

Abstract
The evolutionary history of Plasmodium vivax has recently been addressed in terms of its origin as a parasite of humans and the age of extant populations. The consensus is that P. vivax originated as a result of a host switch from a non-human primate to hominids and that the extant populations did not originate as recently as previously proposed. Here, we show that, in a comparison of parasite isolates from across the world, Asian populations of P. vivax are the oldest. We discuss how this result, together with the phylogenetic evidence that P. vivax derived from Plasmodium found in Southeast Asian macaques, is most simply explained by assuming an Asian origin of this parasite. Nevertheless, the available data show only the tip of the iceberg. We discuss how sampling might affect time estimates to the most recent common ancestor for P. vivax populations and suggest that spatially explicit estimates are needed to understand the demographic history of this parasite better.
A brief introduction to malarial parasites
Human malaria is a parasitic disease that is endemic in most tropical and subtropical ecosystems worldwide [1]. Malarial parasites belong to the genus Plasmodium and infect many vertebrate hosts, including several species of non-human primate [2,3]. These parasitic protozoa have complex life-cycles that involve sexual reproduction in the mosquito vector and asexual stages in the vertebrate host. In primate malarias, the host is inoculated with sporozoites by the bite of an infected Anopheles mosquito during its blood meal. These sporozoites migrate to the liver, where they invade parenchymal cells and differentiate into hepatic merozoites. These hepatic merozoites are released into the bloodstream, where they attach to and invade red blood cells. A subpopulation of merozoites develops into gametocytes, which can be taken up by a mosquito. The life cycle is completed by sexual reproduction in the mosquito, followed by the development and invasion of haploid sporozoites into the mosquito salivary glands, from which they can inoculate a vertebrate host [3].
Four Plasmodium species are parasitic to humans: Plasmodium falciparum, Plasmodium malariae, Plasmodium ovale and Plasmodium vivax. Of these, P. falciparum and P. vivax are associated with most malaria morbidity and mortality [3]. There is great genetic diversity in the antigen-encoding genes of malarial parasites, whereas there is a relatively low level of polymorphism in genes encoding other proteins [4]. Evolutionary genetic approaches can be used to investigate this pattern by assessing the effect of positive selection on the observed polymorphism and estimating when the extant parasite populations originated [4–7]. Until recently, evolutionary studies focused on P. falciparum because it is the most important malarial parasite of humans in terms of its associated mortality, especially in sub-Saharan Africa [4–7]. However, in other areas of the world, malaria is often caused by P. vivax, the second most important malarial parasite of humans in terms of morbidity. Currently, there are 70–80 million cases of P. vivax malaria per year and it has re-emerged in many regions of the world from which malaria was eliminated in the 1950–1960s [8]. P. vivax has extraordinary phenotypic diversity, especially in its relapse patterns, and is found in a broad range of ecotypes, from Russia to the tropical regions of Asia, the Pacific, and South and Central America [3,8].
Plasmodium vivax: from monkeys to humans
Researchers are beginning to understand the evolutionary history of P. vivax. Recent phylogenetic studies [9,10] have shown that this parasite originated from a malarial parasite of non-human primates as a result of a host switch, probably from a macaque. These results receive additional support when the combined data of complete mitochondrial genomes reported in two independent studies are analyzed using Bayesian phylogenetic methods (see Glossary) [11,12] (Figure 1). The species included in the phylogeny are described in Box 1. In our analysis of the complete mitochondrial data, Plasmodium cynomolgi, a parasite found in macaques, is the closest sister species to P. vivax. We also include the Plasmodium simium sequence to show that it is identical to that of P. vivax, supporting previous claims that this South American species originated from a host switch from humans to monkeys [9–11]. Overall, this molecular phylogeny is consistent with others [10] and with early evolutionary inferences based on morphological traits and the G+C content of the genomes of the parasites [2,13]. However, phylogenetic analyses are limited by a relatively poor sample of malarial parasites of primates. Several species that are parasitic to gibbons and orangutans have yet to be included in molecular phylogenetic analyses, which would increase knowledge of the origin of P. vivax. Nevertheless, based on the available data, the most parsimonious explanation is that P. vivax derived from a parasite of non-human primates that is closely related to the extant Plasmodium species found in Southeast Asian macaques.
Figure 1.
