

Abstract
We present a Bayesian framework for inferring spatial preferences of recombination from multiple putative recombinant nucleotide sequences. Phylogenetic recombination detection has been an active area of research for the last 15 years. However, only recently attempts to summarize information from several instances of recombination have been made. We propose a hierarchical model that allows for simultaneous inference of recombination breakpoint locations and spatial variation in recombination frequency. The dual multiple change-point model for phylogenetic recombination detection resides at the lowest level of our hierarchy under the umbrella of a common prior on breakpoint locations. The hierarchical prior allows for information about spatial preferences of recombination to be shared among individual data sets. To overcome the sparseness of breakpoint data, dictated by the modest number of available recombinant sequences, we a priori impose a biologically relevant correlation structure on recombination location log odds via a Gaussian Markov random field hyperprior. To examine the capabilities of our model to recover spatial variation in recombination frequency, we simulate recombination from a predefined distribution of breakpoint locations. We then proceed with the analysis of 42 human immunodeficiency virus (HIV) intersubtype gag recombinants and identify a putative recombination hotspot.
RECOMBINATION is a well-studied phenomenon that occurs in the genomes of many organisms through the exchange or transfer of genomic fragments demarcated by recombination breakpoints. Although recombination is ubiquitous, the rate of recombination varies across species and spatially along genomes within species. In the presence of spatial variation in recombination frequencies, recombination breakpoints are not distributed uniformly, tending to cluster in hotspots, leaving other cold regions intact (Smith 2001; Kauppi et al. 2004; Myers et al. 2005). Here, we consider the problem of identifying recombination hotspots along the human immunodeficiency virus (HIV) genome.
Rapid HIV mutation rates and infrequent recombination between genetically distinct viral genomes allow for recombination detection from evolutionary histories (phylogenies) of a recombinant and its putative parental sequences (Awadalla 2003). Such phylogenetic-based recombination detection (Hein 1990; Salminen et al. 1995; Grassly and Holmes 1997; McGuire et al. 1997; Suchard et al. 2002; Husmeier 2005) relies on the observation that genomic sequences experiencing infrequent recombination can be decomposed into breakpoint delimited blocks with distinct evolutionary histories (Li et al. 1988). We illustrate the idea behind all phylogenetic recombination detection methods with a simple example. Figure 1 shows a short multiple sequence alignment divided by recombination into two parts, such that a different phylogeny summarizes the sequence relationships in each part. The presence of alignment sites informative for phylogenetic reconstruction (shown in boldface type) is necessary for successful phylogenetic recombination detection.
Figure 1.—

Illustration of phylogenetic recombination detection. A multiple sequence alignment is divided into two parts by a recombination breakpoint (dashed line). These two parts support distinct phylogenies, shown on either side of the alignment. Sites that provide information about the topology of a phylogenetic tree are shown in boldface type.
Phylogenetic recombination detection is quite different from coalescent-based methods for analyzing recombination (Stumpf and McVean 2003). The latter approaches are most successful in studying frequently occurring recombination among closely related sequences randomly sampled from a neutrally evolving population (Fearnhead et al. 2004; McVean et al. 2004). However, as sequence diversity increases, selection, demographic history, and population structure are more likely to play a role in sequence evolution, making the application of coalescent-based approaches to HIV recombination problematic (McVean et al. 2002). This is especially true for recombination between different HIV subtypes as their evolutionary history reflects the subtype geographical distribution and their adaptation to different host populations (Robertson et al. 1995; Vidal et al. 2000; Rambaut et al. 2001; Choisy et al. 2004; Kalish et al. 2004). In such complicated evolutionary scenarios, phylogenetic recombination detection offers an attractive alternative as it allows for recombination inference without explicitly modeling the details of the process.
Given the myriad phylogenetic methods for inferring recombination events in individual HIV sequences, mapping recombination hotspots appears to be a straightforward task. However, recent attempts of phylogenetic mapping of recombination hotspots in the HIV genome (Magiorkinis et al. 2003; Zhang et al. 2005) run into major difficulties. First, phylogenetically informative sites are sparsely distributed, making estimation of recombination locations somewhat imprecise. Ignoring uncertainty about the number of recombination events and their locations within each recombinant leads to loss of power due to inefficient use of sequence data. Finally, the modest number of recombination events relative to the number of sites in individual alignments results in a sparse breakpoint distribution that prohibits direct estimation of site-specific recombination frequencies.
To address these issues, we propose a Bayesian hierarchical model that allows integration over breakpoint locations and stochastically interpolates site-specific recombination probabilities with the help of a smoothing prior shared by all recombinants. To specify the distribution of breakpoint locations, conditional on sequence data, we begin with a dual multiple change-point (DMCP) model (Minin et al. 2005). The DMCP model operates on a multiple sequence alignment of a putative recombinant and its “parental” strains and models recombination as a change-point process. We achieve information sharing among recombinants by assuming that homologous sites of all alignments have the same prior probability of being a recombination breakpoint. Estimation of such site-specific recombination probabilities is the key to identifying recombination hot-/coldspots. To handle the sparse breakpoint information, we recruit Gaussian Markov random fields (GMRFs), a popular class of distributions used to model temporal or spatial dependence (Besag 1974; Besag et al. 1991; Rue and Held 2005). Normally distributed vector 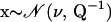 is called a GMRF with respect to a graph 𝒢 with nodes 𝒱 and edges ℰ, provided that Qij ≠ 0 if and only if
is called a GMRF with respect to a graph 𝒢 with nodes 𝒱 and edges ℰ, provided that Qij ≠ 0 if and only if 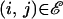 or i = j. To impose a biologically relevant correlation structure on site-specific recombination log odds (transformed probabilities), we use a GMRF prior on a linear graph 𝒢 connecting adjacent sites in a multiple sequence alignment. Such spatial smoothing allows sites where recombination is not observed to borrow information from adjacent sites where recombination is observed.
or i = j. To impose a biologically relevant correlation structure on site-specific recombination log odds (transformed probabilities), we use a GMRF prior on a linear graph 𝒢 connecting adjacent sites in a multiple sequence alignment. Such spatial smoothing allows sites where recombination is not observed to borrow information from adjacent sites where recombination is observed.
We approximate the posterior distribution of all model parameters via Markov chain Monte Carlo (MCMC) simulation. Since the number of change points in individual DMCP models is random, we use reversible-jump MCMC sampling to move between spaces with different dimensions (Green 1995). On the population level, we explore a high-dimensional (of the order 103–104) space of recombination log odds via a block updating scheme using Metropolis–Hastings transition kernels with multivariate Gaussian proposals as implemented in the freely distributed GMRFLib library (Rue 2001; Rue et al. 2004). In contrast to typical spatial applications of GMRFs (Elliott et al. 2000), we apply smoothing to probabilities of recombination breakpoints that themselves are random rather than directly observed as data. To our knowledge, this is the first use of GMRF priors in a random environment. We demonstrate the need for a nonlinear constraint on the GMRF to control the total number of breakpoints and provide a computationally efficient implementation of such a constraint.
We test our model through a simulation study, where recombination events are generated by permuting sequences in an alignment of primate mitochondrial DNA genes. The ability of the model to reconstruct several “true” recombination probability profiles is examined under different simulation conditions. Next, we apply our hierarchical model to 42 publicly available putative recombinants between HIV subtypes A and G that span the gag coding region of the viral genome. We find strong evidence for an ∼300-nucleotide recombination hotspot in the Capsid gene. In the discussion, we summarize our findings and propose further extensions to the smoothing prior on recombination locations.
