

Abstract
Contrasting patterns of X-linked vs. autosomal diversity may be indicative of the mode of selection operating in natural populations. A number of observations have shown reduced X-linked (or Z-linked) diversity relative to autosomal diversity in various organisms, suggesting a large impact of genetic hitchhiking. However, the relative contribution of other forces such as population bottlenecks, variation in reproductive success of the two sexes, and differential introgression remains unclear. Here, we survey 13 loci, 6 X-linked and 7 autosomal, in natural populations of the house mouse (Mus musculus) subspecies complex. We studied seven populations of three different subspecies, the eastern house mouse M. musculus castaneus, the central house mouse M. m. musculus, and the western house mouse M. m. domesticus, including putatively ancestral and derived populations for each. All populations display lower diversity on the X chromosomes relative to autosomes, and this effect is most pronounced in derived populations. To assess the role of demography, we fit the demographic parameters that gave the highest likelihood of the data using coalescent simulations. We find that the reduction in X-linked diversity is too large to be explained by a simple demographic model in at least two of four derived populations. These observations are also not likely to be explained by differences in reproductive success between males and females. They are consistent with a greater impact of positive selection on the X chromosome, and this is supported by the observation of an elevated KA and elevated KA/KS ratios on the rodent X chromosome. A second contribution may be that the X chromosome less readily introgresses across subspecies boundaries.
IN an effort to understand the nature of positive selection, its frequency, and its relative impact on variability in natural populations, recent studies in the era of population genomics have focused on comparisons between ancestral and recently colonized populations. The expectation is that the colonization of novel environments will involve novel selective pressures. In the well-studied cases of Drosophila and humans, recent colonization events out of sub-Saharan Africa are thought to have occurred in the last 10,000 and 50,000–150,000 years before present, respectively (David and Capy 1988; Lachaise et al. 1988; Cavalli-Sforza et al. 1994; Excoffier 2002). Numerous studies provide evidence of positive selection in non-African populations of Drosophila and humans (Harr et al. 2002; Payseur et al. 2002; Glinka et al. 2003; Kauer et al. 2003; Orengo and Aguade 2004; Storz et al. 2004; Ometto et al. 2005; Stajich and Hahn 2005; Pool et al. 2006). However, disentangling the effects of selection from those of the demographic history of a species presents a considerable challenge (Wall et al. 2002; Haddrill et al. 2005; Ometto et al. 2005).
One way to identify signatures of selection in natural populations is to compare patterns of X-linked and autosomal diversity. As proposed by Aquadro et al. (1994), the hemizygous state of the X chromosome in males provides a means by which to distinguish between alternative models of selection, namely those of background selection (Charlesworth et al. 1993) and genetic hitchhiking (Maynard Smith and Haigh 1974). The background selection model attributes reductions in diversity due to selection against deleterious mutations, while the hitchhiking model predicts reductions in diversity due to selection favoring beneficial mutations. The predictions for X chromosomes and autosomes differ, however. Because recessive X-linked deleterious mutations are efficiently purged, background selection results in a relatively greater reduction of linked neutral variation on autosomes (Charlesworth et al. 1993; Charlesworth 1996). In contrast, genetic hitchhiking causes a relatively large reduction in X-linked neutral variation. This is because, first, the rate of adaptive mutations (provided they are, on average, recessive) should be higher on the X chromosome (Charlesworth et al. 1987) and, second, shorter sojourn times of beneficial mutations on the X chromosome mean that relatively large blocks of linked genes are affected (Avery 1984; Aquadro et al. 1994). An increased rate of adaptive substitution on the X chromosome compared to the autosomes also requires that adaptive mutations are not ancestrally deleterious and segregating at mutation–selection balance (Orr and Betancourt 2001). When these conditions are met, hitchhiking is expected to leave the X chromosome less polymorphic than autosomes. Intriguingly, many studies in Drosophila and humans find that ancestral African populations of both species display levels of X-linked diversity comparable to those of autosomal diversity, while non-African populations display relatively low levels of X-linked diversity (reviewed in Betancourt et al. (2004)).
Despite the availability of its genome sequence and status as a major genetic model organism, a relative paucity of data on DNA sequence polymorphism exists for natural populations of the house mouse (Mus musculus). The house mouse complex consists of at least three subspecies. The commonest of these is M. m. domesticus (found from the Near East to western Europe and North Africa), M. m. musculus (north of the Caucasus and the Himalayas from far eastern Eurasia to eastern Europe), and M. m. castaneus (Southeast Asia). Two models have been proposed to explain the evolution of these house mouse subspecies. The “centrifugal” model assumes that all subspecies diverged more or less simultaneously from a common ancestral population in northern Indo-Pakistan between 300,000 and 800,000 years ago (Boursot et al. 1996; Din et al. 1996). In the “linear” model (Prager et al. 1998), house mice originated in west-central Asia within the present-day range of domesticus, radiated south through the Arabian peninsula, then east and north into south-central Asia, and, from there, north into north-central Asia and Eurasia (giving rise to the subspecies musculus) and into Southeast Asia (giving rise to subspecies castaneus).
In this study, we find that the mouse X chromosome has substantially reduced variability relative to autosomes, especially in derived populations. Although one explanation for the reduced variability on the X chromosome is the occurrence of frequent selective sweeps, another explanation may be differential rates of admixture between the X and the autosome. Studies in the European hybrid zone of two M. musculus subspecies have shown restricted movement of the X chromosome across the subspecies boundary (Tucker et al. 1992; Dod et al. 1993), which may be caused by a disproportionately large number of loci contributing to reproductive isolation (i.e., hybrid sterility and inviability) on the X chromosome (Coyne and Orr 1998). Thus, we discuss the selectively restricted movement of the X chromosome as an alternative explanation for these observations.
MATERIALS AND METHODS
Animal material:
We surveyed multiple populations of each of the three subspecies (in total seven populations) of the house mouse M. musculus. All mice were directly sampled from the wild. Sampling strategies are given in Ihle et al. (2006). Mice from Taiwan were kindly provided by A. Yu, Taiwan University. These samples are described in detail in Huang and Yu (2003). Mice from Iran were kindly provided by F. Bonhomme. These samples are described in detail by H. Rajabi-Maham, A. Orth and F. Bonhomme (unpublished data). Eight individuals per population were chosen for sequencing. The geographic origin and subspecies status of each population are given in Table 1. A single M. famulus individual from India (kindly provided by F. Bonhomme) served as an outgroup. The taxonomic status of samples was confirmed by sequencing ∼930 bp of the mitochondrial d-loop for all individuals. Although house mice in the region of northwest (NW) India are the least well defined and have previously been classified as M. m. bactrianus or a new unnamed subspecies, our mtDNA sequences from this region cluster strongly (98% bootstrap support) with the M. m. castaneus individuals from Taiwan (data not shown). Thus, we consider the NW Indian individuals to belong to the subspecies M. m. castaneus.
TABLE 1.
