans were higher than those estimated at the homologous syntenic region of D. melanogaster (πSIL = 0.0021). The intraspecific nucleotide variability distribution along the OS region was also quite different between species: (1) D. simulans does not show the gradient in nucleotide diversity observed in D. melanogaster (Sánchez-Gracia et al. 2003) and (2) the HKA test (Hudson et al. 1987) was not significant. Indeed, in D. simulans, levels of polymorphism and divergence correlated, as expected by the neutral model, both for the complete region and for the different functional parts (P > 0.10, Figure 4). We also analyzed putative departures from the neutral frequency spectrum using a variety of neutrality tests. No significant results were obtained by Tajima's D or by Fu and Li's D and F, in spite of the atypical nucleotide structure observed in the sample (see below). Nevertheless, all statistics presented positive values reflecting the presence of a number of substitutions segregating at high frequency.
Figure 4.—

Sliding window of silent nucleotide polymorphism and divergence in the OS region. πSIL, silent nucleotide diversity; KSIL, silent nucleotide divergence between D. simulans and D. melanogaster. Window size, 750 bp; step size, 50 bp.
Using a relative rate test approach, we previously found that the OS-E gene evolved in the D. melanogaster lineage at a nonsynonymous substitution rate higher than that in D. simulans or D. mauritiana (Sánchez-Gracia et al. 2003). The analysis of the relative levels of synonymous and nonsynonymous substitutions within D. melanogaster and between D. simulans and D. melanogaster (MK test) was, however, not significant. Using the whole DNA polymorphism data set (including all European D. melanogaster, n = 14, and D. simulans, n = 11, sequences) to recalculate the MK test, significant results were found (χ2 test; P = 0.029); moreover, using all D. simulans lines (European and African lines, n = 22), we obtained a much more significant P-value (χ2 test; P = 0.005).
Linkage disequilibrium and recombination:
In the European sample, the ZZ statistic value was positive and statistically significant (P = 0.001), evidencing the major role of intragenic recombination in shuffling nucleotide variation among DNA sequences. Table 2 shows the estimates of the recombination parameter C obtained by different methods. The unusual haplotype structure detected at the OS region (see below) could be responsible for the discrepancy between the CH and CR estimates (which are much more dependent on departures from the neutral equilibrium assumptions) and those based on the recombination map, CM (Andolfatto and Przeworski 2000); present discrepancy might also reflect some uncertainties in Ne estimates. Nevertheless, recombination levels in D. simulans were clearly higher than those for the syntenic region of D. melanogaster. For the total sample, 35% (1506) of the pairwise comparisons showed significant linkage disequilibrium values, although none of them were significant after applying the Bonferroni procedure. This method, however, is very conservative for a large number of comparisons and small sample sizes.
TABLE 2.
Estimates of the recombination parameter C for the OS-region
| D. simulans (n = 11) | D. melanogaster (n = 14) | |
|---|---|---|
| RM (CR) | 6 (25) | 2 (12) |
| CH | 3.7 | 12.5 |
| CL | 11.7 | 4.5 |
| CM | 192.2 | 14.7 |
Estimates are from European population data. n, sample size.
Haplotype structure:
The present data show a highly structured nucleotide variation pattern in the Montblanc population of D. simulans. Of 11 sequences, we identified 6 identical or nearly identical sequences (differing by a single nucleotide substitution; Figure 3). This group of sequences was named haplogroup H1. The rest of the sequences (haplogroup H2) harbored, on the contrary, four different haplotypes with 83 segregating sites (85 mutations) and 10 indels. These sequences likely contain preexisting neutral variation (see below). In addition, two of the latter sequences (S18 and S28) show a chimerical pattern, likely caused by a recombination event between members of these two divergent haplogroups. This strong haplotype structure extends along the whole OS region, including all their functional regions. We performed computer simulations based on the coalescent process to investigate whether this pattern might be compatible with the neutral equilibrium model. The results show that this haplotype structure is clearly unlikely under the neutral model, even after using the conservative CL value in the simulations (Table 3). In particular, we found that both the number of haplotypes and the haplotype diversity levels were significantly reduced. This reduction in the haplotype diversity also generates positive and highly significant Fs (Fu 1997) values. We also made coalescent simulations to estimate the probability of observing a given number of identical sequences or of differing by only one segregating site in the sample l1. This probability value is a function of θ, C, and the sample size. Again, the results were highly significant [P (l1 ≥ 6) < 0.001]. These tests were still highly significant even following the strong conservative criteria of no recombination (Table 3). The neighbor-joining tree (Figure 5) clearly illustrates two separate clusters (H1 and H2 haplogroups) with a strong reduced level of variation in the H1 group. Branch lengths also reflect the substantial differences in the population mutational parameter and in the number of synonymous and nonsynonymous substitutions between D. melanogaster and D. simulans.
TABLE 3.