METHODS
Synchronizing recombinant and parental sequences:
We begin with a master alignment of K putative recombinants and P candidate parental sequences. Represented by a matrix Y = {Yns}, 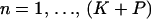 ,
, 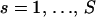 , the alignment is composed of nucleotide base names (A, adenine; G, guanine; T, thymine; C, cytosine) and gap characters (-). To eliminate unnecessary information in Y, we consider only columns where at least one of the recombinants possesses a nucleotide base. For each
, the alignment is composed of nucleotide base names (A, adenine; G, guanine; T, thymine; C, cytosine) and gap characters (-). To eliminate unnecessary information in Y, we consider only columns where at least one of the recombinants possesses a nucleotide base. For each 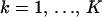 , we create individual, recombinant-specific alignments Y(k) by preserving the rows of Y that correspond to recombinant k and its N(k) − 1 candidate parental sequences (possibly different for each recombinant) and removing the other rows. Sites where the recombinant sequence has a gap are not informative for recombination detection via the DMCP model and are removed from the individual alignments Y(k). Such gap removal establishes an identity between the lengths of putative recombinants and the number of sites in the individual alignments,
, we create individual, recombinant-specific alignments Y(k) by preserving the rows of Y that correspond to recombinant k and its N(k) − 1 candidate parental sequences (possibly different for each recombinant) and removing the other rows. Sites where the recombinant sequence has a gap are not informative for recombination detection via the DMCP model and are removed from the individual alignments Y(k). Such gap removal establishes an identity between the lengths of putative recombinants and the number of sites in the individual alignments, 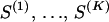 , and simplifies information sharing among individual data sets. We map individual alignments onto the master alignment with functions
, and simplifies information sharing among individual data sets. We map individual alignments onto the master alignment with functions
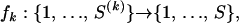 |
(1) |
where fk(i) identifies the site in the master alignment Y that contains the ith nucleotide of recombinant k. Since fk is a “one-to-one” mapping, fk(i) ≠ fk(j) for any i ≠ j, the inverse fk−1 is defined on the range of fk that represents the set of sites in the master alignment where the kth recombinant has no deletions.
Dual multiple change-point model:
We assume that conditional on model parameters Φ(k), each alignment Y(k) is drawn independently from a DMCP model, i.e.,
 |
(2) |
We first describe the model for evolution of individual alignment sites and then define the model structure across sites.
Columns 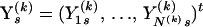 of each individual alignment Y(k) are assumed to evolve independently as a continuous-time Markov chain on the state space {A, G, C, T} (Felsenstein 2004). For each site
of each individual alignment Y(k) are assumed to evolve independently as a continuous-time Markov chain on the state space {A, G, C, T} (Felsenstein 2004). For each site 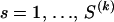 , we parameterize the infinitesimal rate matrix
, we parameterize the infinitesimal rate matrix 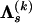 of the Markovian substitution process in terms of its stationary distribution
of the Markovian substitution process in terms of its stationary distribution 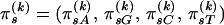 and a transition/transversion rate ratio
and a transition/transversion rate ratio 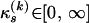 following Hasegawa et al. (1985). To reduce the number of nuisance parameters in the model, we fix all
following Hasegawa et al. (1985). To reduce the number of nuisance parameters in the model, we fix all 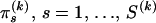 to the overall observed nucleotide frequencies in Y(k) (Li et al. 2000). This leaves us with one free parameter
to the overall observed nucleotide frequencies in Y(k) (Li et al. 2000). This leaves us with one free parameter 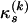 defining the substitution matrix
defining the substitution matrix 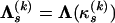 . To complete the phylogenetic model specification, we need a bifurcating tree topology
. To complete the phylogenetic model specification, we need a bifurcating tree topology 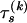 describing the historical relationships among nucleotides, with branch lengths
describing the historical relationships among nucleotides, with branch lengths  representing the expected number of substitutions between the bifurcation events. We further reduce the number of free parameters in the model by integrating
representing the expected number of substitutions between the bifurcation events. We further reduce the number of free parameters in the model by integrating 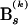 out of the likelihood through assuming an exponential prior on each branch length
out of the likelihood through assuming an exponential prior on each branch length 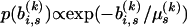 for all
for all  . Therefore, the likelihood of site s in recombinant k is a function of three phylogenetic parameters
. Therefore, the likelihood of site s in recombinant k is a function of three phylogenetic parameters 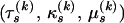 .
.
To model variation of the phylogenetic parameters along the columns of Y(k), we assume that the parameters are piecewise constant in s with jumps occurring at unknown change points. We first introduce a set of topology breakpoints 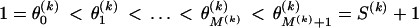 , where M(k) is the unknown number of recombination breakpoints for recombinant k, and
, where M(k) is the unknown number of recombination breakpoints for recombinant k, and 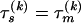 , for all
, for all 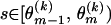 . Since topologies can attain only a finite set of values we require that
. Since topologies can attain only a finite set of values we require that 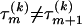 , for all
, for all 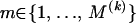 . Similarly we introduce a set of change points
. Similarly we introduce a set of change points 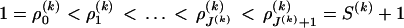 for substitution process parameters and assume that
for substitution process parameters and assume that 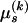 and
and  are constant between change points. In summary, our DMCP model for each recombinant k is defined by a set of parameters Φ(k) = (τ(k), θ(k), μ(k), κ(k), ρ(k)), where
are constant between change points. In summary, our DMCP model for each recombinant k is defined by a set of parameters Φ(k) = (τ(k), θ(k), μ(k), κ(k), ρ(k)), where 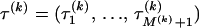 ,
, 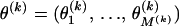 ,
, 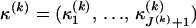 ,
,  , and
, and 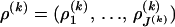 , and the varying dimensionality of the parameter space is determined by M(k) and J(k).
, and the varying dimensionality of the parameter space is determined by M(k) and J(k).
Priors for nuisance parameters:
Since our interest in this article is the recombination breakpoints θ(k), we collect all other parameters for each recombinant into a vector Ψ(k) = (τ(k), μ(k), κ(k), ρ(k)) and refer to them as nuisance parameters. We define a prior distribution for nuisance parameters by assuming substantial prior independence, specifically  . We assume a noninformative prior for
. We assume a noninformative prior for 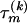 over E(k) possible tree topologies, relating recombinant k with its potential “parents.” The space of topologies permissible under the DMCP model is formed as described in Minin et al. (2005). Constraints on adjacent topologies are incorporated using a simple Markovian structure
over E(k) possible tree topologies, relating recombinant k with its potential “parents.” The space of topologies permissible under the DMCP model is formed as described in Minin et al. (2005). Constraints on adjacent topologies are incorporated using a simple Markovian structure
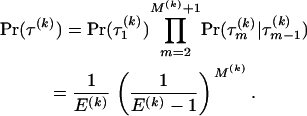 |
(3) |
The prior distribution for ρ(k) is specified by first assuming that J(k) follows a truncated Poisson distribution with a predefined, constant intensity λ and then giving equal prior probabilities to all possible draws of J(k) integers from the set 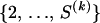 ,
,
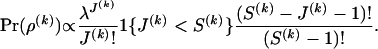 |
(4) |
We use one value of λ for all individual alignments as putative recombinant sequences are derived from the same genomic region and therefore should have an approximately equal number of changes in evolutionary pressure. Substitution parameters are a priori log-normally distributed, 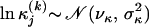 ,
, 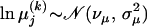 , where νκ, σκ, νμ, and σμ are either estimated in a hierarchical framework or fixed according to our prior knowledge about sequence variability in the genomic region under study. For more details on specifying the prior distribution for nuisance parameters Ψ(k) see Minin et al. (2005).
, where νκ, σκ, νμ, and σμ are either estimated in a hierarchical framework or fixed according to our prior knowledge about sequence variability in the genomic region under study. For more details on specifying the prior distribution for nuisance parameters Ψ(k) see Minin et al. (2005).