Population samples of M. musculus subspecies and populations used in this study
| Sampling location | Country | Subspecies | Geographic coordinates |
|---|---|---|---|
| Katrain, Kunihar, and Dehradun | India | M. m. castaneus | lat 30°19′32′′ N–32°7′0′′ N lon 76°58′51′′ E–78°0′9′′ E |
| Hsinpu and Taichung | Taiwan | M. m. castaneus | Huang and Yu (2003) |
| Around Almaty | Kazakhstan | M. m. musculus | Ihle et al. (2006) |
| Around Prague | Czech Republic | M. m. musculus | lat 49°7′0′′ N–49°12′0′′ N lon 16°11′0′′ E–16°13′0′′ E |
| Around Ahvaz | Iran | M. m. domesticus | H. Rajabi-Maham, A. Orth and F. Bonhomme (unpublished data) |
| Massif Central | France | M. m. domesticus | Ihle et al. (2006) |
| Around Cologne/Bonn | Germany | M. m. domesticus | Ihle et al. (2006) |
For each subspecies, we define the more ancestral population as that closest to the geographic center of origin of the respective subspecies. These are NW India, Iran, and Kazakhstan for M. m. castaneus, M. m. domesticus, and M. m. musculus, respectively. The NW India and Iran subspecies are ancestral populations for the subspecies castaneus and domesticus under both the linear and the centrifugal models. However, the Kazakhstan population cannot be considered truly ancestral for the musculus subspecies under either model. Instead, the origin for the musculus subspecies is inferred to be in NW Afghanistan under the linear model (Prager et al. 1998) and somewhere in northern India under the centrifugal model (Boursot et al. 1996; Din et al. 1996). Kazakhstan is closer to ancestral musculus than the assumed derived population in the Czech Republic. Thus, in the absence of a truly ancestral musculus sample we use it in the comparison of a more ancestral with a derived population. For the subspecies domesticus we include two derived populations, one from France and one from Germany.
Loci:
We sequenced six loci located on the X chromosome and seven loci from chromosomes 4 and 8. The loci consisted entirely of noncoding DNA, either 5′ or 3′ flanking or intronic sequence (Table 2). The loci were randomly selected, but subjected to the constraint that the local recombination rates and gene density should be similar for the X and autosomal loci. We sequenced an average of 9.2 chromosomes per population for X-linked loci and 15.8 chromosomes for autosomal loci. The size of the fragments ranged from 351 to 1425 bp (mean 762 bp). The local recombination rate of each locus was estimated for a 10-Mb window centered on the locus by regressing the genetic position of markers against their physical position. Genetic positions were obtained from the Mouse Genome Informatics map (www.informatics.jax.org) and physical positions from NCBI build 36. Estimates of gene density were obtained by counting the number of genes in a 1-Mb window centered on each locus. The local recombination rate for the X-linked loci ranges from 0.22 to 1.24 cM/Mb (mean = 0.50 cM/Mb) and for the autosomal loci from 0.39 to 1.32 cM/Mb (mean = 0.68 cM/Mb). The difference is not significant (Mann–Whitney U-test; P = 0.475). Estimates of gene density between the six X-linked and seven autosomal loci do not significantly differ (average 13.7 vs. 11.3 genes/Mb, respectively, Mann–Whitney U-test; P = 0.351). Thus, the loci should experience roughly equal opportunity for selection at linked sites. In addition, we note that one autosomal locus, Ggh, which was clearly present as single copy in all NCBI builds prior to build 36, is now listed as being contained within a duplicated region. The evidence for this duplication and whether it extends to natural populations is unclear. However, we note that this locus represents only ∼7% of the total nucleotides sequenced on the autosomes and is always within the range of heterozygosity found at the other six autosomal loci. Thus, there is no indication of this locus being a significant outlier.
TABLE 2.
Characteristics of sequenced fragments
| Gene symbol | Chromosome | Location (relative to gene) | Recombination rate (cM/Mb) | Gene density (genes/Mb) | No. of sites | Position, NCBI build 36 (bp) |
|---|---|---|---|---|---|---|
| Sfrp1 | 8 | >5 kb upstream | 0.39 | 11 | 1144 | 24874380–24875524 |
| Fut10 | 8 | Intron 2–3 | 1.14 | 9 | 496 | 32662925–32663421 |
| Nkd1 | 8 | >5 kb upstream | 0.52 | 13 | 1177 | 91403263–91404440 |
| Nudt7 | 8 | >5 kb upstream | 0.77 | 6 | 1397 | 117013893–117015290 |
| Ggh | 4 | Intron 1–2 | 0.15 | 6 | 499 | 20166795–20167294 |
| Melk | 4 | Intron 6–7 | 1.32 | 16 | 917 | 44331964–44332881 |
| Pum1 | 4 | Intron 11–12 | 0.42 | 18 | 1183 | 130022004–130023187 |
| Gm719 | X | Intron 9 | 1.24 | 18 | 351 | 135356826–135357177 |
| Lamp2 | X | Intron 7 | 0.26 | 17 | 391 | 34669560–34669951 |
| Fate1 | X | Intron 2 | 0.43 | 15 | 573 | 68236312–68236885 |
| Arx | X | >10 kb upstream | 0.54 | 8 | 365 | 89525496–89525861 |
| Tex16 | X | >1 kb downstream | 0.22 | 7 | 431 | 108245208–108245639 |
| Hadh2 | X | >5 kb upstream | 0.31 | 17 | 1024 | 147335787–147336811 |
PCR and DNA sequencing:
For each locus a 30-μl PCR containing 100 ng of genomic DNA, 1.5 mm MgCl2, 200 μm dNTPs, 1 μm of each primer, and 0.5 unit Taq polymerase (Eppendorf Master Taq, Hamburg, Germany) was performed. A typical cycling profile consisted of 30 cycles of 50 sec at 94°, 50 sec at 60°, and 2 min at 72°. PCR products were purified using Millipore (Bedford, MA) Montagé plates according to the manufacturer's protocol. PCR products were sequenced in both directions using BigDye version 3.1 on an ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA). Individuals heterozygous for multiple insertions or deletions either were sequenced with additional primers or alleles were cloned (Topo TA cloning kit; Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. At least seven clones per locus and individual were sequenced for all cloned PCR products, which essentially eliminated all potential PCR artifacts. Sequencing reactions were purified using Sephadex columns. Sequences have been submitted to GenBank under accession nos. EF435057–EF436227. All PCR and sequencing primers are available from the authors upon request.
Data analysis:
Summary statistics:
DNA sequences were aligned using muscle (Edgar 2004). In the case of heterozygous base positions within the sequence of one individual (stemming from directly sequencing the autosomal loci) we randomly assigned one nucleotide state to each of the two alleles (i.e., produced “pseudohaplotypes”). We calculated three summary statistics of within-population diversity levels: (1) Watterson's θW (Watterson 1975), based on the number of segregating (polymorphic) sites in the sample; (2) π (Nei and Li 1979), the average number of pairwise differences in the sample; and (3) a modification of Tajima's D-statistic (Tajima 1989). Tajima's D-statistic contrasts low-frequency and intermediate-frequency sites in a sample, with negative values indicative of population expansion, strong purifying selection, or recovery after a selective sweep, whereas positive values may indicate the presence of population structure, balancing selection, or weak/incomplete bottlenecks (Ometto et al. 2005). Tajima's D may be influenced by the number of segregating sites (Schaeffer 2002) and the number of alleles sampled (A. Eyre-Walker and S. Schaeffer, personal communication). To make valid comparisons in the skew of the site frequency spectrum between groups of loci with systematic differences in both the amount of variation and the number of alleles sampled (i.e., the X-linked vs. autosomal loci), we employed the method of Schaeffer (2002), which divides Tajima's D by its minimum value, termed D/Dmin. Interpretations of D/Dmin remain the same as interpretations based on the uncorrected D. To compare X-linked loci where as few as 8 chromosomes were sampled vs. autosomal loci where typically 16 chromosomes were sampled, we randomly resampled eight pseudohaplotypes 100 times (A. Eyre-Walker and S. Schaeffer, personal communication), separately for each locus. For each resampled set we calculated D/Dmin and took the average across these resampled data sets. This procedure was performed for all loci/populations where >8 chromosomes were sampled using software kindly provided by A. Eyre-Walker. All other polymorphism indexes and their significance were calculated using scripts written in perl.