Haplotype-based tests
| Probability
|
||||
|---|---|---|---|---|
| Observed value | C = 0 | C = CL | C = CM | |
| Montblanca | ||||
| l1 | 6 | <0.001 | <0.001 | <0.001 |
| h | 0.855 | 0.008 | 0.003 | <0.001 |
| Fu's Fs | 7.594 | 0.009 | 0.003 | <0.001 |
| Maputob | ||||
| l1 | 3 | 0.438 | 0.146 | 0.009 |
| h | 0.982 | 1.000 | 1.000 | 0.484 |
| Fu's Fs | 0.049 | 1.000 | 0.507 | 0.078 |
m, number of sites. P-values of l1 were based on a one-tailed test while that of h and Fu's Fs were based on a two tailed-test.
n = 11, m = 4622, and CL = 11.7.
n = 11, m = 1733, and CL = 35.1.
Figure 5.—

Neighbor-joining tree of the OS region reconstructed using Jukes and Cantor corrected distances. D. simulans and D. melanogaster sequences are depicted as open circles and solid diamonds, respectively. Bootstrap values >90% are shown under the branches, and X/Y values represent the maximum-likelihood estimates of the number of synonymous and nonsynonymous changes, respectively. H#1, haplogroup 1; H#2, haplogroup; Rec, putative recombinant sequences.
African sample:
To determine whether the unusual haplotype structure detected in the European sample of D. simulans was also present in other populations of this species, we extended the analysis to 11 additional lines from a population of their putative ancestral geographical area, the east African population of Maputo (Mozambique) (Lachaise et al. 1988). Table 4 summarizes the nucleotide variation estimates for the ∼2-kb comparable sequenced regions (fragment 2, Figure 1). In agreement with previous reports, levels of nucleotide diversity were lower in the European (derived) sample. The majority of the polymorphisms found in the European sample are a subset of those segregating in African sequences, and no fixed differences between populations were observed (Figure 6). Nevertheless, the Snn statistic (Hudson 2000) indicates that the two populations are genetically differentiated (P = 0.012); this differentiation might be caused by the peculiar haplotype structure of the European population but also by a putative substructure in the Maputo sample. In the African sample, we did not find the unusual haplotype structure detected in the European one. The African sample presents 10 different haplotypes although, interestingly, one line has information (including indels) identical to that in the H1 haplogroup (Figure 6). In contrast to the Montblanc population, no statistical test of neutrality was significant in the African sample. The neighbor-joining tree (Figure 7) clearly reflects the different pattern of variation between the two samples.
TABLE 4.
Summary of the nucleotide variation at the fragment 2 of D. simulans
| Maputo | Total | |
|---|---|---|
| Sample size, n | 11 | 22 |
| S (η) | 71 (72) | 81 (83) |
| No. of sites | 1733 | 1733 |
| No. of silent sites | 1398.4 | 1398.4 |
| πSIL | 0.0200 | 0.0176 |
| KSIL | 0.0594 | 0.0606 |
S, number of segregating sites; η, total number of mutations; πSIL, silent nucleotide diversity; KSIL, silent nucleotide divergence between D. simulans and D. melanogaster.
Figure 6.—

Nucleotide polymorphisms detected at fragment 2 of the two populations of D. simulans. S and MZ sequences correspond to the lines from Montblanc (Europe) and Maputo (Africa), respectively.
Figure 7.—

Neighbor-joining tree of the OS region (fragment 2) using the sequences of the two D. simulans populations. Open and solid circles indicate Montblanc and Maputo sequences, respectively. H#1, haplogroup 1.
Selective and demographic models:
At first glance, the present pattern of intraspecific DNA variation seems to reflect the footprint of positive natural selection (i.e., a selective sweep) caused by the increase in frequency of a favorable mutation located on, or near to, the OS-E and OS-F gene region. Fay and Wu (2000) proposed a statistical test sensible to the excess of high-frequency variants produced by the hitchhiking effect in the presence of recombination. Despite the negative value of this statistic in the European population (H = −5.472), it is not enough to produce a significant excess of high-frequency-derived polymorphisms (P = 0.209). Furthermore, results of the composite-likelihood method of Kim and Stephan (2002) (see also Meiklejohn et al. 2004), which considers both the frequency of derived variants and their spatial distribution, also indicate that neither a simple selective-sweep model nor a partial-sweep scenario fit the data significantly better than the standard neutral model.