Spatially smoothed prior for recombination locations:
To specify prior probabilities for recombination breakpoint locations, we first switch from their point-process representation to site-specific recombination indicators 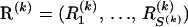 , where
, where 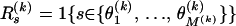 ,
, 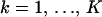 ,
, 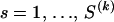 , and 1{·} is the indicator function. For clarity of presentation we ignore the fact that the first site of an alignment cannot be a topology breakpoint according to our definition. Such reparameterization allows us to introduce recombination probabilities
, and 1{·} is the indicator function. For clarity of presentation we ignore the fact that the first site of an alignment cannot be a topology breakpoint according to our definition. Such reparameterization allows us to introduce recombination probabilities 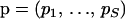 on the master alignment and then map them onto individual recombinants using functions (1) to define a prior distribution for breakpoint locations,
on the master alignment and then map them onto individual recombinants using functions (1) to define a prior distribution for breakpoint locations,
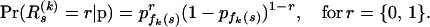 |
(5) |
In other words, we determine the prior probability of a site being a recombination breakpoint by finding its position in the master alignment and retrieving the corresponding component from the vector of common recombination probabilities p. Conditional on recombination probabilities p, we assume that breakpoint locations are independent within and between recombinants, so
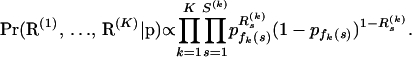 |
(6) |
If we denote the number of recombinants that do not have gaps at site s of the master alignment by 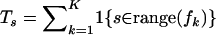 and define the total number of recombination breakpoints at site s,
and define the total number of recombination breakpoints at site s,  , for
, for 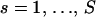 , then Equation 6 simplifies to
, then Equation 6 simplifies to
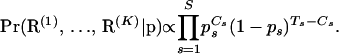 |
(7) |
Because in practice the total number of observed breakpoints is smaller than the number of sites S by one to two orders of magnitude, estimation of the common recombination probabilities p is unrealistic without further assumptions about their prior distribution. Since HIV recombination is mediated by the enzyme reverse transcriptase that processes nucleotides sequentially (Negroni and Buc 2001), we argue that recombination probabilities should have similar values at adjacent locations. To model such spatial dependency among components of p, we first obtain recombination log odds  , where
, where
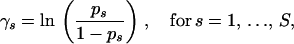 |
(8) |
and then use a GMRF prior that penalizes large differences between recombination log odds at neighboring sites,
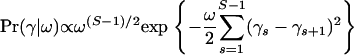 |
(9) |
It is easy to see that distribution (9) is improper if we reexpress 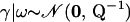 , where the precision matrix
, where the precision matrix
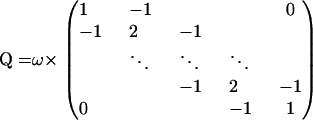 |
(10) |
satisfies the identity Q1 = 0. In the context of small area estimation, Ghosh et al. (1998) and Sun et al. (1999) show that despite the singularity of matrix Q such autoregressive priors lead to a proper posterior distribution under mild conditions on the model likelihood function. Our “pseudolikelihood” (7) does not satisfy these conditions when Cs = 0 for all s, or when Cs = Ts for all s. Although very unlikely, such values of recombination counts do have strictly positive probability mass a posteriori. Therefore, we cannot guarantee propriety of the posterior distribution of all model parameters and must replace density (9) by a proper approximation, assuming a priori that 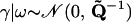 , where
, where 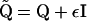 , I is the S × S identity matrix, and ε is a small positive constant. Note that the addition of a positive constant to the diagonal elements of Q preserves the precision matrix sparseness, but forces
, I is the S × S identity matrix, and ε is a small positive constant. Note that the addition of a positive constant to the diagonal elements of Q preserves the precision matrix sparseness, but forces 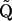 to be diagonally dominant and therefore positive definite. The proper approximation introduces an additional term,
to be diagonally dominant and therefore positive definite. The proper approximation introduces an additional term, 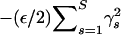 , to the exponent of density (9). In all examples, we use ε = 10−6 such that this term ≈0.05, assuming
, to the exponent of density (9). In all examples, we use ε = 10−6 such that this term ≈0.05, assuming 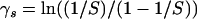 for all s.
for all s.
In addition to providing spatial preferences for breakpoint locations, the vector of recombination probabilities p defines the prior distribution for the total number of breakpoints 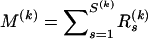 for each alignment k. It is important to put more prior mass on small values of M(k) to avoid inferring spurious breakpoints from noisy sequence data. The original DMCP model assumes that M(k) is truncated-Poisson distributed with a rate chosen in such a way that Pr(M(k) > 0) is equal to a predefined constant, usually 0.5. Similarly, in our hierarchical formulation, we want to control the overall probability of at least one recombination breakpoint in all individual alignments by imposing certain constraints on p. We first note that our site-specific prior on R(k) imposes a Poisson-binomial distribution for M(k) with small probabilities of success, usually on the order
for each alignment k. It is important to put more prior mass on small values of M(k) to avoid inferring spurious breakpoints from noisy sequence data. The original DMCP model assumes that M(k) is truncated-Poisson distributed with a rate chosen in such a way that Pr(M(k) > 0) is equal to a predefined constant, usually 0.5. Similarly, in our hierarchical formulation, we want to control the overall probability of at least one recombination breakpoint in all individual alignments by imposing certain constraints on p. We first note that our site-specific prior on R(k) imposes a Poisson-binomial distribution for M(k) with small probabilities of success, usually on the order 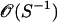 . Therefore, le Cam's theorem implies that the distribution of M(k) is approximately Poisson with rate
. Therefore, le Cam's theorem implies that the distribution of M(k) is approximately Poisson with rate 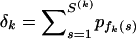 (le Cam 1960). For some constant c, we can set δk = −ln(1 − c), so that
(le Cam 1960). For some constant c, we can set δk = −ln(1 − c), so that 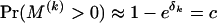 . Because restricting recombination probabilities for each recombinant individually is impractical, we impose our constraint on the population-level recombination probabilities,
. Because restricting recombination probabilities for each recombinant individually is impractical, we impose our constraint on the population-level recombination probabilities, 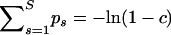 . Since
. Since 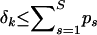 , this population-level restriction implies a more conservative prior distribution for the number of breakpoints in each individual data set k with Pr(M(k) > 0) ≤ c.
, this population-level restriction implies a more conservative prior distribution for the number of breakpoints in each individual data set k with Pr(M(k) > 0) ≤ c.
We complete our model specification by assuming a priori that ω ∼ Γ(α, β). Following Bernardinelli et al. (1995) we express our prior belief about ω in terms of a ratio of recombination probabilities 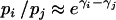 . On the basis of in vitro HIV recombination detection experiments (Moumen et al. 2001; Dykes et al. 2004; Galetto et al. 2004) we expect that site recombination probabilities should not vary more than sevenfold or equivalently that recombination log odds should not differ by >2. Since our smoothing prior implies that
. On the basis of in vitro HIV recombination detection experiments (Moumen et al. 2001; Dykes et al. 2004; Galetto et al. 2004) we expect that site recombination probabilities should not vary more than sevenfold or equivalently that recombination log odds should not differ by >2. Since our smoothing prior implies that 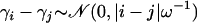 , setting ω = S − 1 ensures that even the most physically distant log odds do not deviate from each other by >2 with probability 0.95. Therefore, we fix the prior mean α/β = S − 1 and choose β to be a small constant (0.01 in the simulation study and 0.02 in the analysis of HIV recombinants).
, setting ω = S − 1 ensures that even the most physically distant log odds do not deviate from each other by >2 with probability 0.95. Therefore, we fix the prior mean α/β = S − 1 and choose β to be a small constant (0.01 in the simulation study and 0.02 in the analysis of HIV recombinants).
Inference via MCMC simulation:
To approximate the analytically intractable posterior distribution of all model parameters
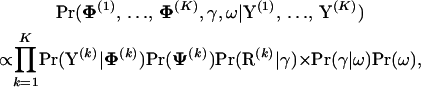 |
(11) |
we sample from (11) using MCMC simulation. During MCMC iterations, we use a Metropolis-within-Gibbs scheme to update the model parameters in two major blocks.
In the first block, we simulate from the full conditional distribution of all individual alignment parameters. The hierarchical structure of our model immediately implies the conditional independence of Φ(k)s,
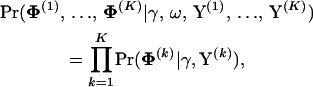 |
(12) |
making it possible to cycle through recombinants for each k and simulate from
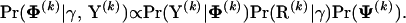 |
(13) |
Minin et al. (2005) describe a reversible-jump MCMC sampler to simulate from the posterior distribution of the DMCP model parameters under a uniform prior on recombination locations. Here, we use a similar algorithm to sample from the distributions in (13) with appropriate modifications of acceptance ratios to incorporate the shared prior over recombination locations. We refer interested readers to Suchard et al. (2003) and Minin et al. (2005) for a more detailed description of the DMCP sampling scheme.