Evaluation of potential sampling biases:
Due to inbreeding within nests, we included only a single individual per nest (Ihle et al. 2006). Males were usually chosen to simplify sequencing at X-linked loci (although a few female individuals were used when samples were insufficient); thus typically only a single X chromosome is sampled per nest. In the case of autosomal loci, two chromosomes from a single nest are always included. To check for an effect of including two chromosomes per nest for autosomes, but usually only one per nest for X-linked loci, we randomly used only one pseudohaplotype from all individuals where two chromosomes per locus were sequenced. In this manner we produced data sets for X-linked and autosomal loci of the same sample size (i.e., eight chromosomes, which is equivalent to the lowest number of chromosomes sequenced for the X chromosome in any given population). Polymorphism analyses were repeated separately for each of these random haplotype data sets. We detected no significant differences in estimates of nucleotide diversity between those using the full sample and conclude that sampling two alleles per individual has minimal effect.
Phylogenetic trees based on pairwise population GST:
We calculated pairwise population average GST trees on the basis of the autosomal data to evaluate the possibility of gene flow at the autosomal loci. For each autosomal locus, we calculated the average (over all polymorphic sites within a fragment) GST-value (Nei 1973) and generated a distance matrix from these values. This distance matrix was then transformed into a phylogenetic tree using the program neighbor (http://bioweb.pasteur.fr/seqanal/interfaces/neighbor-simple.html) and graphically represented in Treeview (Page 1996).
Rates of protein evolution on the X chromosome and autosomes:
If selective sweeps are commoner on the X chromosome, i.e., the rate of adaptive mutations is higher, this may be reflected in an increased rate of amino acid substitutions. To test for this we downloaded the KA- and KS-values for mouse–rat orthologs from one randomly selected transcript per gene from ENSEMBL (www.ensembl.org). Only genes that were X-linked in both species or autosomal in both species were considered, which yielded a gene set of 559 X-linked and 16,240 autosomal orthologs.
Coalescent simulations:
Coalescent times under neutrality are very variable, and some previous studies in Drosophila have found that reduced X-linked relative to autosome variability is consistent with a simple demographic bottleneck during the origin of the population (Wall et al. 2002; Haddrill et al. 2005). We used coalescent simulations to test for this using the program ms (Hudson 2002). The demographic scenario is depicted in Figure 1. The basic idea behind the simulation is to test the effect of a potential bottleneck during the course of the origin of each of the seven populations, while still allowing for some amount of gene flow. The focal population is population 2 in Figure 1. During the course of the origin of this population we first assume a deep split between a pair of subspecies (one subspecies is represented by population 3, and the other by populations 1 and 2) and second a split of the focal population (population 2) from the rest of the subspecies (population 1). Note that this model is general enough to be compatible with both the centrifugal and the linear model of evolution of house mice. Under both models a subspecies split predates a potential population bottleneck. We fixed one parameter: migration rates between populations 1 and 2 were assumed to be three times higher than migration rates between populations 1 and 3. The following parameters were varied: Ne in the base population (N0 in Figure 1), the mutation rate, recombination rate, time points of the two splits, migration rates, and the strength of the bottleneck (the fraction, x, by which population size is reduced in the origin of population 2) (see Table 3). We included the cases where the migration rate was zero and there was no bottleneck (x = 0).
Figure 1.—

Framework of demographic models used in coalescent simulations. N0 denotes the effective population size of the ancestor leading to the Mus musculus subspecies complex. The focal population is population 2, which is assumed to have split from a more ancestral population 1 of the same subspecies at a certain time in the past (split time 1). Populations 1 and 2 share a most recent common ancestor with another subspecies, population 3, at a certain time in the past (split time 2). Arrows denote gene flow between subspecies and between subpopulations of the same subspecies. The factor “x” denotes a reduction in the size of population 2. For details see materials and methods and Tables 3 and 5.
TABLE 3.
Range of parameters used to estimate the most likely demographic scenario
| Parameter | Range | No. of intervals used |
|---|---|---|
| Reduction factor from ancestral population size | 1/1,000–1/1.25 | 16 |
Mutation rate ( /bp) /bp) |
2.5 × 10−10–1.75 × 10−9 | 7 |
| Split 1 (yra) | 1,000–200,000 | 5 |
| Split 2 (yra) | 500,000; 1,000,000 | 2 |
| Migration rate (m) | 0; 5 × 10−7; 2.5 × 10−7 | 3 |
| Recombination rate (r/bp) | 2.5 × 10−9; 2.5 × 10−10 | 2 |
| Effective population size (Ne) | 0.5 × 106; 1 × 106; 2 × 106 | 3 |
Assuming two generations per year.
Altogether we simulated 21,420 different parameter combinations with 5000 simulations each. For each parameter combination we simulated seven autosomal loci. For each locus the mutation rate was treated as a random variable drawn from a uniform distribution with mean, 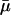 , equal to the parameter value for that simulation and variance =
, equal to the parameter value for that simulation and variance = 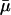 2/75. We calculated θW for each simulated locus using the program HKAanalyzer (Andolfatto 2005) and then determined the mean
2/75. We calculated θW for each simulated locus using the program HKAanalyzer (Andolfatto 2005) and then determined the mean  and variance
and variance 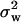 across the seven loci. Adopting procedures similar to those used by Enard et al. (2002) we recorded whether the output from each simulation fulfilled the conditions |
across the seven loci. Adopting procedures similar to those used by Enard et al. (2002) we recorded whether the output from each simulation fulfilled the conditions |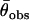 −
−  | <
| < 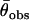 /5 and |
/5 and |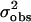 −
− 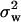 | <
| <  , where the subscript obs refers to the value for the autosomal loci obtained from the empirical data set. We estimated the relative likelihood of a particular parameter set as the proportion of all simulations for that parameter set that fulfilled these conditions.
, where the subscript obs refers to the value for the autosomal loci obtained from the empirical data set. We estimated the relative likelihood of a particular parameter set as the proportion of all simulations for that parameter set that fulfilled these conditions.
We used the parameter combination with the highest likelihood in each population against which to evaluate observed ratios of X-linked to autosomal polymorphism (X/A ratios). We simulated six X-linked loci and seven autosomal loci with these parameter values and recorded the corresponding X/A ratio for each of the 5000 runs (see below for details of correcting the values of the X chromosome). The significance (P-value) of an observed X/A ratio in a population is obtained as two times the fraction of simulations that are equal to or smaller than the observed value (the factor of two accounts for a two-tailed test). Since we tested seven different populations we performed a sequential Bonferroni correction (Rice 1989).