We further investigated whether the reduction in nucleotide variability and the haplotype structure detected in the European sample was compatible with a population bottleneck event. We simulated random DNA sequence samples under different bottleneck scenarios (null hypothesis) using the coalescent framework. To capture most of the information included in the gene genealogies, we summarized the observed and simulated data in terms of the number of identical lines, l1, Wall's Q (Wall 1999) statistic, and also in terms of the new statistic, Λ, which combines their probabilities. We found that only recent bottlenecks (Tb < 0.020; i.e., 16,000 years; 10 generations/year) are compatible with the data (Figure 8A, using CL = 11.7 as the population recombination parameter). Only these conditions generate a number of identical lines similar to those found in the data without affecting significantly the genetic structure of the whole sample. Simulations using higher recombination rate values (CL is likely an underestimate of the true recombination value), however, produce systematically lower probability values, i.e., reducing the bottleneck scenarios compatible with the data. In this case, only an extremely recent bottleneck would fit the Montblanc data (Figure 8B). In contrast, a lower value of the parameter f (f = 0.5) affects the results in the opposite way; that is, it increases slightly the number of bottleneck scenarios compatible with the data (results not shown). On the other hand, the computer simulations have also shown that, for low values of T0 (the time for recovering the population size), the effects of the bottleneck on patterns of nucleotide variation are much more dependent on the parameters Td and b; in particular, for the same severity, models with T0 → 0 are much more compatible with this demographic scenario (results not shown). In these conditions, the atypical haplotype structure detected in the sample might have been generated by older and smaller reductions of the ancestral population size. Since the ad hoc choice of l1 for defining haplogroups might not be conservative, we also generated all empirical distributions of the statistics using the conservative l statistic and, although the number of bottleneck scenarios compatible with the data increased slightly, this does not change the main conclusions of the article (results not shown).
Figure 8.—

P-values (one-tailed test) of the Λ statistic under different bottleneck scenarios (A) C = 11.7, f = 1. (B) C = 50, f = 1. Contours represent the confidence region of parameter space with P-values from P < 0.01 (darker shading) and increasing in units of 0.01 (to the lighter shading). Times are scaled in units of 4Ne generations, 10 generations/year. The standard constant-size neutral model is rejected by the Λ statistical test (C = 11.7, P = 0.002; C = 50, P < 10−4).
DISCUSSION
Nucleotide diversity at the OS region in D. simulans and D. melanogaster:
Previous studies at the OS region of D. melanogaster have shown major differences in the evolutionary history of OS-E and OS-F genes. Here, we find that D. melanogaster and D. simulans also exhibit dissimilar evolutionary patterns. Levels of silent nucleotide variation, as well as estimates of the recombination parameter, were higher in D. simulans than in D. melanogaster (Tables 1 and 2). These results cannot be attributed to putative changes in the chromosomal location of the OS region between these species; although there is a fixed inversion between D. melanogaster and D. simulans (3R chromosomal arm: 85F1-93F6), it does not include the OS region and thus we can assume that the OS genes are in a syntenic conserved segment (see Aulard et al. 2004 for a review).
There are evidences that levels of silent polymorphism in autosomal regions are significantly higher in D. simulans—in both ancestral and derived populations—than in D. melanogaster, which might represent a global higher effective population size of D. simulans (Baudry et al. 2004, 2006; Mousset and Derome 2004). In addition, comparisons of the genetic and cytological maps between these species revealed marked differences in the recombination rate at the OS region (True et al. 1996). These factors might contribute to increasing the linkage selection effects in D. melanogaster (Hill and Robertson 1966; Maynard Smith and Haigh 1974; Charlesworth et al. 1993). The significant results of the HKA test at the OS region in this species (Sánchez-Gracia et al. 2003), but not in D. simulans, would be in agreement with the linkage selection effect hypothesis and might explain the significant gradient of silent nucleotide variation detected along the OS region in D. melanogaster (Figure 4).
A reduction in Ne might affect the fixation probability of slightly deleterious mutations (e.g., nonsynonymous mutations). In fact, we have detected a nearly significant excess of nonsynonymous fixations (six) at the OS-E gene of the D. melanogaster lineage (since the split of the two Drosophila species), while there was only a single fixed replacement in the D. simulans lineage (Figure 5) and the MK test was not significant (Sánchez-Gracia et al. 2003). Therefore, the data might be interpreted as a relaxation of the strength of selection in D. melanogaster, caused by the reduction of population size. This factor, nevertheless, should affect both fixed and polymorphic changes; however, we did not detect any amino acid replacement segregating at the OS-E (or OS-F) gene in these species, although the number of polymorphic sites is very small. The inclusion of the D. simulans polymorphism data in the MK tests, however, would suggest a deficit of nonsynonymous polymorphisms, supporting the relaxation hypothesis. Although a putative Ne reduction might explain part of the data (differences in the levels of nucleotide variation and in the HKA and MK results), they cannot account for the discrepant behavior of the two OS genes of D. melanogaster (we have not detected any reduction of the selective pressure at the OS-F gene), suggesting that both the OS-E and OS-F genes are evolving under different selective evolutionary forces.
Haplotype structure in D. simulans:
In D. simulans, we have detected a strong and atypical genetic structure, caused by the presence of a number of lines with nearly identical sequence (haplogroup H1). Several studies have shown that African (putative ancestral) and European (derived) populations of these species are genetically structured [e.g., Pgd (Begun and Aquadro 1994), runt (Labate et al. 1999), In(2L)t breakpoint (Andolfatto and Kreitman 2000), vermilion and G6pd genes (Hamblin and Veuille 1999; Veuille et al. 2004), and the rp49-jan-ocn region (Parsch et al. 2001; Rozas et al. 2001; Quesada et al. 2003)]. None of these surveys, however, revealed a continuous structured genomic region as long as that observed in the OS region. These results likely reflect some demographic and/or selective effects.