The second block of parameters consists of the recombination log-odds vector γ and the GMRF precision ω. Conditioning on the parameters of the individual alignments yields
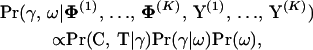 |
(14) |
where recombination counts 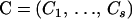 and trials
and trials 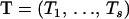 are as defined in Equation 7, and
are as defined in Equation 7, and
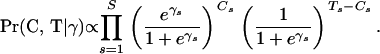 |
(15) |
Note that the sum of recombination probabilities constraint translates into the nonlinear algebraic identity
 |
(16) |
We first describe a sampling procedure on the unconstrained space of recombination log odds and then show how to approximate (16) with a linear constraint that can be incorporated into the sampling algorithm with very little computational burden.
To sample from distribution (14), we rely on the strategy introduced by Rue (2001) and Knorr-Held and Rue (2002) and update (ω, γ) simultaneously. Following their scheme, we first propose a new value for the precision parameter ω* = ωu, where ω is the current precision and u is a random variable with density Pr(u) ∝ 1 + 1/u, defined on the interval [1/U, U], U > 1. This proposal is symmetric and can be tuned by the constant U that controls the “length” of proposal jumps. Given a new value of the precision, we then generate a proposal for the vector of log-odds γ* from a multivariate Gaussian distribution that approximates Pr(γ | C, T, ω*) near its mode, where
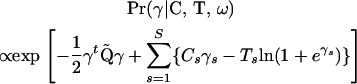 |
(17) |
The Newton–Raphson algorithm is used to locate this mode γ′. Log concavity of density (17) guarantees at most one mode. Then, a second-order Taylor approximation of ln Pr(γ | C, T, ω*) around γ′ generates the proposal mean and precision matrix and concludes the Gaussian proposal construction. The proposed values (ω*, γ*) are accepted or rejected jointly with probability given by the Metropolis–Hastings acceptance ratio. The computational efficiency of this multivariate proposal follows from the special shape of density (17). Note that, during construction of the Gaussian approximation, it is sufficient to apply the Taylor approximation only to the function 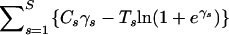 . Since all mixed derivatives of this function are zero, the off-diagonal elements of
. Since all mixed derivatives of this function are zero, the off-diagonal elements of  are equal to the off-diagonal entries of the Gaussian proposal precision matrix. Therefore, the multivariate normal proposals retain the same sparseness of Q and can be efficiently realized using fast methods of Cholesky decomposition for sparse matrices. For more details on approximating densities of the form similar to (17), see Rue (2001) and Rue et al. (2004).
are equal to the off-diagonal entries of the Gaussian proposal precision matrix. Therefore, the multivariate normal proposals retain the same sparseness of Q and can be efficiently realized using fast methods of Cholesky decomposition for sparse matrices. For more details on approximating densities of the form similar to (17), see Rue (2001) and Rue et al. (2004).
Implementing prior constraints:
We now turn to the problem of incorporating the imposed restrictions on recombination probabilities into our MCMC algorithm. Implementing a proposal that approximates (17) well while satisfying nonlinear constraint (16) is difficult. However, if we can replace constraint (16) with a linearized form  for some vector
for some vector 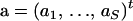 and scalar e, then we can use unconstrained Gaussian proposals as before to generate a candidate state
and scalar e, then we can use unconstrained Gaussian proposals as before to generate a candidate state 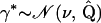 and recenter the proposal to satisfy the linear constraint via
and recenter the proposal to satisfy the linear constraint via
 |
(18) |
where ν and 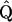 are obtained via a Taylor expansion of ln Pr(γ | C, T, ω*). Such recentering comes at minimal computational cost as the Cholesky factorization of
are obtained via a Taylor expansion of ln Pr(γ | C, T, ω*). Such recentering comes at minimal computational cost as the Cholesky factorization of 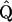 , needed in the unconditional proposal, can be reused to perform the algebraic operations in Equation 18 (Rue and Held 2005).
, needed in the unconditional proposal, can be reused to perform the algebraic operations in Equation 18 (Rue and Held 2005).
To arrive at specific values of a and e, we linearize the function  around an arbitrary point
around an arbitrary point 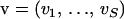 . Plugging this linearization into Equation 16 yields
. Plugging this linearization into Equation 16 yields
 |
(19) |
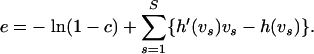 |
(20) |
Choosing v is less straightforward since we particularly need the linear approximation of (16) to be accurate near a posteriori probable values of γ. To make an intelligent guess about posterior support of recombination log odds, we generate a short “training chain” prior to running our MCMC sampler. During these training iterations, we alternate between sampling from the full conditional distributions defined by (12) and (14) with one heuristic modification that allows us to control the overall recombination probability implicitly. To understand the motivation behind this heuristic, we first point out that 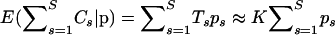 , where the latter approximation holds when the number of gaps in the master alignment is small. Therefore, if a new state of γ(i) is accepted at the ith iteration, then a new value of
, where the latter approximation holds when the number of gaps in the master alignment is small. Therefore, if a new state of γ(i) is accepted at the ith iteration, then a new value of  should be close to
should be close to  . To heuristically control the overall recombination probability via the binomial pseudolikelihood (15), we update the vector of trials such that
. To heuristically control the overall recombination probability via the binomial pseudolikelihood (15), we update the vector of trials such that 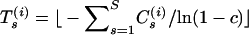 for all s at iteration i, where ⌊x⌋ denotes the largest integer that does not exceed x. After training runs are complete we set the approximation point v to an arithmetic average of simulated components of γ.
for all s at iteration i, where ⌊x⌋ denotes the largest integer that does not exceed x. After training runs are complete we set the approximation point v to an arithmetic average of simulated components of γ.
In all following analyses we set the prior probability of at least one recombination c = 0.5, and therefore we aim at preserving the condition 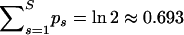 . Table 1 shows posterior medians and 95% Bayesian credible intervals (BCIs) of the overall recombination probability,
. Table 1 shows posterior medians and 95% Bayesian credible intervals (BCIs) of the overall recombination probability, 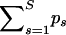 , for all analyzed data sets. Although in each case the posterior distribution of the overall recombination is slightly shifted to the right from 0.693, this shift and the spread of the distribution are quite small. Therefore, we conclude that our linear approximation performs well.
, for all analyzed data sets. Although in each case the posterior distribution of the overall recombination is slightly shifted to the right from 0.693, this shift and the spread of the distribution are quite small. Therefore, we conclude that our linear approximation performs well.
TABLE 1.
Constraining the overall probability of recombination
| Data set | Posterior median | 95% BCI |
|---|---|---|
| Simulation, A = 0.9 | 0.76 | (0.72, 0.88) |
| Simulation, A = 0.7 | 0.76 | (0.72, 0.89) |
| Simulation, A = 0.5 | 0.72 | (0.70, 0.80) |
| Simulation, A = 0.3 | 0.72 | (0.70, 0.78) |
| HIV gag | 0.73 | (0.71, 0.80) |
We report posterior medians and 95% BCIs of the sum of recombination probabilities 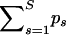 for all analyzed data sets.
for all analyzed data sets.
RESULTS
Simulation study:
To test our model in the presence of a “known” recombination hotspot, we design a small simulation study. We start with an 888-site long alignment of four primate DNA sequences from humans (H), orangutans (O), squirrel monkeys (S), and lemurs (L), previously used to assess the accuracy of the DMCP model (Minin et al. 2005). This data set strongly supports phylogeny (H, O, (S, L)) as demonstrated by several research groups (Yang and Rannala 1997; Larget and Simon 1999; Suchard et al. 2001). We set the true (under simulation conditions) recombination probabilities for 887 sites (excluding the first site) of this alignment in such a way that sites in the interval [401, 600] are more likely to be breakpoint locations. The sum of recombination probabilities in the hotspot interval is denoted by A. All other sites are assigned a probability of recombination (1 − A)/687, such that recombination probabilities sum to one and define a probability mass function for a discrete random variable attaining values from 2 to 888. We then generate 30 realizations, 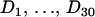 , of this random variable. For each Di,
, of this random variable. For each Di,  , we create a new sequence alignment by permuting the nucleotides of H and L in sites Di through 888. In these newly formed alignments, sites from 1 to Di − 1 should support the phylogeny (H, O, (S, L)), while the other portion of the alignment should favor the phylogeny (L, O, (S, H)), obtained by exchanging H and L in the original tree.