Choice of parameters:
To guide our choice of N0 (the effective population size in the ancestral population, see Figure 1) we employed a multilocus Bayesian Markov chain Monte Carlo method implemented in the MCMCcoal program (Yang 2002; Rannala and Yang 2003). We estimated the multilocus parameter 4Neμ from the autosomal data separately for the Iranian and the Indian populations. The Markov chain was run for 106 generations. A 95% confidence interval for 4Neμ was calculated by sampling from the output at the end of the run. Each analysis was run three times and with different random number seeds to check for convergence. Ne was estimated from these values by dividing by 4μ, where μ was assumed to be 1.67 × 10−9–2.98 × 10−9/bp/generation (Eyre-Walker et al. 2002). We took the average of the estimates of the three independent runs. We estimate Ne to be 786,400 for the Iranian population (95% C.I.: 594,500–1,019,200) and 2,100,300 (95% C.I.: 1,739,700–2,486,200) for the Indian population, assuming a mutation rate of 1.67 × 10−9. With a higher mutation rate of 2.98 × 10−9 these values are 441,300 (95% C.I.: 333,100–571,100) and 1,170,000 (95% C.I.: 974,900–1,393,300), respectively. A recent survey of nucleotide diversity in M. musculus, based on a pooled sample of laboratory inbred strains across different subspecies, yielded an estimate of 4Neμ of 0.0054 (Ideraabdullah et al. 2004), implying an Ne to be between 450,000 and 810,000. Thus, the combined evidence suggests that the population size of the house mouse lies between 500,000 and 2,000,000. In our simulations we used three different values for N0 (i.e., 0.5 million, 1 million, and 2 million).
Other parameters (mutation, migration, and recombination) were chosen on the basis of empirical observations (see Table 3).
Correcting parameter values on the X chromosome:
The X chromosome and autosomes differ in a number of characteristics, including their effective population size, mutation rate, and recombination rate (Jensen-Seaman et al. 2004). For the simulations, we applied the following corrections for the X chromosome. First, we corrected X/A ratios for differences in population size by assuming that the effective size of the X chromosome is 0.75 that of the autosomes (thus, in the simulations, we assume that males and females have the same variance in reproductive success). Second, in rodents the mutation rate in the male germline is higher than that in the female germline (Malcom et al. 2003; Sandstedt and Tucker 2005), resulting in a lower mutation rate on the X chromosome. For the loci included in our study, the divergence between the M. musculus and M. famulus X chromosome was 83% of that observed between the autosomes of these two species, and we used this as an estimate of the difference in mutation rate. Thus, in total, the X chromosomal 4Neμ values used in the simulations are multiplied by 0.62 (0.75 × 0.83).
Third, the single X chromosome in males does not recombine. Recombination on the autosomes is also lower in males than in females, by a factor γ = 0.8 (Dod et al. 1993 and references therein). Combining these observations, a freely recombining pair of genes on an autosome experiences 1.35 times more recombination than a freely recombining pair of genes on the X chromosome. To correct for this, we multiplied the recombination rate for the X chromosome by 0.74.
Male-to-female ratio of effective population size:
Although we assumed that the male and female effective population sizes were the same in the simulations, a formal possibility for generally reduced variability on the X chromosome is a smaller effective population size of females than males, because a higher fraction of X chromosomes reside in females each generation than of autosomes (Caballero 1994). To test for this, we compared polymorphism and divergence between autosomal loci and the mitochondrial d-loop region. All animals, including the outgroup M. famulus, were sequenced for the d-loop using primers F: 5′-CATTATTCTGGTCTTGTAAACC-3′ and R: 5′-GCCAGGACCAAACCTTTGTGT-3′ (Prager et al. 1993). PCR and sequencing conditions were as above. Under neutrality and assuming equal effective population sizes of males and females, the level of nucleotide polymorphism at the mtDNA locus should be one-quarter of the diversity at an autosomal locus. The mutation rate for mtDNA is higher than the autosomal mutation rate by a factor x (Miyata et al. 1982). We estimated x as KmtDNA/Kautosomes, where Kautosomes is the average divergence to M. famulus across the seven autosomal loci. Thus, the expected ratio of  . Larger values than
. Larger values than 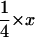 indicate a higher effective population size of females, while smaller values than
indicate a higher effective population size of females, while smaller values than 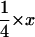 indicate a smaller effective population size of females than males.
indicate a smaller effective population size of females than males.
RESULTS
Polymorphism and divergence for six X-linked and seven autosomal loci are summarized in Table 4 and detailed in supplemental Tables 1 and 2 at http://www.genetics.org/supplemental/. Averaging over all loci in the putative ancestral populations (India, Iran, and Kazakhstan) reveals the subspecies M. m. castaneus as the most polymorphic (for both the X and the autosomes), followed by M. m. domesticus and finally M. m. musculus (Figure 2). At the autosomes, no significant correlation across loci between nucleotide diversity and recombination rate exists for any population surveyed here, but a positive trend is present in most populations, consistent with previous observations in natural populations of house mice (Nachman 1997). When pooled over all populations the positive trend is nearly significant (F1,41 = 3.02, P = 0.09). At the X chromosome we were unable to detect any such association (F1,34 = 0.088, P = 0.768); the low diversity on the X reduces the power of this test.
TABLE 4.
Summary of polymorphism and divergence
| Mean π (SE) | Mean θ (SE) | Mean K (JC) (SE) | Mean θ/Ka (SE) | Mean D (SE) | Mean D/Dmin (SE) | |
|---|---|---|---|---|---|---|
| X-Linked (n = 6) | ||||||
| India | 0.00265 (0.00074) | 0.00322 (0.00093) | 0.03134 (0.00535) | 0.16700 (0.05546) | −0.631 (0.320) | −0.427 (0.192) |
| Taiwan | 0.00038 (0.00020) | 0.00039 (0.00018) | 0.03111 (0.00535) | 0.01685 (0.00930) | −0.118 (0.642) | −0.172 (0.420) |
| Kazakhstan | 0.00095 (0.00028) | 0.00105 (0.00032) | 0.03358 (0.00682) | 0.04074 (0.01264) | −0.067 (0.530) | 0.000 (0.474) |
| Czech Republic | 0.00025 (0.00018) | 0.00023 (0.00016) | 0.03291 (0.00687) | 0.01081 (0.00755) | 0.334 (0.000) | 0.316 (0.000) |
| Iran | 0.00123 (0.00039) | 0.00159 (0.00065) | 0.03247 (0.00542) | 0.07019 (0.03221) | −0.439 (0.468) | -0.249 (0.250) |
| France | 0.00010 (0.00010) | 0.00015 (0.00015) | 0.03218 (0.00562) | 0.00331 (0.00331) | −1.055 (NA) | −1.000 (NA) |
| Germany | 0.00049 (0.00024) | 0.00053 (0.00020) | 0.03299 (0.00551) | 0.02606 (0.00989) | −0.487 (0.551) | −0.473 (0.527) |
| Autosomal (n = 7) | ||||||
| India | 0.00821 (0.00153) | 0.00794 (0.00121) | 0.03882 (0.00142) | 0.21111 (0.03801) | 0.035 (0.308) | 0.026 (0.087) |
| Taiwan | 0.00608 (0.00114) | 0.00503 (0.00102) | 0.03867 (0.00136) | 0.13402 (0.02846) | 0.997 (0.401) | 0.246 (0.111) |
| Kazakhstan | 0.00168 (0.00069) | 0.00164 (0.00055) | 0.03869 (0.00159) | 0.04481 (0.01596) | −0.287 (0.379) | −0.275 (0.134) |
| Czech Republic | 0.00142 (0.00064) | 0.00157 (0.00075) | 0.03868 (0.00158) | 0.04446 (0.02231) | −0.131 (0.678) | −0.101 (0.236) |
| Iran | 0.00325 (0.00068) | 0.00345 (0.00073) | 0.03784 (0.00175) | 0.09202 (0.01904) | −0.147 (0.303) | −0.028 (0.109) |
| France | 0.00260 (0.00115) | 0.00233 (0.00068) | 0.03798 (0.00242) | 0.06558 (0.01892) | −0.205 (0.609) | −0.199 (0.235) |
| Germany | 0.00126 (0.00044) | 0.00129 (0.00056) | 0.03770 (0.00181) | 0.03535 (0.01660) | 0.036 (0.467) | −0.052 (0.191) |
π, the average number of pairwise differences in a sample (Nei and Li 1979); θ, Watterson's estimator (Watterson 1975); K, divergence (Jukes–Cantor corrected); D, Tajima's D-statistic (Tajima 1989); D/Dmin, ratio of Tajima's D to its theoretical minimum (Schaeffer 2002).