Selective factors:
The existence of a genomic region depleted (completely or partially) of variation is a distinctive fingerprint of the increase in frequency of an advantageous mutation in or close to the affected region, i.e., a selective sweep (Maynard Smith and Haigh 1974). This selective scenario has been proposed to explain an unusual, and extremely similar to that found in Montblanc, haplotype structure observed in the rp49 gene (Rozas et al. 2001) and in the neighbor-linked regions of D. simulans (Parsch et al. 2001; Quesada et al. 2003; Meiklejohn et al. 2004). These authors found the same haplotype structure in both African and European populations, with no significant genetic differentiation between them. Here, nevertheless, we found no significant departures of the standard neutral model using the statistical tests especially designed to detect the action of positive selection. Nonetheless, Rozas et al. (2001) failed to detect a significant excess of high-frequency-derived variants in the initial 1.8-kb survey at the rp49 region, even though Quesada et al. (2003) determined that the haplotype structure gradually decayed at both sides of the most structured stretch, as expected for selective sweeps on a recombining region. In another survey at the same genomic region, Meiklejohn et al. (2004) also concluded that the unusual haplotype feature was likely promoted by a partial selective sweep and estimated the strength of selection associated with the advantageous mutation and the approximate location. In addition, Kim (2006) showed that under recurrent selective sweeps no significant excess of high-frequency-derived alleles is expected; therefore, it cannot be rejected that the lack of significant results at the OS region was caused by a reduced statistical power of the tests or because the current selective scenario is more complex than that assumed in the statistical tests. If true, since OS and rp49 regions are clearly unlinked (they are located 16 polytene bands apart), the rp49 and OS region surveys might be detecting different selective events.
In contrast to Rozas et al. (2001) data, we detected the unusual haplotype structure only in the European population; therefore, the putative selective event might represent some local adaptation process. In a microsatellite variability screen of D. simulans, Schofl and Schlotterer (2004) found that the number of beneficial mutations is higher in derived populations of D. simulans, likely reflecting the adaptive process to new environments. Interestingly, many of the accurate single-locus studies in D. simulans (i.e., ignoring polymorphism data form surveys with fewer than eight sequences within a single population) of samples from derived populations point to selective forces as a major way of explaining the data (e.g., Zurovcova and Eanes 1999; Kern et al. 2002, 2004; Schlenke and Begun 2003, 2004, 2005; Derome et al. 2004; DuMont et al. 2004; Lazzaro 2005; but see also Irvin et al. 1998; Schmid et al. 1999; Duvernell and Eanes 2000). Although these studies did not provide a formal test contrasting adaptive and demographic hypotheses, it seems reasonable to consider that adaptive evolution might occur in derived populations of D. simulans.
We cannot exclude the possibility of a selective sweep originating in Africa. In fact, we have detected one haplogroup H1 line (with identical genetic information, including indels) in the African population. Given the relatively low number of surveyed lines (11 sequences, Figure 6), this European major haplotype would also be present in Africa at a relatively high frequency, i.e., a higher frequency than that expected in a panmictic population. In this case, current patterns of variation might be explained by the action of positive natural selection (in the African population) followed by a further demographic event (such as a bottleneck caused by the out-of-Africa spread). Indeed, present estimates of the date of the putative hitchhiking event (∼10,000 years ago for the OS region) are consistent with this hypothesis.
Demographic factors:
There are a number of demographic scenarios that, a priori, might explain this unusual haplotype configuration. D. simulans, and D. melanogaster, originated in tropical areas and, with the rise of agriculture (i.e., in historical times), spread worldwide by commensalism with humans (Dobzhansky 1965; Lachaise et al. 1988). The species, therefore, experienced a number of founder events, and likely adaptive changes, through a period much shorter than the within-species most recent common ancestor time (4Ne as average). Therefore, the signature of the historical events should still be present in patterns of molecular evolution, and hence the assumption of neutral stationary equilibrium can be unjustified. Hamblin and Veuille (1999) suggested that D. simulans-derived populations could have been generated by a recent admixture of genetically differentiated African populations. Unfortunately, most of the multilocus surveys in D. simulans have been conducted in North American populations, the European samples being exceptional. Andolfatto and Przeworski (2000) compared the population parameters C and θ in 16 independent loci of D. simulans and found a greater-than-expected intralocus linkage disequilibrium. They showed that the data do not fit with a symmetric island model and would need more complex demographic scenarios to be explained. Andolfatto (2001) reexamined the available data and concluded that, although congruent with a simple bottleneck caused by the out-of-Africa, it might be also explained by the presence of an ancient (African) population structure. In addition, Wall et al. (2002) found that, under reasonable conditions, no simple evolutionary model (a simple hitchhiking or a bottleneck) could explain the North American D. simulans data set of Begun and Whitley (2000). Recently, Baudry et al. (2006)—analyzing X-linked nucleotide variation at four loci in nine populations of this species—concluded that their data were consistent with a demographic bottleneck in derived populations.