, we create a new sequence alignment by permuting the nucleotides of H and L in sites Di through 888. In these newly formed alignments, sites from 1 to Di − 1 should support the phylogeny (H, O, (S, L)), while the other portion of the alignment should favor the phylogeny (L, O, (S, H)), obtained by exchanging H and L in the original tree.
We generate only 30 recombinants since this quantity represents well the number of recombinant sequences typically available for analysis. When sample size is small relative to the number of sites covered by putative recombinants, the strength of a hotspot plays a critical role in our ability to recover the region. Therefore, we examine performance of our model for different hotspot probability mass values A = 0.9, 0.7, 0.5, and 0.3. The top left plot in Figure 2 shows the artificially generated probabilities used to simulate recombination events. The remaining plots in the left column of Figure 2 depict posterior medians (solid lines) and 95% BCIs (shaded areas) of recombination probabilities estimated under different true values of hotspot strength A. Solid dots mark true recombination sites where sequences H and L begin their permutation. We see that our model successfully identifies hotspots in the presence of a strong signal. On the other hand, when A = 0.3, simulated breakpoint locations are hardly distinguishable from a random sample from all 888 sites, and our method aptly detects no hotspots. This conservative behavior of our estimation procedure is adequate and, moreover, desirable to avoid erroneous detection of recombination hotspots.
Figure 2.—

Simulation study. The left top plot shows recombination probabilities used to simulate recombination events in primate mitochondrial DNA sequences. The letter A denotes the probability mass over the region [401, 600]. The rest of the plots on the left depict the sites at which recombination was simulated (solid dots) and the posterior median (solid line) and 95% BCIs (shading) of inferred site-specific recombination probabilities. The right top plot depicts the prior density of GMRF precision ω. Posterior densities of ω are plotted underneath the prior.
Figure 2, right, shows the prior density (top histogram) and the marginal posteriors of the GMRF precision ω for each value of A. Note that when significant clustering of breakpoints is observed, the posterior mass of ω concentrates closer to zero. We expect such behavior since greater variability in recombination log odds, supported by data, leads to a decrease of smoothness. Additionally, the bottom plot shows that the prior of ω dominates the posterior when true breakpoints are distributed nearly uniformly.
Finally, we test the ability of our model to recover the strength of a hotspot A. For each value of A, Table 2 reports the true proportion of simulated breakpoints contained in the interval [401, 600], the posterior median and 95% BCI of the normalized probability masses, 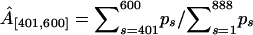 and
and 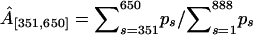 . The normalization is necessary for comparison of the simulated and estimated recombination probabilities as the former sum to one by construction and the latter sum to ∼ln 2 due to the enforced constraint. Mass
. The normalization is necessary for comparison of the simulated and estimated recombination probabilities as the former sum to one by construction and the latter sum to ∼ln 2 due to the enforced constraint. Mass 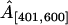 consistently underestimates the strength of the hotspot with a 95% BCI covering the true value of A only when A = 0.7. However, if we expand the region by 50 sites in both directions, the posterior of
consistently underestimates the strength of the hotspot with a 95% BCI covering the true value of A only when A = 0.7. However, if we expand the region by 50 sites in both directions, the posterior of 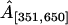 more accurately reflects the true strength of the hotspot. This indicates that uncertainty in estimated breakpoint locations leads to an overestimation of the size of the simulated hotspot region.
more accurately reflects the true strength of the hotspot. This indicates that uncertainty in estimated breakpoint locations leads to an overestimation of the size of the simulated hotspot region.
TABLE 2.
Recombination hotspot strength
| Inferred Â[401,600]
|
Inferred Â[351,650]
|
|||
|---|---|---|---|---|
| Simulated A | Posterior median | 95% BCI | Posterior median | 95% BCI |
| 0.9 | 0.66 | (0.47, 0.82) | 0.76 | (0.58, 0.88) |
| 0.7 | 0.57 | (0.37, 0.75) | 0.67 | (0.48, 0.83) |
| 0.5 | 0.32 | (0.24, 0.48) | 0.48 | (0.36, 0.62) |
| 0.3 | 0.26 | (0.18, 0.35) | 0.38 | (0.28, 0.49) |
The first column contains the sum of recombination probabilities 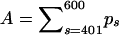 , used to simulate breakpoint locations in primate mitochondrial DNA alignment. The remaining columns report posterior medians and 95% BCIs of the normalized recombination probability masses in the regions [401, 600] and [351, 650].
, used to simulate breakpoint locations in primate mitochondrial DNA alignment. The remaining columns report posterior medians and 95% BCIs of the normalized recombination probability masses in the regions [401, 600] and [351, 650].
A newly observed HIV recombination hotspot:
We apply our model to detect spatial recombination preferences in the gag coding region of the HIV genome. We select 42 sequences from the Los Alamos HIV Sequence Database, all of which have been previously classified as recombinants of pure subtypes A and G (see supplemental information at http://www.genetics.org/supplemental/ for accession numbers). We focus our attention on these two subtypes to limit variation in breakpoint locations due to different subtype composition. Although the effects of such variation remain unknown, experimental evidence is emerging that highlights the importance of subtype composition in the biochemistry of recombination (Chin et al. 2005). The recombinant sequences that we select for our analysis come from several different epidemiological studies (Guo et al. 1993; Durali et al. 1998; Peeters et al. 2000; Barlow et al. 2001; Tebit et al. 2002; Vidal et al. 2003) and therefore should represent a diverse set of recombination events. Besides the recombinant sequences, individual alignments contain representative sequences of subtypes A, G, and B, where the latter serves as an outgroup. Lengths of the alignments range from 562 to 820 nucleotides covering 1118 bp of the HIV genome.
In the top plot of Figure 3 we show the locations of gene products Matrix and Capsid in the master alignment of gag and indicate the position of one of the HIV instability elements (INSs). INSs are RNA sequence motifs involved in post-transcriptional regulation of the HIV gene expression (Mikaélian et al. 1996). It is possible that INS primary or secondary structure promotes recombination. Below the gene map we depict the posterior medians and 95% BCIs of the population-level recombination probabilities. This recombination profile strongly suggests an ∼300-nucleotide-long hotspot near the beginning of the Capsid coding region. The posterior median of the GMRF ω precision amounts to 418, and the 95% BCI of ω is (225, 721).
Figure 3.—

Analysis of HIV recombinants. The top plot illustrates the locations of gene products in the HIV gag coding region and marks the position of an instability element (INS) in the Capsid reading frame (hatched box). Below the gene map we show posterior medians (solid line) and 95% BCIs (shading) of population-level recombination probabilities. In the bottom two plots we depict averaged individual-level recombination probabilities (vertical bars), estimated jointly with the hierarchical model (plot second from bottom) and independently with the DMCP model (bottom plot). Solid circles mark breakpoint locations in individual recombinants as estimated by the joint and independent approaches.
The bottom two plots of Figure 3 contain individual-level recombination characteristics, estimated jointly with the hierarchical approach and independently with the DMCP model. In both plots, vertical bars represent naive estimates of site-specific posterior recombination probabilities, obtained by taking the posterior mean of Cs/Ts for all s, where counts Cs and trials Ts retain their definitions in the joint and independent analyses. Solid circles mark point estimates of breakpoint locations in individual alignments. Point estimates are defined as sites where the topology with maximum posterior probability is not equal to the topology with maximum probability at the preceding site. We see that in the joint analysis breakpoints and higher recombination probabilities cluster more tightly in the Capsid region, when compared with the independent DMCP analysis. Moreover, several breakpoints in the “cold” regions of the gag do not receive substantial posterior support during the joint analysis. Under the hierarchical model such shrinkage of individual-level recombination probabilities and breakpoint estimates results from sharing spatial breakpoint information among individual recombinants via the common recombination prior.