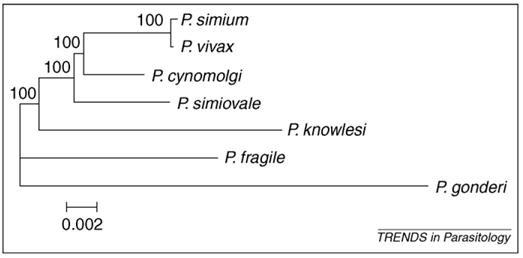
Bayesian phylogenetic tree using complete mitochondrial genomes for parasites of Southeast Asian primates and Plasmodium simium, using Plasmodium gonderi as the outgroup. This phylogenetic tree was inferred under the Hasegawa–Kishino–Yano (HKY) substitution model with MrBayes [28]. The search was performed with 1 000 000 generations, and the first 10 000 trees were discarded in the burning (see Ref. [28] for details). Values above branches are posterior probabilities expressed as percentages. A neighbor-joining tree [29] inferred under the Tamura and Nei substitution model with the software MEGA [30] was used as a prior (see Ref. [31] for information about DNA substitution models). P. simium is identical to Plasmodium vivax, indicating a transfer from humans to South American monkeys (see discussion in Refs [10,11]). The tree indicates that P. vivax is closely related to Plasmodium cynomolgi. The P. vivax–P. cynomolgi group diverged from other species of Plasmodium found in non-human primates. This phylogeny indicates that P. vivax originated from a Plasmodium species related to those currently found in Southeast Asian macaques. Branch length is representative of number of nucleotide substitutions per site, as indicated by the scale bar.
The origin of P. vivax could be linked to the relative ages of the extant populations of this parasite around the world; however, such inferences should be made with caution. The time to the most recent common ancestor (TMRCA) estimates of the extant parasite populations do not necessarily provide evidence about the location of the ancestral population In addition, some considerations must be made when estimating the TMRCA in species in which genetic geographic structure is expected (see later).
Plasmodium vivax geographic mitochondrial variation
Estimating the TMRCA for the extant P. vivax populations has both basic and practical implications. For example, an ancient Asian origin could support the notion that P. vivax selected for the fixation of Duffy-negative genotypes in African human populations. In such a scenario, complex migration patterns of hominids could be evidence of an early introduction of P. vivax into Africa [12]. In addition, the TMRCA estimates depend on the effective population size, which is proportional to the amount of genetic diversity maintained in the population. Thus, knowing the evolutionary history of malarial parasite populations is essential for detecting genetic adaptive variation such as that expected in potential antigens and for understanding patterns of linkage disequilibrium that could enable the identification of genes associated with, for example, drug resistance. Understanding the geographic population structure is also important in epidemiological surveillance studies because the source of parasites could be established more easily in cases of imported malaria [14–16].
Two comprehensive published analyses estimate the TMRCA in P. vivax based on mitochondrial genomes [11,12]. The study by Mu et al. [11] includes 176 sequences (GenBank accession numbers AY791517.1–AY791692.1) and the study by Jongwutiwes et al. [12] includes 106 sequences (GenBank accession numbers AY598035.1–AY598140.1). There is some overlap in the estimates of TMRCA: Mu et al. [11] concluded that P. vivax originated between 53 [12] estimated that such an event occurred between 217 000 and 304 000 years ago. These studies differ in the analytical methods used and have some differences in the geographic origin of their samples. Jongwutiwes et al. [12] included few divergent sequences from Melanesia (N = 4) and an extensive sample from Asia (N = 69), whereas Mu et al. [11] included a more comprehensive sample from Melanesia (N = 69), mostly from Papua New Guinea, and sequences from Africa (N = 12). Given such differences in sampling, a better estimate of TMRCA can be achieved by analyzing the combined dataset.
Box 1. Plasmodium species included in our phylogenetic analysis
Thirteen species of Plasmodium have been identified in non-human primates in Southeast Asia [2,3]: seven in macaques, four in gibbons and two in orangutans (their description and major biological characteristics can be found in Refs [2,3]). The species considered in this article are those for which complete mitochondrial genomes are available. Other analyses have included a more comprehensive group of species [9,10]; however, the taxonomic sampling is still incomplete.
Plasmodium gonderi: this species is found in Central Africa and is used as our outgroup [9,10]. Its natural hosts are Cercocebus atys and Cercopithecus spp.
Plasmodium fragile: its natural hosts are Macaca radiata, Macaca mulatta and Presbytis spp. It is found in Southern India and Sri Lanka.
Plasmodium cynomolgi: its natural hosts are Macaca nemestrina, Macaca fascicularis, M. mulatta, M. radiata, Macaca sinica, Presbytis entrellus and Presbytis citratus. It is widely distributed in Southeast Asia.
Plasmodium simiovale: its natural host is M. sinica. It is found in Sri Lanka.
Plasmodium knowlesi: its natural hosts are M. fascicularis, Macaca nigra and M. nemestrina. It is widely distributed in Southeast Asia.
Plasmodium simium: it is found in a restricted area of South America. Molecular evidence demonstrates that it is identical to Plasmodium vivax and is considered to have originated from a host switch from humans to non-human primates in that region [9–11].