Several pieces of the European D. simulans OS region results are congruent with those of Baudry et al. (2006). First, Montblanc and Maputo are genetically differentiated populations. Second, levels of silent nucleotide variation in the European population were lower than in the African sample (Table 3). Third, the strong haplotype structure detected in the European sample departs significantly from the neutral expectation (Figure 3). Fourth, the statistical tests used in Baudry et al. (2006) behave similarly in Montblanc and in the two derived populations surveyed by these authors. These tests, however, gave very different results in Maputo and in the populations postulated to be the geographic origin of D. simulans (populations centered on Madagascar), but results similar to those obtained for Zimbabwe and Cameroon populations. To ascertain whether a bottleneck scenario can be responsible for the observed haplogroup pattern, we performed coalescent simulations under different population-size reduction models. Results demonstrate that a recent bottleneck (≤16,000 years ago) is sufficient to account for current departures from the standard neutral model detected in the European sample of D. simulans. Nonetheless, as the true recombination rate was likely higher than the CL conservative value, the bottleneck times compatible with the data would be smaller (e.g., between 4000 and 8000 years ago for C = 50 and f = 1; Figure 8B). Even so, the collections of bottleneck times are in agreement with biogeographic evidences of the European expansion of this species, and hence the pattern of nucleotide variation observed at the OS region might reflect the founder effect caused by the recent colonization process. Although the range of putative bottleneck severities affecting the European population of D. simulans is similar to those estimates from derived populations of D. melanogaster (Baudry et al. 2004; Orengo and Aguade 2004; Thornton and Andolfatto 2006; but see Li and Stephan 2006), current bottleneck times are consistent with the hypothesis that D. simulans would have spread worldwide more recently than its sibling species (Morton et al. 2004; Baudry et al. 2006).
We have also found that reductions with very small recovery times (T0 → 0) or with bottlenecks with f < 1 (i.e., population decline-like scenarios) are scenarios more compatible with the data than other bottlenecks of the same severity. This feature might indicate that the Montblanc population might have not completely recovered the population size existing before the bottleneck event caused by the colonization process or also that the recovery time was fairly short. Finally, it should be noted that present analyses were not conducted by a maximum-likelihood approach—they represent a range of bottleneck times and strengths for which a bottleneck model cannot be rejected, rather than the true likelihood surface; therefore, they should be interpreted with caution.
The present DNA polymorphism pattern might also be generated by more complex demographic scenarios, such as the recent admixture of two differentiated populations (e.g., Hamblin and Veuille 1999), or by a funding effect from an African pool (including a low frequency of haplogroup 1). The former scenario requires, however, that one of those populations must contribute with sequences that harbor no variation. Although spatial or temporal fluctuations in local effective population sizes (Gravot et al. 2004) could contribute to this pattern, it is not clear if this might have occurred in the evolutionary history of the Montblanc population. Therefore, it will be necessary to build null models combining population structure and bottleneck effects.
In conclusion, although the unusual pattern of nucleotide variation observed at the OS region of the European population of D. simulans could be promoted by positive selection, it might be explained solely by demographic factors. Distinguishing between the effect of selective sweeps and demographic factors, such as a recent bottleneck, is a fundamental, yet complex, question. Both a large-scale (multilocus) genomic survey in geographically distinct populations of D. simulans (African and derived populations) and a contrasting among competing hypotheses using powerful statistical methods (e.g., Galtier et al. 2000; Li and Stephan 2006) would be needed to unambiguously determine the role of the different evolutionary forces shaping nucleotide variation in this species.
Acknowledgments
We are particularly indebted to Sebastian E. Ramos-Onsins for sharing unpublished software for computer simulations as well as for insightful discussions. We thank Filipe G. Vieira for his assistance with computer programming and S. O. Kolokotronis for his comments on the manuscript. We also thank M. Veuille and two anonymous reviewers for helpful comments and suggestions on the manuscript. We thank Serveis Científico-Tècnics, Universitat de Barcelona, for automated sequencing facilities. A.S. was supported by a predoctoral fellowship from the Universitat de Barcelona. This work was funded by grants BMC2001-2906 and BFU2004-02253 from the Dirección General de Investigación Científica y Técnica (Spain) and by grant 2001SGR-00101 from Comissió Interdepartamental de Recerca i Innovació Tecnològica (Spain).