A cluster of several breakpoints at the end of Capsid does not substantially elevate the corresponding population-level recombination probabilities. Recombination signal in this region comes only from six recombinants. All of these breakpoints are located at the very end of individual alignments and some of them represent noise, associated with topological uncertainty, rather than recombination events. Figure 3 demonstrates that the posterior support of all breakpoints in this cluster decreases during the joint analysis, resulting in either a shift of their estimates or an elimination of weakly supported breakpoints.
We also compare the joint and independent analyses with respect to their estimates of the total number of breakpoints in each recombinant. We plot the posterior mean number of breakpoints, obtained using both approaches, in Figure 4. Great variability in M(k) among individual recombinants highlights the importance of allowing flexibility in the number of breakpoints. Most data sets exhibit a slight increase in a posteriori supported number of breakpoints during the joint analysis, but the overall pattern remains unchanged between the two types of analysis. To investigate the cause of the increased support, we compare recombination profiles (data not shown) of all individual recombinants, obtained via the joint and independent approaches. We find that in all cases the increase occurs due to higher values of population-level recombination probabilities in the “hot” portion of gag boosting the posterior confidence in breakpoints located in this region that are weakly or moderately supported under the independent analysis with a flat recombination prior. Therefore, the informative recombination prior does not introduce false breakpoints, but rather amplifies the existing signal inside recombination hotspots. This amplification can be clearly seen by comparing the “skylines” in the bottom two plots of Figure 3.
Figure 4.—

Number of breakpoints in individual recombinants. The top plot shows independently estimated posterior mean numbers of breakpoints plotted against jointly estimated posterior mean numbers of breakpoints for the 42 HIV gag individual recombinants. In the two bottom bar plots, we show the posterior mean numbers of breakpoints for the two types of recombination analysis that correspond to the x- and y-axes of the top plot.
Diagnostics of MCMC performance:
To assess the performance of our sampler we first examine parameters at the individual recombinant level of the DMCP models. The total number of breakpoints M(k) for each alignment k is a pivotal parameter in the DMCP model as its time evolution demonstrates how well our reversible-jump MCMC sampler moves between spaces of different dimension. Since M(k) is a discrete-valued parameter it is natural to examine the regeneration times ti, 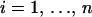 , the time steps at which the Markov chain visits a predefined state (or a set of states), where n is the random number of total visits observed during an MCMC run of fixed length. Mykland et al. (1995) note that the behavior of a renewal process defined by regeneration times of a Markov chain may be used to test the performance of an MCMC sampler. The authors suggest plotting ti/tn against i/n. According to the law of large numbers for renewal processes, this scaled regeneration quantile (SRQ) plot should be close to a line passing through points (0, 0) and (1, 1). Since the total number of breakpoints is only a marginalization of the complete Markov chain state, regeneration times of M(k) are not independent and identically distributed (i.i.d.). However, Li et al. (2000) show that the same interpretation of SRQ plots remains useful even when regeneration times are not strictly i.i.d.
, the time steps at which the Markov chain visits a predefined state (or a set of states), where n is the random number of total visits observed during an MCMC run of fixed length. Mykland et al. (1995) note that the behavior of a renewal process defined by regeneration times of a Markov chain may be used to test the performance of an MCMC sampler. The authors suggest plotting ti/tn against i/n. According to the law of large numbers for renewal processes, this scaled regeneration quantile (SRQ) plot should be close to a line passing through points (0, 0) and (1, 1). Since the total number of breakpoints is only a marginalization of the complete Markov chain state, regeneration times of M(k) are not independent and identically distributed (i.i.d.). However, Li et al. (2000) show that the same interpretation of SRQ plots remains useful even when regeneration times are not strictly i.i.d.
We evaluate the performance of our sampler on the HIV data set with 42 recombinants. For each 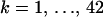 , we choose the posterior median of M(k) to be the renewal state for defining regeneration times ti. We show 42 superimposed SRQ plots in Figure 5, left. Since all SRQ plots in Figure 5 are concentrated around the line y = x, we conclude that our MCMC chains are running long enough to sufficiently sample the posterior distributions of the individual-level parameters.
, we choose the posterior median of M(k) to be the renewal state for defining regeneration times ti. We show 42 superimposed SRQ plots in Figure 5, left. Since all SRQ plots in Figure 5 are concentrated around the line y = x, we conclude that our MCMC chains are running long enough to sufficiently sample the posterior distributions of the individual-level parameters.
Figure 5.—

Convergence diagnostics. The left plot depicts 42 scaled regeneration quantile (SRQ) plots, where ti denotes an iteration, at which the total number of breakpoints in individual alignments returns to its posterior median for the ith time. Gelman–Rubin potential scale reduction factors (PSRFs, solid line) and their corresponding 97.5% quantiles (dashed line) for recombination log odds 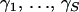 are plotted against site indexes on the right.
are plotted against site indexes on the right.
To monitor convergence of population-level parameters, we use a Gelman–Rubin potential scale reduction factor (PSRF) (Gelman and Rubin 1992). This statistic tests whether multiple Markov chains, started at different values, converge to the same distribution. The PSRF is approximately equal to the square root of the variance estimated by combining all chains, divided by an average of within-chain variances. If all chains reach stationarity, the PSRF approaches 1. If we assume that the stationary distribution is normal, we also can compute confidence bounds for the t-distributed PSRF.
We calculate PSRFs for the recombination log odds from five chains, each started with different values of γ and ω. We generate initial values ω(0) from a uniform distribution over the interval (0, 10,000). We then sample a value for 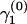 from a normal distribution with mean
from a normal distribution with mean 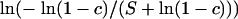 and variance of 2. Conditional on ω(0) and
and variance of 2. Conditional on ω(0) and 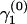 , we initialize the remaining recombination log odds through a random-walk realization,
, we initialize the remaining recombination log odds through a random-walk realization,  , for
, for 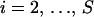 . Such a distribution of starting values for (ω, γ) should be overdispersed with respect to the posterior, as recommended by Gelman and Rubin (1992). Figure 5, right, depicts the PSRFs (solid line) with their 97.5% quantiles for the HIV example recombination log odds. The PSRF and its 95% quantile for ln ω are 1.04078 and 1.09845, respectively. Close proximity of all estimated PSRFs to 1 suggests that all chains reach stationarity.
. Such a distribution of starting values for (ω, γ) should be overdispersed with respect to the posterior, as recommended by Gelman and Rubin (1992). Figure 5, right, depicts the PSRFs (solid line) with their 97.5% quantiles for the HIV example recombination log odds. The PSRF and its 95% quantile for ln ω are 1.04078 and 1.09845, respectively. Close proximity of all estimated PSRFs to 1 suggests that all chains reach stationarity.
DISCUSSION
We present a new Bayesian model for estimating spatial preferences of breakpoints when multiple instances of recombination are observed. The hierarchical framework is built on an individual-level multiple change-point model and a population-level prior for breakpoint locations. Spatial smoothing of population-level recombination probabilities facilitates their estimation when the number of recombination events is small compared to the total number of sites covered by the sequences. Moreover, such smoothing has a meaningful biological interpretation. In retroviruses, recombination occurs during template switching by reverse transcriptase as it linearly copies the viral RNA genome into DNA (Negroni and Buc 2001). Therefore, we expect adjacent sites to have similar log odds of recombination. We realize smoothing by placing a GMRF hyperprior on recombination log odds. GMRFs offer a unified and flexible framework for imposing complex correlation structures in high-dimensional parameter (sub)spaces. Additionally, fast algorithms, available for simulation of GMRFs, allow us to sample efficiently the space of model parameters during MCMC simulation.
Breakpoints in the DMCP model require special attention as their total number needs to be individually controlled in the presence of noisy sequence information. The common prior distribution provides such oversight. We constrain the sum of recombination probabilities to impose an approximately Poisson distribution with a fixed rate on the total number of recombination events in each data set. Such a seemingly trivial modification considerably complicates our MCMC implementation as the modification changes the a priori correlation structure of the recombination log odds, which without constraints is dictated solely by the GMRF hyperprior. To overcome this difficulty, we introduce a linearized constraint for the recombination log odds that approximates our original restrictions on the recombination probabilities. The advantage of such a linear approximation is the ease and computational efficiency of incorporating it into our MCMC transition kernels. We demonstrate that our linear constraint achieves desirable behavior both for the recombination probabilities and for the total number of breakpoints in individual alignments.