X loci multiplied by four-thirds.
Figure 2.—

Mean (±SE) ratios of polymorphism (θW) to divergence (K) for X-linked and autosomal loci. X-linked values of θW are corrected by multiplying by four-thirds.
Autosomes vs. X chromosomes:
The autosomes show higher diversity (θW) than the X chromosome in all seven populations. However, to directly compare levels of polymorphism between the X and autosomes it is necessary to account for (1) differences in effective population size and (2) differences in mutation rates. Male-driven molecular evolution is a well-documented phenomenon in mammals (Li et al. 2002; Sandstedt and Tucker 2005), a fact that generally results in the X chromosome experiencing a reduced mutation rate relative to autosomes. In our study, we observe that the average divergence (K) of X-linked loci is only 83% that of autosomes. Thus, we compared the average ratio of polymorphism (multiplied by four-thirds for X loci) to divergence for X-linked vs. autosomal loci (Figure 2; Table 4). In all cases, these ratios are lower on the X chromosome relative to the autosomes (N = 7, Wilcoxon signed rank test, P = 0.018).
Ancestral vs. derived populations:
In each of the surveyed subspecies, the putative ancestral population has higher levels of both X-linked and autosomal diversity compared to the samples from the derived populations (Table 4 and Figure 2). This is especially striking for X-linked polymorphism, where average differences between the ancestral and the derived population range from 3- to 10-fold. The comparisons in the Taiwanese and French populations are individually significant when using locus as a replicate in a Mann–Whitney U-test (P = 0.006 and 0.008, respectively). Thus, while both autosomal and X chromosome diversity are reduced in the derived populations, the reduction on the X chromosome is more extreme.
Frequency spectra:
With the exception of the Czech Republic population, the average values of Tajima's D (Tajima 1989) and D/Dmin (Schaeffer 2002; see materials and methods) on the X chromosome are negative, implying an excess of rare variants on the X chromosome (Table 4 and supplemental Table 1 at http://www.genetics.org/supplemental/). Although many fragments on the X chromosome contain no segregating sites in the derived populations (and hence the values are based on only one or two loci) the more polymorphic India and Iran populations also display strong negative values. In contrast, the average values of the autosomal loci are very close to zero (India, Iran, Germany; Table 4 and supplemental Table 2 at http://www.genetics.org/supplemental/). Other populations have negative values (Kazakhstan, Czech Republic, and France), although the absolute values are smaller than those observed on the X chromosome. The Taiwanese population has consistent positive values across the autosomal loci.
Coalescent simulations:
To evaluate the impact of demography on observed X/A ratios, we performed coalescent simulations (Hudson 2002). The range and intervals of parameter values are given in Table 3. For each population we took the demographic parameters that best fit the autosomal data (Table 5) and asked whether the X chromosomal data would be compatible with such a model. The probability of the observed X/A ratio under the most likely demographic model for each population is given in Table 5. After sequential Bonferroni correction, the Taiwanese and the French populations have a significantly lower X/A ratio than expected. To assess the relative merit of the chosen models, we applied the Δ-Akaike information criterion (ΔAIC) method (Burnham and Anderson 2002; see discussion).
TABLE 5.
Most likely demographic scenarios
| Population | Ne | 4Nr | 4Nμ | 4Nm | Reduction | Reduction factor | Split time 1 (yra) | Split time 2 (yra) | No. of simulations fitting the data | Likelihood of model | Two-tailed P-values of ratios of X-linked to autosomal diversity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Kazakhstan | 2 × 106 | 20 | 6 | 0 | 0.001 | 1000 | 2,000 | 1,000,000 | 734 | 0.1468 | 0.6232 |
| Czech Republic | 2 × 106 | 20 | 10 | 0 | 0.006 | 167 | 20,000 | 500,000 | 555 | 0.111 | 0.5864 |
| Iran | 1 × 106 | 10 | 7 | 0 | 0.02 | 50 | 10,000 | 1,000,000 | 930 | 0.186 | 0.4864 |
| Germany | 2 × 106 | 20 | 6 | 0 | 0.004 | 250 | 10,000 | 1,000,000 | 638 | 0.1276 | 0.9364 |
| France | 2 × 106 | 20 | 4 | 2 | 0.02 | 50 | 20,000 | 500,000 | 759 | 0.1518 | <0.0001* |
| India | 2 × 106 | 20 | 7 | 2 | 0.1 | 10 | 2,000 | 1,000,000 | 1193 | 0.2386 | 0.0028* |
| Taiwan | 2 × 106 | 20 | 6 | 2 | 0.008 | 125 | 2,000 | 500,000 | 1009 | 0.2018 | <0.0001* |
These remain significant after sequential Bonferroni correction.
Assuming two generations per year.
Past and ongoing gene flow:
Comparatively high variation on the autosomes could also reflect recent gene flow, if gene flow at the X chromosome is restricted. Evidence consistent with gene flow would be indicated if (1) populations of different subspecies cluster with each other or (2) the presumed more ancestral population within each subspecies occupies a more derived position in a phylogenetic tree. We constructed neighbor-joining trees on the basis of the GST-statistic for each autosomal locus separately (Figure 3). Two loci (Sfrp1 and Pum) are consistent with the inferred population history. For other loci, however, we found some evidence that populations cluster in unexpected ways. The Taiwanese population is located very close to the root at locus Nkd and groups with the subspecies musculus at locus Nudt7, instead of with the Indian population. Similarly, the domesticus French population groups with musculus populations at locus Ggh (even though this grouping is supported only by one polymorphic site) and is located ancestrally with respect to the Iranian and German populations at loci Fut10 and Melk. Thus, evidence from the Taiwanese and the French populations, and possibly the Indian population, is consistent with some amount of gene flow. However, while consistent with gene flow, these differences could also reflect ancestral polymorphism (see discussion).
Figure 3.—

Phylogenetic grouping of house mouse populations for seven autosomal loci on the basis of GST. Populations in boldface type indicate misgrouping along the phylogenetic tree.