References
- Aguadé, M., 1998. Different forces drive the evolution of the Acp26Aa and Acp26Ab accessory gland genes in the Drosophila melanogaster species complex. Genetics 150: 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguadé, M., 1999. Positive selection drives the evolution of the Acp29AB accessory gland protein in Drosophila. Genetics 152: 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., 2001. Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol. Biol. Evol. 18: 279–290. [DOI] [PubMed] [Google Scholar]
- Andolfatto, P., and M. Kreitman, 2000. Molecular variation at the In(2L)t proximal breakpoint site in natural populations of Drosophila melanogaster and D. simulans. Genetics 154: 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., and M. Przeworski, 2000. A genome-wide departure from the standard neutral model in natural populations of Drosophila. Genetics 156: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner, M., 1989. Drosophila: A Laboratory Handbook. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Aulard, S., L. Monti, N. Chaminade and F. Lemeunier, 2004. Mitotic and polytene chromosomes: comparisons between Drosophila melanogaster and Drosophila simulans. Genetica 120: 137–150. [DOI] [PubMed] [Google Scholar]
- Baudry, E., Viginier and M. Veuille, 2004. Non African populations of Drosophila melanogaster have a unique origin. Mol. Biol. Evol. 21: 1482–1491. [DOI] [PubMed] [Google Scholar]
- Baudry, E., N. Derome, M. Huet and M. Veuille, 2006. Contrasted polymorphism patterns in a large sample of populations from the evolutionary genetics model Drosophila simulans. Genetics 173: 759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun, D. J., and C. F. Aquadro, 1994. Evolutionary inferences from DNA variation at the 6-phosphogluconate dehydrogenase locus in natural populations of Drosophila: selection and geographic differentiation. Genetics 136: 155–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun, D. J., and P. Whitley, 2000. Reduced X-linked nucleotide polymorphism in Drosophila simulans. Proc. Natl. Acad. Sci. USA 97: 5960–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirera, S., and M. Aguadé, 1997. Evolutionary history of the sex-peptide (Acp70A) gene region in Drosophila melanogaster. Genetics 147: 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A. G., S. Glanowski, R. Nielsen, P. D. Thomas, A. Kejariwal et al., 2003. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science 302: 1960–1963. [DOI] [PubMed] [Google Scholar]
- Derome, N., K. Metayer, C. Montchamp-Moreau and M. Veuille, 2004. Signature of selective sweep associated with the evolution of sex-ratio drive in Drosophila simulans. Genetics 166: 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky, T., 1965. Wild and domestic species of Drosophila, pp. 533–547 in The Genetics of Colonizing Species, edited by H. G. Baker and G. L. Stebbins. Academic Press, New York.
- DuMont, V. B., J. C. Fay, P. P. Calabrese and C. F. Aquadro, 2004. DNA variability and divergence at the Notch locus in Drosophila melanogaster and D. simulans: a case of accelerated synonymous site divergence. Genetics 167: 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvernell, D. D., and W. F. Eanes, 2000. Contrasting molecular population genetics of four hexokinases in Drosophila melanogaster, D. simulans and D. yakuba. Genetics 156: 1191–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emes, R. D., S. A. Beatson, C. P. Ponting and L. Goodstadt, 2004. Evolution and comparative genomics of odorant- and pheromone-associated genes in rodents. Genome Res. 14: 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J. C., and C. I. Wu, 1999. A human population bottleneck can account for the discordance between patterns of mitochondrial versus nuclear DNA variation. Mol. Biol. Evol. 16: 1003–1005. [DOI] [PubMed] [Google Scholar]
- Fay, J. C., and C. I. Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., and W. H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo, K., and D. P. Smith, 2001. A large family of divergent Drosophila odorant-binding proteins expressed in gustatory and olfactory sensilla. Genetics 159: 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier, N., F. Depaulis and N. H. Barton, 2000. Detecting bottlenecks and selective sweeps from DNA sequence polymorphism. Genetics 155: 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad, Y., C. D. Bustamante, D. Lancet and S. Paabo, 2003. Natural selection on the olfactory receptor gene family in humans and chimpanzees. Am. J. Hum. Genet. 73: 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelbrant, A. A., H. Skaletsky and A. Chess, 2004. Selective pressures on the olfactory receptor repertoire since the human-chimpanzee divergence. Proc. Natl. Acad. Sci. USA 101: 9019–9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravot, E., M. Huet and M. Veuille, 2004. Effect of breeding structure on population genetic parameters in Drosophila. Genetics 166: 779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin, M. T., and M. Veuille, 1999. Population structure among African and derived populations of Drosophila simulans: evidence for ancient subdivision and recent admixture. Genetics 153: 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe, D. S., R. L. Dorit and J. R. Carlson, 2000. Molecular evolution of odorant-binding protein genes OS-E and OS-F in Drosophila. Genetics 155: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe, D. S., C. R. Scafe, A. J. McKinney and M. A. Tanouye, 2002. Genome-wide analysis of the odorant-binding protein gene family in Drosophila melanogaster. Genome Res. 12: 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and A. Robertson, 1966. The effect of linkage on limits to artificial selection. Genet. Res. 8: 269–294. [PubMed] [Google Scholar]
- Hudson, R. R., 1987. Estimating the recombination parameter of a finite population model without selection. Genet. Res. 50: 245–250. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1990. Gene genealogies and the coalescent process, pp. 1–44 in Oxford Surveys in Evolutionary Biology, edited by J. Antonovics and D. Futuyma. Oxford University Press, Oxford.