The analysis of the HIV gag genomic region strongly suggests a recombination hotspot near the beginning of the Capsid coding region. Since local sequence motifs have been long suspected to promote HIV recombination (Balakrishnan et al. 2001; Moumen et al. 2001, 2003; Negroni and Buc 2001; Galetto et al. 2004), we examine this part of the HIV genome for the presence of known motifs. One of the HIV instability elements, denoted as INS2-M6 by Schneider et al. (1997), covers sites [564, 609] of our master alignment (see Figure 3). We hypothesize that either primary or secondary structure of this RNA segment promotes formation of a recombination hotspot in the Capsid coding region. This hypothesis grows even more promising in light of preliminary experimental results confirming an increased rate of in vitro reverse transcriptase strand transfer in the Capsid hotspot (S. Carpenter, personal communication).
Selection of recombinant sequences for hotspot mapping can bias results and therefore should be performed with caution. For example, several sequences may be descendants of the same ancestral recombinant. Including such recombinants into the analysis would violate our assumption of independence among recombination events, leading to overcounting of breakpoints in some regions of the master alignment. Researchers should pay particular attention to circulating recombinant forms (CRFs) since by definition they may be overrepresented in a population sample. To check for this possibility, we examined CRFs with recombination between A and G subtypes in the gag coding region and found that no known CRF contributes breakpoint signal at the hotspot that we identified from the 42 HIV gag recombinants (data not shown). Another danger comes from the fact that individual recombinants usually cover different portions of the master alignment. Although site-specific trials T account for such uneven coverage, the breakpoint noise, often seen at the boundaries of individual alignments, may be amplified if many recombinants start or end in close proximity to each other.
Since the factors promoting HIV recombination in vivo are largely unknown, it is natural to capitalize on the flexible GMRF structure and incorporate covariates into the prior of recombination log odds, using a generalized linear model framework. Such an extension will not only improve estimation of hotspot locations by injecting additional information into the model, but also enable the testing of the role of specific sequence features in producing a nonuniform distribution of breakpoint locations along the HIV genome. Our model augmented with covariates should be superior to previous approaches that use phylogenetic recombination detection to test spatial association of recombination hotspots with local genomic RNA properties (Magiorkinis et al. 2003; Zhang et al. 2005), as the hierarchical approach allows for integration over all breakpoint locations supported by molecular sequence data.
Finally, we outline future opportunities for bridging phylogenetic and coalescent-based methods for studying recombination. These two approaches are often considered competitors (Awadalla 2003). In our opinion, phylogenetic and coalescent-based methods for studying recombination do not compete, but rather complement each other. Both frameworks provide sensible tools for analyzing recombination among sequences, but differ in the recombination/mutation rate ratio most appropriate for the chosen method. Moreover, it is not hard to envision a Bayesian model with a phylogenetic change-point likelihood controlling breakpoint locations and a coalescent-based prior forcing phylogenies to obey the laws of population genetics. This unified framework is particularly promising for studying recombination during HIV intrahost evolution as both phylogenetic and coalescent-based approaches have advantages to contribute when analyzing such sequence data.
Acknowledgments
We thank Benjamin Redelings, Robert Rovetti, and two anonymous reviewers for their comments that greatly improved the manuscript. This work was supported by National Institutes of Health grant GM068955, by the University of California Los Angeles AIDS Institute, and by the James B. Pendleton Charitable Trust.
References
- Awadalla, P., 2003. The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 4: 50–60. [DOI] [PubMed] [Google Scholar]
- Balakrishnan, M., P. Fay and R. A. Bambara, 2001. The kissing hairpin sequence promotes recombination within the HIV-I 5′ leader region. J. Biol. Chem. 276: 36482–36492. [DOI] [PubMed] [Google Scholar]
- Barlow, K., I. Tatt, P. Cane, D. Pillay and J. Clewley, 2001. Recombinant strains of HIV type 1 in the United Kingdom. AIDS Res. Hum. Retroviruses 17: 467–474. [DOI] [PubMed] [Google Scholar]
- Bernardinelli, L., D. Clayton and C. Montomoli, 1995. Bayesian estimates of disease maps: How important are priors? Stat. Med. 14: 2411–2431. [DOI] [PubMed] [Google Scholar]
- Besag, J., 1974. Spatial interaction and the statistical analysis of lattice systems (with discussion). J. R. Stat. Soc. Ser. B 36: 192–236. [Google Scholar]
- Besag, J., J. York and A. Mollié, 1991. Bayesian image restoration, with two applications in spatial statistics (with discussion). Ann. Inst. Stat. Math. 43: 1–59. [Google Scholar]
- Chin, M., T. Rhodes, J. Chen, W. Fu and W. Hu, 2005. Identification of a major restriction in HIV-1 intersubtype recombination. Proc. Natl. Acad. Sci. USA 102: 9002–9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choisy, M., C. Woelk, J. Guegan and D. Robertson, 2004. Comparative study of adaptive molecular evolution in different human immunodeficiency virus groups and subtypes. J. Virol. 78: 1962–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durali, D., J. Morvan, F. Letourneur, D. Schmitt, N. Guegan et al., 1998. Cross-reactions between the cytotoxic T-lymphocyte responses of human immunodeficiency virus-infected African and European patients. J. Virol. 72: 3547–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykes, C., M. Balakrishnan, V. Planelles, Y. Zhu, R. Bambara et al., 2004. Identification of a preferred region for recombination and mutation in HIV-1 gag. Virology 326: 262–279. [DOI] [PubMed] [Google Scholar]
- Elliott, P., J. Wakefield, N. Best and D. Briggs (Editors), 2000. Spatial Epidemiology: Methods and Applications. Oxford University Press, London/New York/Oxford.
- Fearnhead, P., R. Harding, J. Schneider, S. Myers and P. Donnelly, 2004. Application of coalescent methods to reveal fine-scale rate variation and recombination hotspots. Genetics 167: 2067–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 2004. Inferring Phylogenies. Sinauer Associates, Sunderland, MA.