Male and female effective population size:
Levels of nucleotide polymorphism at the mtDNA d-loop region and divergence to M. famulus are given in Table 6. The observed θmtDNA/θautosome ratios are compared to those expected under equal male and female effective sizes in Table 7. Although a majority of populations display ratios consistent with a higher female effective population size (i.e., ratios greater than expected) and the deviations in this direction are greater in magnitude, this trend is not significant (N = 7, Wilcoxon signed rank test, P = 0.176).
TABLE 6.
Nucleotide polymorphism at the mtDNA d-loop region in populations of house mice
| Population | Sample size (n) | No. of sites | Segregating sites | π | θ | Tajima's D | Divergence (K) |
|---|---|---|---|---|---|---|---|
| Kazakhstan | 8 | 882 | 3 | 0.0014 | 0.0013 | 0.20 | 0.138 |
| Czech Republic | 8 | 882 | 3 | 0.0015 | 0.0013 | 0.46 | 0.137 |
| India | 8 | 880 | 14 | 0.0066 | 0.0061 | 0.40 | 0.132 |
| Taiwan | 7 | 883 | 12 | 0.0063 | 0.0055 | 0.70 | 0.137 |
| Iran | 8 | 883 | 20 | 0.0071 | 0.0087 | −0.99 | 0.140 |
| Germany | 8 | 882 | 19 | 0.0093 | 0.0083 | 0.63 | 0.140 |
| France | 8 | 881 | 8 | 0.0026 | 0.0035 | −1.25 | 0.142 |
TABLE 7.
Observed and expected (under equal population size of males and females) ratios of mitochondrial to autosomal diversity
| Population | Expected | Observed | Deviation |
|---|---|---|---|
| Kazakhstan | 0.875 | 0.80 | −0.075 |
| Czech Republic | 0.875 | 0.83 | −0.045 |
| India | 0.875 | 0.77 | −0.105 |
| Taiwan | 0.875 | 1.10 | +0.225 |
| Iran | 0.875 | 2.53 | +1.655 |
| Germany | 0.875 | 6.42 | +5.545 |
| France | 0.875 | 1.50 | +0.625 |
Rates of protein evolution:
In comparisons of the mouse with the rat, the average KA/KS-value is significantly higher for X-linked orthologs compared to autosomal ones [P = 0.0047, d.f. = 16,797, t-test, KA/KS (autosome) = 0.12, KA/KS (X) = 0.26], suggesting a higher rate of adaptive substitutions on the X chromosome. Because the rate of synonymous substitutions is likely biased toward lower values on the X chromosome, inflating KA/KS (i.e., smaller effective population size of the X, male-driven evolution, and weak selection on synonymous sites; see Lu and Wu 2005), we considered only replacement sites. We found that KA alone is also significantly higher on the X chromosome [P = 0.011, t-test, KA (autosome) = 0.040, KA (X) = 0.050].
DISCUSSION
Our study represents the first to describe the level and pattern of nucleotide polymorphism in natural populations of the M. musculus subspecies on a broad scale. The overall observations meet our general predictions, namely that the presumed ancestral populations are more variable. Furthermore, although almost all populations display some degree of reduced X-linked relative to autosomal variability (even after correcting for differences in population size and mutation rate), this pattern is generally more pronounced in derived populations and in two cases dramatically so. These observations mirror the pattern seen in numerous, unrelated organisms [i.e., human (Payseur and Nachman 2002), Drosophila (Begun and Whitley 2000; Kauer et al. 2002), flycatcher (Borge et al. 2005), and chicken (Sundstrom et al. 2004)]. Recurrent genetic hitchhiking is expected to influence X-linked vs. autosomal diversity under a wider range of conditions in mammals, where recombination occurs in both sexes (Betancourt et al. 2004). However, alternative hypotheses such as population demography (Wall et al. 2002), differences in the variance of male and female reproductive success (Caballero 1994; Charlesworth 2001), or differential introgression at X chromosomes and autosomes may also explain or contribute to this pattern, and we discuss each in turn.
To assess the impact of demography on the observed X/A ratios, we performed coalescent simulations to obtain a more appropriate neutral model. Naturally, we have considered only a small subset of possible demographic scenarios, and the actual colonization history of the populations is likely much more complex than assumed here. Within the constraint of the parameters we selected, we used the most likely demographic scenario for each population against which to evaluate the reduction in X chromosome diversity. We showed that X-linked diversity in two derived populations (Taiwan and France) and one ancestral population (India) is too low to be explained by demography alone. Although we have not considered a wide range of possible parameters, of the 21,420 parameter combinations we did evaluate for each population, between 980 and 2400 (5–13%, dependent on the population) were not significantly different from the maximum-likelihood combination [using the criterion that the ΔAIC < 2; (Burnham and Anderson 2002)]. This indicates that many other parameter combinations including varying degrees of migration, mutation, Ne, and time since populations split give similar levels of support. However, cursory examination of the more extreme combinations among these indicates that alternative models would still be rejected as an explanation for the low X/A ratio for the same populations.
The social organization of house mouse populations (Bronson 1979) suggests a higher female effective population size than male effective population size, which should result in relatively high X-linked diversity, i.e., in the opposite direction to that observed. By comparing autosomal and mtDNA variability we found that a majority of populations display ratios of mitochondrial to autosomal diversity consistent with a higher female effective population size, whereas the opposite would be expected if differences in effective population size between the sexes were to explain the observed X/A ratios. In addition, populations with the most significant reductions in X-linked relative to autosomal variability are among those consistent with a higher effective size of females. A recent large-scale compilation of nuclear and mitochondrial diversity across animal species suggests that the influence of adaptive evolution on the nonrecombining mitochondrial genome is pervasive (Bazin et al. 2006), which, in our case, would make this interpretation more conservative, as the female Ne would appear smaller than the actual number. Thus, taken together, a difference in reproductive success between the sexes seems an unlikely alternative explanation.
Our observation of reduced X-linked variation in derived populations of house mice is consistent with theoretical predictions about the relative impact of positive selection operating on the X chromosome and autosomes. This may be due to (1) a higher rate of adaptive evolution on the X chromosome due to the average recessiveness of beneficial mutations, or (2) decreased sojourn times on the X chromosome, or both. In particular, a recent theoretical study predicts a greater role for the latter in mammals, where, unlike in Drosophila, recombination occurs in both sexes (Betancourt et al. 2004).
If the notion of a stronger effect of genetic hitchhiking on the X is correct, one would expect to observe a higher rate of protein evolution on the X (Charlesworth et al. 1987). Ancestral and derived populations of Drosophila display a similar pattern of X-linked vs. autosomal diversity as we observe in mice, but a recent study found no evidence of an increased rate of protein evolution on the X chromosome in Drosophila (Thornton et al. 2006). This result has been interpreted as evidence for alternative explanations such as demographic effects (Wall et al. 2002) or other confounding factors such as chromosomal inversions and different effective population sizes in males and females (Andolfatto 2001; Charlesworth 2001).
In contrast, human–chimp comparisons provide evidence of an elevated rate of adaptive substitution on the mammalian X chromosome, both on the level of whole-genome comparisons (Nielsen et al. 2005) and on the level of comparisons of X-linked vs. autosomal sperm-expressed genes (Torgerson and Singh 2006). Our finding of an elevated rate of amino acid evolution in rodents as well is thus consistent with reduced variability on the X chromosome and a significant role of positive selection.