- Hudson, R. R., 2000. A new statistic for detecting genetic differentiation. Genetics 155: 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111: 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguadé, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., K. Bailey, D. Skarecky, J. Kwiatowski and F. J. Ayala, 1994. Evidence for positive selection in the superoxide dismutase (sod) region of Drosophila melanogaster. Genetics 136: 1329–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvin, S. D., K. A. Wetterstrand, C. M. Hutter and C. F. Aquadro, 1998. Genetic variation and differentiation at microsatellite loci in Drosophila simulans: evidence for founder effects in New World populations. Genetics 150: 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes, T. H., and C. R. Cantor, 1969. Evolution of protein molecules, pp. 21–123 in Mammalian Protein Metabolism, edited by H. N. Munro. Academic Press, New York.
- Kern, A. D., C. D. Jones and D. J. Begun, 2002. Genomic effects of nucleotide substitutions in Drosophila simulans. Genetics 162: 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern, A. D., C. D. Jones and D. J. Begun, 2004. Molecular population genetics of male accessory gland proteins in the Drosophila simulans complex. Genetics 167: 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., 2006. Allele frequency distribution under recurrent selective sweeps. Genetics 172: 1967–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160: 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger, M. J., and K. G. Ross, 2002. Identification of a major gene regulating complex social behavior. Science 295: 328–332. [DOI] [PubMed] [Google Scholar]
- Krieger, M. J., and K. G. Ross, 2005. Molecular evolutionary analyses of the odorant-binding protein gene Gp-9 in fire ants and other Solenopsis species. Mol. Biol. Evol. 22: 2090–2103. [DOI] [PubMed] [Google Scholar]
- Kumar, S., K. Tamura and and M. Nei, 2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinformatics 5: 150–163. [DOI] [PubMed] [Google Scholar]
- Labate, J. A., C. H. Biermann and W. F. Eanes, 1999. Nucleotide variation at the runt locus in Drosophila melanogaster and Drosophila simulans. Mol. Biol. Evol. 16: 724–731. [DOI] [PubMed] [Google Scholar]
- Lachaise, D., M. L. Cariou, J. R. David, F. Lemeunier, L. Tsacas et al., 1988. Historical biogeography of the Drosophila melanogaster species subgroup. Evol. Biol. 22: 159–225. [Google Scholar]
- Lazzaro, B. P., 2005. Elevated polymorphism and divergence in the class C scavenger receptors of Drosophila melanogaster and D. simulans. Genetics 169: 2023–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., and W. Stephan, 2006. Inferring the demographic history and rate of adaptive substitution in Drosophila. PloS Genet. 2: e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison, W. P., and D. R. Maddison, 1992. MacClade: Analysis of Phylogeny and Character Evolution, Version 3.0. Sinauer Associates, Sunderland, MA.
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- Meiklejohn, C. D., Y. Kim, D. L. Hartl and J. Parsch, 2004. Identification of a locus under complex positive selection in Drosophila simulans by haplotype mapping and composite-likelihood estimation. Genetics 168: 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, R. A., M. Choudhary, M. L. Cariou and R. S. Singh, 2004. A reanalysis of protein polymorphism in Drosophila melanogaster, D. simulans, D. sechellia and D. mauritiana: effects of population size and selection. Genetica 120: 101–114. [DOI] [PubMed] [Google Scholar]
- Mousset, S., and N. Derome, 2004. Molecular polymorphism in Drosophila melanogaster and D. simulans: What have we learned from recent studies? Genetica 120: 79–86. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- Ngai, J., M. M. Dowling, L. Buck, R. Axel and A. Chess, 1993. The family of genes encoding odorant receptors in the channel catfish. Cell 72: 657–666. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., C. Bustamante, A. G. Clark, S. Glanowski, T. B. Sackton et al., 2005. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 3: e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo, D. J., and M. Aguade, 2004. Detecting the footprint of positive selection in a European population of Drosophila melanogaster: multilocus pattern of variation and distance to coding regions. Genetics 167: 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, C. A., R. A. Watts, R. G. Gregg, M. A. McCall, L. D. Houck et al., 2005. Lineage-specific differences in evolutionary mode in a salamander courtship pheromone. Mol. Biol. Evol. 22: 2243–2256. [DOI] [PubMed] [Google Scholar]
- Parsch, J., C. D. Meiklejohn and D. L. Hartl, 2001. Patterns of DNA sequence variation suggest the recent action of positive selection in the janus-ocnus region of Drosophila simulans. Genetics 159: 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelosi, P., and R. Maida, 1995. Odorant-binding proteins in insects. Comp. Biochem. Physiol. B, Biochem. Mol. Biol. 111: 503–514. [DOI] [PubMed] [Google Scholar]