- Galetto, R., A. Moumen, V. Giacomoni, M. Veron, P. Charneau et al., 2004. The structure of HIV-1 genomic RNA in the gp120 gene determines a recombination hot spot in vivo. J. Biol. Chem. 279: 36625–36632. [DOI] [PubMed] [Google Scholar]
- Gelman, A., and D. Rubin, 1992. Inference from iterative simulation using multiple sequences. Stat. Sci. 7: 457–511. [Google Scholar]
- Ghosh, M., K. Natarajan, T. Stroud and B. Carlin, 1998. Generalized linear models for small-area estimation. J. Am. Stat. Assoc. 93: 273–282. [Google Scholar]
- Grassly, N., and E. Holmes, 1997. A likelihood method for the detection of selection and recombination using nucleotide sequences. Mol. Biol. Evol. 14: 239–247. [DOI] [PubMed] [Google Scholar]
- Green, P., 1995. Reversible jump Markov chain Monte Carlo computation and Bayesian model determination. Biometrika 82: 711–732. [Google Scholar]
- Guo, H. G., M. S. Reitz, R. C. Gallo, Y. C. Ko and K. S. Chang, 1993. A new subtype of HIV-1 gag sequence detected in Taiwan. AIDS Res. Hum. Retroviruses 9: 925–927. [DOI] [PubMed] [Google Scholar]
- Hasegawa, M., H. Kishino and T. Yano, 1985. Dating the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22: 160–174. [DOI] [PubMed] [Google Scholar]
- Hein, J., 1990. Reconstructing evolution of sequences subject to recombination using parsimony. Math. Biosci. 98: 185–200. [DOI] [PubMed] [Google Scholar]
- Husmeier, D., 2005. Discriminating between rate heterogeneity and interspecific recombination in DNA sequence alignments with phylogenetic factorial hidden Markov models. Bioinformatics 21: ii166–ii172. [DOI] [PubMed] [Google Scholar]
- Kalish, M., K. Robbins, D. Pieniazek, A. Schaefer, N. Nzilambi et al., 2004. Recombinant viruses and early global HIV-1 epidemic. Emerg. Infect. Dis. 10: 1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi, L., A. Jeffreys and S. Keeney, 2004. Where the crossovers are: recombination distributions in mammals. Nat. Rev. Genet. 5: 413–424. [DOI] [PubMed] [Google Scholar]
- Knorr-Held, L., and H. Rue, 2002. On block updating in Markov random field models for desease mapping. Scand. J. Stat. 29: 597–614. [Google Scholar]
- Larget, B., and D. Simon, 1999. Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Mol. Biol. Evol. 16: 750–759. [Google Scholar]
- le Cam, L., 1960. An approximation theorem for the Poisson binomial distribution. Pac. J. Math. 10: 1181–1197. [Google Scholar]
- Li, W., M. Tanimura and P. Sharp, 1988. Rates and dates of divergence between AIDS virus nucleotide sequences. Mol. Biol. Evol. 5: 313–330. [DOI] [PubMed] [Google Scholar]
- Li, S., D. Pearl and H. Doss, 2000. Phylogenetic tree construction using Markov chain Monte Carlo. J. Am. Stat. Assoc. 95: 493–508. [Google Scholar]
- Magiorkinis, G., D. Paraskevis, A. Vandamme, E. Magiorkinis, V. Sypsa et al., 2003. In vivo characteristics of human immunodeficiency virus type 1 intersubtype recombination: determination of hot spots and correlation with sequence similarity. J. Gen. Virol. 84: 2715–2722. [DOI] [PubMed] [Google Scholar]
- McGuire, G., F. Wright and M. Prentice, 1997. A graphical method for detecting recombination in phylogenetic data sets. Mol. Biol. Evol. 14: 1125–1131. [DOI] [PubMed] [Google Scholar]
- McVean, G., P. Awadalla and P. Fearnhead, 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160: 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, G., S. Myers, S. Hunt, P. Deloukas, D. Bentley et al., 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304: 581–584. [DOI] [PubMed] [Google Scholar]
- Mikaélian, I., M. Krieg, M. Gait and J. Karn, 1996. Interactions of INS (CRS) elements and the splicing machinery regulate the production of Rev-responsive mRNAs. J. Mol. Biol. 257: 246–264. [DOI] [PubMed] [Google Scholar]
- Minin, V., K. Dorman, F. Fang and M. Suchard, 2005. Dual multiple change-point model leads to more accurate recombination detection. Bioinformatics 21: 3034–3042. [DOI] [PubMed] [Google Scholar]
- Moumen, A., L. Polomack, B. Roques, H. Buc and M. Negroni, 2001. The HIV-1 repeated sequence R as a robust hot-spot for copy-choice recombination. Nucleic Acids Res. 29: 3814–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moumen, A., L. Polomack, T. Unge, M. Veron, H. Buc et al., 2003. Evidence for a mechanism of recombination during reverse transcription dependent on the structure of the acceptor RNA. J. Biol. Chem. 278: 15973–15978. [DOI] [PubMed] [Google Scholar]
- Myers, S., L. Bottolo, C. Freeman, G. McVean and P. Donnelly, 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324. [DOI] [PubMed] [Google Scholar]
- Mykland, P., L. Tierney and B. Yu, 1995. Regeneration in Markov chain samplers. J. Am. Stat. Assoc. 90: 233–241. [Google Scholar]
- Negroni, M., and H. Buc, 2001. Mechanisms of retroviral recombination. Annu. Rev. Genet. 35: 275–302. [DOI] [PubMed] [Google Scholar]
- Peeters, M., E. Esu-Williams, L. Vergne, C. Montavon, C. Mulanga-Kabeya et al., 2000. Predominance of subtype A and G HIV type 1 in Nigeria, with geographical differences in their distribution. AIDS Res. Hum. Retroviruses 16: 315–325. [DOI] [PubMed] [Google Scholar]
- Rambaut, A., D. Robertson, O. Pybus, M. Peeters and E. Holmes, 2001. Human immunodeficiency virus: phylogeny and the origin of HIV-1. Nature 410: 1047–1048. [DOI] [PubMed] [Google Scholar]
- Robertson, D., P. Sharp, F. McCutchan and B. Hahn, 1995. Recombination in HIV-1. Nature 374: 124–126. [DOI] [PubMed] [Google Scholar]
- Rue, H., 2001. Fast sampling of Gaussian Markov random fields. J. R. Stat. Soc. Ser. B 63: 325–338. [Google Scholar]
- Rue, H., and L. Held, 2005. Gaussian Markov Random Fields: Theory and Applications (Monographs on Statistics and Applied Probability, Vol. 104). Chapman & Hall, London.
- Rue, H., I. Steinsland and S. Erland, 2004. Approximating hidden Gaussian Markov random fields. J. R. Stat. Soc. Ser. B 66: 877–892. [Google Scholar]
- Salminen, M., J. Carr, D. Burke and F. McCutchan, 1995. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retroviruses 11: 1423–1425. [DOI] [PubMed] [Google Scholar]
- Schneider, R., M. Campbell, G. Nasioulas, B. Felber and G. Pavlakis, 1997. Inactivation of the human immunodeficiency virus type 1 inhibitory elements allows Rev-independent expression of gag and gag/protease and particle formation. J. Virol. 71: 4892–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, G., 2001. Homologous recombination near and far from DNA breaks: alternative roles and contrasting views. Annu. Rev. Genet. 35: 243–274. [DOI] [PubMed] [Google Scholar]
- Stumpf, M., and G. McVean, 2003. Estimating recombination rates from population genetic data. Nat. Rev. Genet. 4: 959–968. [DOI] [PubMed] [Google Scholar]
- Suchard, M., R. Weiss and J. Sinsheimer, 2001. Bayesian selection of continuous-time Markov chain evolutionary models. Mol. Biol. Evol. 18: 1001–1013. [DOI] [PubMed] [Google Scholar]
- Suchard, M., R. Weiss, K. Dorman and J. Sinsheimer, 2002. Oh brother, where art thou? A Bayes factor test for recombination with uncertain heritage. Syst. Biol. 51: 715–728. [DOI] [PubMed] [Google Scholar]
- Suchard, M., R. Weiss, K. Dorman and J. Sinsheimer, 2003. Inferring spatial phylogenetic variation along nucleotide sequences: a multiple change-point model. J. Am. Stat. Assoc. 98: 427–437. [Google Scholar]
- Sun, D., R. Tsutakawa and P. Speckman, 1999. Posterior distribution of hierarchical models using CAR(1) distributions. Biometrika 86: 341–350. [Google Scholar]
- Tebit, D., L. Zekeng, L. Kaptué, M. Salminen, H. Kräusslich et al., 2002. Genotypic and phenotypic analysis of HIV type 1 primary isolates from western Cameroon. AIDS Res. Hum. Retroviruses 18: 39–48. [DOI] [PubMed] [Google Scholar]
- Vidal, N., M. Peeters, C. Mulanga-Kabeya, N. Nzilambi, D. Robertson et al., 2000. Unprecedented degree of human immunodeficiency virus type 1 (HIV-1) group M genetic diversity in the Democratic Republic of Congo suggests that the HIV-1 pandemic originated in Central Africa. J. Virol. 74: 10498–10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal, N., D. Koyalta, V. Richard, C. Lechiche, T. Ndinaromtan et al., 2003. High genetic diversity of HIV-1 strains in Chad, west central Africa. J. Acquired Immune Defic. Syndr. 33: 239–246. [DOI] [PubMed] [Google Scholar]
- Yang, Z., and B. Rannala, 1997. Bayesian phylogenetic inference using DNA sequences. Mol. Biol. Evol. 14: 717–724. [DOI] [PubMed] [Google Scholar]
- Zhang, C., J. Wei and S. He, 2005. The key role for local base order in the generation of multiple forms of China HIV-1 B′/C intersubtype recombinants. BMC Evol. Biol. 5: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]