One alternative explanation is that the autosomes more readily flow across subspecies barriers than do X chromosomes. The house mouse subspecies are only partially reproductively isolated from each other and hybridize whenever geographically possible (Karn et al. 2002). However, in the most thoroughly studied hybrid zone of central Europe (between the subspecies domesticus and musculus), it was shown that different regions of the genome introgress at different rates, with the X chromosome being severely restricted in its movement across the hybrid zone, while some autosomal genes may move far across the subspecies border (Tucker et al. 1992; Dod et al. 1993). The restricted movement of the X chromosome across the subspecies boundary along the European hybrid zone may be caused by a disproportionately large number of loci contributing to reproductive isolation (i.e., hybrid sterility and inviability) on the X chromosome (Coyne and Orr 1998).
At several autosomal loci we found evidence that could be consistent with gene flow in the Taiwanese and French and possibly the Indian populations, although the pattern could equally well reflect ancestral polymorphism or a difference in the frequency of derived polymorphic sites among populations within a subspecies. In contrast, these populations show no evidence for gene flow at X-linked loci (data not shown). The Taiwanese and French populations are those for which we find the strongest deviation of the X/A ratio under the most likely demographic model. Whether differential gene flow between X chromosomes and autosomes is the cause of the pattern is not clear. The Indian population is located close to an area of very complex history of house mouse populations (Karn et al. 2002), but neither the Taiwanese nor French population is located close to any known hybrid zones. In Japan, admixture between musculus and castaneus has resulted in a population that is sometimes referred to as M. m. molossinus. Thus, it is possible that the musculus subspecies has also contributed to the Taiwanese autosomal gene pool, and the consistent positive Tajima's D and D/Dmin-values in this population would be consistent with such a scenario. Additional samples collected over a wider geographic range would be necessary to clarify this. A definitive statement about the relative roles of selective sweeps vs. gene flow and/or ancestral polymorphism in shaping the pattern of X-linked and autosomal variability in house mice awaits further study including larger numbers of loci and wider sampling areas, but simple demographic models can clearly be rejected.
Acknowledgments
We thank B. Schmitz for expert technical help, F. Bonhomme and A. Orth for providing unpublished sequence data from the mitochondrial d-loop region in the Iranian population, and A. Eyre-Walker for providing software to calculate and resample D/Dmin. F. Bonhomme and A. Yu kindly provided DNA samples from Iranian and Taiwanese mice, respectively. J. Pialek, S. Ihle, and S. Kipp kindly provided samples from European mice. A. Betancourt, S. Hutter, L. Ometto, J. Parsch, T. Price, D. Tautz, and two anonymous reviewers gave helpful comments on the manuscript. This study was supported by an Emmy-Noether fellowship from the Deutsche Forschungsgemeinschaft to B.H. and by an International Graduate School in Genetics and Functional Genomics—Cologne postdoctoral fellowship to J.F.B.
References
- Andolfatto, P., 2001. Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol. Biol. Evol. 18: 279–290. [DOI] [PubMed] [Google Scholar]
- Andolfatto, P., 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437: 1149–1152. [DOI] [PubMed] [Google Scholar]
- Aquadro, C. F., D. J. Begun and E. C. Kindahl, 1994. Selection, recombination and DNA polymorphism in Drosophila, pp. 45–56 in Non-Neutral Evolution: Theories and Molecular Data, edited by B. Golding. Chapman & Hall, New York.
- Avery, P. J., 1984. The population genetics of haplo-diploids and X-linked genes. Genet. Res. 44: 321–341. [Google Scholar]
- Bazin, E., S. Glemin and N. Galtier, 2006. Population size does not influence mitochondrial genetic diversity in animals. Science 312: 570–572. [DOI] [PubMed] [Google Scholar]
- Begun, D. J., and P. Whitley, 2000. Reduced X-linked nucleotide polymorphism in Drosophila simulans. Proc. Natl. Acad. Sci. USA 97: 5960–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt, A. J., Y. Kim and H. A. Orr, 2004. A pseudohitchhiking model of X vs. autosomal diversity. Genetics 168: 2261–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borge, T., M. T. Webster, G. Andersson and G. P. Saetre, 2005. Contrasting patterns of polymorphism and divergence on the Z chromosome and autosomes in two Ficedula flycatcher species. Genetics 171: 1861–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursot, P., W. Din, R. Anand, D. Darviche, B. Dod et al., 1996. Origin and radiation of the house mouse: mitochondrial DNA phylogeny. J. Evol. Biol. 9: 391–415. [Google Scholar]
- Bronson, F. H., 1979. The reproductive ecology of the house mouse. Q. Rev. Biol. 54: 265–299. [DOI] [PubMed] [Google Scholar]
- Burnham, K. P., and D. R. Anderson, 2002. Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach. Springer-Verlag, New York.
- Caballero, A., 1994. Developments in the prediction of effective population-size. Heredity 73: 657–679. [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza, L. L., P. Menozzi and A. Piazza, 1994. The History and Geography of Human Genes. Princeton University Press, Princeton, NJ.