- Quesada, H., U. E. M. Ramirez, J. Rozas and M. Aguadé, 2003. Large-scale adaptive hitchhiking upon high recombination in Drosophila simulans. Genetics 165: 895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J., M. Gullaud, G. Blandin and M. Aguadé, 2001. DNA variation at the rp49 gene region of Drosophila simulans: evolutionary inferences from an unusual haplotype structure. Genetics 158: 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sánchez-Delbarrio, X. Messeguer and R. Rozas, 2003. DnaSP: DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Saiki, R. K., D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi et al., 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239: 487–491. [DOI] [PubMed] [Google Scholar]
- Saitou, N., and M. Nei, 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4: 406–425. [DOI] [PubMed] [Google Scholar]
- Sánchez-Gracia, A., M. Aguadé and J. Rozas, 2003. Patterns of nucleotide polymorphism and divergence in the odorant-binding protein genes OS-E and OS-F: analysis in the melanogaster species subgroup of Drosophila. Genetics 165: 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke, T. A., and D. J. Begun, 2003. Natural selection drives Drosophila immune system evolution. Genetics 164: 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke, T. A., and D. J. Begun, 2004. Strong selective sweep associated with a transposon insertion in Drosophila simulans. Proc. Natl. Acad. Sci. USA 101: 1626–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke, T. A., and D. J. Begun, 2005. Linkage disequilibrium and recent selection at three immunity receptor loci in Drosophila simulans. Genetics 169: 2013–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, K. J., L. Nigro, C. H. Aquadro and D. Tautz, 1999. Large number of replacement polymorphisms in rapidly evolving genes of Drosophila: implications for genome-wide surveys of DNA polymorphism. Genetics 153: 1717–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofl, G., and C. Schlotterer, 2004. Patterns of microsatellite variability among X chromosomes and autosomes indicate a high frequency of beneficial mutations in non-African D. simulans. Mol. Biol. Evol. 21: 1384–1390. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré, S., D. J. Balding, R. C. Griffiths and P. Donnelly, 1997. Inferring coalescence times from DNA sequence data. Genetics 145: 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, K., and P. Andolfatto, 2006. Approximate Bayesian inference reveals evidence for a recent, severe bottleneck in a Netherlands population of Drosophila melanogaster. Genetics 172: 1607–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- True, J. R., J. M. Mercer and C. C. Laurie, 1996. Differences in crossover frequency and distribution among three sibling species of Drosophila. Genetics 142: 507–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veuille, M., E. Baudry, M. Cobb, N. Derome and E. Gravot, 2004. Historicity and the population genetics of Drosophila melanogaster and D. simulans. Genetica 120: 61–70. [DOI] [PubMed] [Google Scholar]
- Vogt, R. G., and L. M. Riddiford, 1981. Pheromone binding and inactivation by moth antennae. Nature 293: 161–163. [DOI] [PubMed] [Google Scholar]
- Vogt, R. G., G. D. Prestwich and M. R. Lerner, 1991. Odorant-binding-protein subfamilies associate with distinct classes of olfactory receptor neurons in insects. J. Neurobiol. 22: 74–84. [DOI] [PubMed] [Google Scholar]
- Vogt, R. G., F. E. Callahan, M. E. Rogers and J. C. Dickens, 1999. Odorant binding protein diversity and distribution among the insect orders, as indicated by LAP, an OBP-related protein of the true bug Lygus lineolaris (Hemiptera, Heteroptera). Chem. Senses 24: 481–495. [DOI] [PubMed] [Google Scholar]
- Vogt, R. G., M. E. Rogers, M. D. Franco and M. Sun, 2002. A comparative study of odorant binding protein genes: differential expression of the PBP1-GOBP2 gene cluster in Manduca sexta (Lepidoptera) and the organization of OBP genes in Drosophila melanogaster (Diptera). J. Exp. Biol. 205: 719–744. [DOI] [PubMed] [Google Scholar]
- Voight, B. F., A. M. Adams, L. A. Frisse, Y. Qian, R. R. Hudson et al., 2005. Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc. Natl. Acad. Sci. USA 102: 18508–18513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosshall, L. B., 2000. Olfaction in Drosophila. Curr. Opin. Neurobiol. 10: 498–503. [DOI] [PubMed] [Google Scholar]
- Wall, J. D., 1999. Recombination and the power of statistical test of neutrality. Genet. Res. 74: 65–79. [Google Scholar]
- Wall, J. D., P. Andolfatto and M. Przeworski, 2002. Testing models of selection and demography in Drosophila simulans. Genetics 162: 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Watts, R. A., C. A. Palmer, R. C. Feldhoff, P. W. Feldhoff, L. D. Houck et al., 2004. Stabilizing selection on behavior and morphology masks positive selection on the signal in a salamander pheromone signaling complex. Mol. Biol. Evol. 21: 1032–1041. [DOI] [PubMed] [Google Scholar]
- Willett, C. S., 2000. Evidence for directional selection acting on pheromone-binding proteins in the genus Choristoneura. Mol. Biol. Evol. 17: 553–562. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]
- Zurovcova, M., and W. F. Eanes, 1999. Lack of nucleotide polymorphism in the Y-linked sperm flagellar dynein gene Dhc-Yh3 of Drosophila melanogaster and D. simulans. Genetics 153: 1709–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]