- Charlesworth, B., 1996. Background selection and patterns of genetic diversity in Drosophila melanogaster. Genet. Res. 68: 131–149. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., 2001. The effect of life-history and mode of inheritance on neutral genetic variability. Genet. Res. 77: 153–166. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., J. A. Coyne and N. Barton, 1987. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 130: 113–146. [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne, J. A., and H. A. Orr, 1998. The evolutionary genetics of speciation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 353: 287–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, J. R., and P. Capy, 1988. Genetic variation of Drosophila melanogaster natural populations. Trends Genet. 4: 106–111. [DOI] [PubMed] [Google Scholar]
- Din, W., R. Anand, P. Boursot, D. Darviche, B. Dod et al., 1996. Origin and radiation of the house mouse: clues from nuclear genes. J. Evol. Biol. 9: 519–539. [Google Scholar]
- Dod, B., L. S. Jermiin, P. Boursot, V. H. Chapman, J. T. Nielsen et al., 1993. Counterselection on sex-chromosomes in the Mus-musculus European hybrid zone. J. Evol. Biol. 6: 529–546. [Google Scholar]
- Edgar, R. C., 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard, W., M. Przeworski, S. E. Fisher, C. S. Lai, V. Wiebe et al., 2002. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418: 869–872. [DOI] [PubMed] [Google Scholar]
- Excoffier, L., 2002. Human demographic history: refining the recent African origin model. Curr. Opin. Genet. Dev. 12: 675–682. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., P. D. Keightley, N. G. Smith and D. Gaffney, 2002. Quantifying the slightly deleterious mutation model of molecular evolution. Mol. Biol. Evol. 19: 2142–2149. [DOI] [PubMed] [Google Scholar]
- Glinka, S., L. Ometto, S. Mousset, W. Stephan and D. De Lorenzo, 2003. Demography and natural selection have shaped genetic variation in Drosophila melanogaster: a multi-locus approach. Genetics 165: 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill, P. R., K. R. Thornton, B. Charlesworth and P. Andolfatto, 2005. Multilocus patterns of nucleotide variability and the demographic and selection history of Drosophila melanogaster populations. Genome Res. 15: 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., M. Kauer and C. Schlotterer, 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 99: 12949–12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S. W., and H. T. Yu, 2003. Genetic variation of microsatellite loci in the major histocompatibility complex (MHC) region in the southeast Asian house mouse (Mus musculus castaneus). Genetica 119: 201–218. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Ideraabdullah, F. Y., E. de la Casa-Esperon, T. A. Bell, D. A. Detwiler, T. Magnuson et al., 2004. Genetic and haplotype diversity among wild-derived mouse inbred strains. Genome Res. 14: 1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle, S., I. Ravaoarimanana, M. Thomas and D. Tautz, 2006. An analysis of signatures of selective sweeps in natural populations of the house mouse. Mol. Biol. Evol. 23: 790–797. [DOI] [PubMed] [Google Scholar]
- Jensen-Seaman, M. I., T. S. Furey, B. A. Payseur, Y. Lu, K. M. Roskin et al., 2004. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 14: 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn, R. C., A. Orth, F. Bonhomme and P. Boursot, 2002. The complex history of a gene proposed to participate in a sexual isolation mechanism in house mice. Mol. Biol. Evol. 19: 462–471. [DOI] [PubMed] [Google Scholar]
- Kauer, M., B. Zangerl, D. Dieringer and C. Schlotterer, 2002. Chromosomal patterns of microsatellite variability contrast sharply in African and non-African populations of Drosophila melanogaster. Genetics 160: 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer, M. O., D. Dieringer and C. Schlotterer, 2003. A microsatellite variability screen for positive selection associated with the “out of Africa” habitat expansion of Drosophila melanogaster. Genetics 165: 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaise, D., M.-L. Cariou, J. R. David, F. Lemeunier, L. Tsacas et al., 1988. Historical biogeography of the Drosophila melanogaster species subgroup. Evol. Biol. 22: 159–225. [Google Scholar]
- Li, W. H., S. Yi and K. Makova, 2002. Male-driven evolution. Curr. Opin. Genet. Dev. 12: 650–656. [DOI] [PubMed] [Google Scholar]
- Lu, J., and C. I. Wu, 2005. Weak selection revealed by the whole-genome comparison of the X chromosome and autosomes of human and chimpanzee. Proc. Natl. Acad. Sci. USA 102: 4063–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcom, C. M., G. J. Wyckoff and B. T. Lahn, 2003. Genic mutation rates in mammals: local similarity, chromosomal heterogeneity, and X-versus-autosome disparity. Mol. Biol. Evol. 20: 1633–1641. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favorable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- Miyata, T., H. Hayashida, R. Kikuno, M. Hasegawa, M. Kobayashi et al., 1982. Molecular clock of silent substitution: at least six-fold preponderance of silent changes in mitochondrial genes over those in nuclear genes. J. Mol. Evol. 19: 28–35. [DOI] [PubMed] [Google Scholar]
- Nachman, M. W., 1997. Patterns of DNA variability at X-linked loci in Mus domesticus. Genetics 147: 1303–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., 1973. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70: 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., and W.-H. Li, 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76: 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., C. Bustamante, A. G. Clark, S. Glanowski, T. B. Sackton et al., 2005. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 3: e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ometto, L., S. Glinka, D. De Lorenzo and W. Stephan, 2005. Inferring the effects of demography and selection on Drosophila melanogaster populations from a chromosome-wide scan of DNA variation. Mol. Biol. Evol. 22: 2119–2130. [DOI] [PubMed] [Google Scholar]
- Orengo, D. J., and M. Aguade, 2004. Detecting the footprint of positive selection in a European population of Drosophila melanogaster: multilocus pattern of variation and distance to coding regions. Genetics 167: 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., and A. J. Betancourt, 2001. Haldane's sieve and adaptation from the standing genetic variation. Genetics 157: 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, R. D., 1996. TREEVIEW: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12: 357–358. [DOI] [PubMed] [Google Scholar]
- Payseur, B. A., and M. W. Nachman, 2002. Natural selection at linked sites in humans. Gene 300: 31–42. [DOI] [PubMed] [Google Scholar]
- Payseur, B. A., A. D. Cutter and M. W. Nachman, 2002. Searching for evidence of positive selection in the human genome using patterns of microsatellite variability. Mol. Biol. Evol. 19: 1143–1153. [DOI] [PubMed] [Google Scholar]
- Pool, J. E., V. Bauer DuMont, J. L. Mueller and C. F. Aquadro, 2006. A scan of molecular variation leads to the narrow localization of a selective sweep affecting both Afrotropical and cosmopolitan populations of Drosophila melanogaster. Genetics 172: 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager, E. M., R. D. Sage, U. Gyllensten, W. K. Thomas, R. Hubner et al., 1993. Mitochondrial-DNA sequence diversity and the colonization of Scandinavia by house mice from East Holsten. Biol. J. Linn. Soc. 50: 85–122. [Google Scholar]
- Prager, E. M., C. Orrego and R. D. Sage, 1998. Genetic variation and phylogeography of central Asian and other house mice, including a major new mitochondrial lineage in Yemen. Genetics 150: 835–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannala, B., and Z. Yang, 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164: 1645–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, W. R., 1989. Analyzing tables of statistical tests. Evol. Int. J. Org. Evol. 43: 223–225. [DOI] [PubMed] [Google Scholar]
- Sandstedt, S. A., and P. K. Tucker, 2005. Male-driven evolution in closely related species of the mouse genus Mus. J. Mol. Evol. 61: 138–144. [DOI] [PubMed] [Google Scholar]
- Schaeffer, S. W., 2002. Molecular population genetics of sequence length diversity in the Adh region of Drosophila pseudoobscura. Genet. Res. 80: 163–175. [DOI] [PubMed] [Google Scholar]
- Stajich, J. E., and M. W. Hahn, 2005. Disentangling the effects of demography and selection in human history. Mol. Biol. Evol. 22: 63–73. [DOI] [PubMed] [Google Scholar]
- Storz, J. F., B. A. Payseur and M. W. Nachman, 2004. Genome scans of DNA variability in humans reveal evidence for selective sweeps outside of Africa. Mol. Biol. Evol. 21: 1800–1811. [DOI] [PubMed] [Google Scholar]
- Sundstrom, H., M. T. Webster and H. Ellegren, 2004. Reduced variation on the chicken Z chromosome. Genetics 167: 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, K., D. Bachtrog and P. Andolfatto, 2006. X chromosomes and autosomes evolve at similar rates in Drosophila: no evidence for faster-X protein evolution. Genome Res. 16: 498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgerson, D. G., and R. S. Singh, 2006. Enhanced adaptive evolution of sperm-expressed genes on the mammalian X chromosome. Heredity 96: 39–44. [DOI] [PubMed] [Google Scholar]
- Tucker, P. K., R. D. Sage, J. Warner, A. C. Wilson and E. M. Eicher, 1992. Abrupt cline for sex-chromosomes in a hybrid zone between 2 species of mice. Evolution 46: 1146–1163. [DOI] [PubMed] [Google Scholar]
- Wall, J. D., P. Andolfatto and M. Przeworski, 2002. Testing models of selection and demography in Drosophila simulans. Genetics 162: 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 2002. Likelihood and Bayes estimation of ancestral population sizes in hominoids using data from multiple loci. Genetics 162: 1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]