The combined data from these two studies are used in our investigation, giving a total of 282 sequences with 93 haplotypes covering the known geographical distribution of P. vivax [11,12]. By analyzing this combined dataset, a haplotype network was obtained (Figure 2) and the results are similar to those reported by Mu et al. [11]. Overall, Asian haplotypes were found to be more diverse than those of other regions (45 haplotypes from 114 sequences). We also found an extensive number of haplotypes in Melanesia (32 haplotypes from 73 sequences, with a haplotype diversity of 0.889); indeed, this region forms a separated block that is connected to haplotypes from the Americas. The number of haplotypes per se does not provide evidence of the location of the ancient population or which population shares an older common ancestor. Indeed, both Melanesian and Asian populations have similar numbers of haplotypes after correcting for the number of isolates. Nevertheless, two major observations can be made from this combined dataset. First, the number of haplotypes is correlated with the subpopulation sample size, which indicates that the overall number of haplotypes is still underestimated. Only the samples from the Americas seem to be saturated, with seven haplotypes from 47 sequences. Second, there seems to be an excess of single haplotypes (see later).
Estimating the neutral mutation rate
To estimate the TMRCA, an estimate of the neutral mutation rate is needed [11,12]: that is, a rate at which the mitochondrial genome evolves under the assumption of a molecular clock (i.e. a constant rate of evolution). This assumption was tested by using the alignment that generated the phylogeny depicted in Figure 1 and considered only Southeast Asian primates, with Plasmodium gonderi as the outgroup. Based on this alignment, the assumption of a molecular clock was not rejected when a phylogeny estimated under this assumption was compared with a phylogeny that did not assume a clock and no statistical differences were found in their branch lengths by a likelihood ratio test. It was, therefore, reasonable to assume a constant rate of evolution within this group of species, which includes P. vivax and Plasmodia that are parasitic to Southeast Asian macaques. The divergence between Plasmodium fragile and Plasmodium knowlesi was then used to estimate the neutral rate of evolution for the mitochondrial genome of these Plasmodia, assuming that they diverged as part of the radiation of their hosts between 3.5 and 4.7 million years ago (Mya): that is, the origin of the silenus group of macaques and the divergence of the sinica and fascicularis groups [17]. However, co-speciation cannot be assumed: our premise is that malarial parasites radiated when macaque populations were increasing and overlapping geographically, therefore enabling host switches. These events lead us to estimates of between 4.31 × 10−9 and 3.21 × 10−9 mutations per base pair per year based on the 3.5 and 4.7 Mya scenarios. As an example, by using such rates, the divergence between P. cynomolgi and P. vivax is estimated to be between 1.17 and 1.60 Mya, a timeframe when several Macaca populations, in addition to Homo erectus, were expanding their ranges, resulting in overlapping geographic distributions [18]. However, this time estimate is not when the host switch occurred because information is still lacking about the specific lineage of parasites of non-human primates that gave rise to P. vivax.
Estimating the TMRCA
The next set of assumptions relates to the mode and rate of evolution of P. vivax populations. Although we tested for a molecular clock, we cannot assume that P. vivax populations evolved neutrally as one random mating population with a constant population size. Given the worldwide distribution of this parasite, geographic genetic structure is expected because the probability that two individuals mate will be affected by the geographic barriers and distance between them. Population structure affects the coalescence time of a sample of alleles [19,20]. In a structured population, the subpopulations are expected to diverge differentially by genetic drift. The divergence rate depends on the size of each subpopulation; for example, smaller populations are expected to diverge faster. If two populations are combined that have been evolving separately with limited migration between them, TMRCA will be estimated on a spurious effective population size. It follows that estimates of the TMRCA must consider whether there is geographic structure or one random mating population [19,20].
To check for population structure, an analysis of molecular variance [21] was performed as implemented in Arlequin v3.0 [22]. This was statistically significant, indicating that there is geographic population structure. If there is significant population structure, more-comprehensive sampling is required that considers a series of parasite populations in which each can be assumed to be a unit in the analysis. That is, each of these subpopulations or units can be considered to evolve neutrally with random mating. Given the actual sampling and limited information from each isolate, it is hard to establish how many populations there are in the available dataset.
To illustrate the effect of population structure and sampling on TMRCA estimates, we calculated the TMRCA in the separated and combined datasets using the geographical subdivision proposed by Mu et al. [11]. If sampling were not important, we would expect both datasets to generate similar TMRCA estimates when the same method of analysis is used. Mu et al. [11] calculated coalescent-based maximum-likelihood estimates of the pairwise migration rates among subpopulations (Asia, Melanesia, Africa, America and India). Such estimates can be used to calculate the TMRCA, assuming geographic structure under the infinite-sites model using the Gene-Tree program [23]. This algorithm can estimate both the TMRCA in the overall sample and the TMRCA within each subpopulation.
As a first approximation in this exploratory analysis, polymorphic sites (positions in the alignment) that violated the assumptions of the infinite-sites model were excluded by using GeneTree, leaving 56 sites out of 75. It is important to note that the haplotype structure was maintained and that the sites that violated this assumption were the same in both datasets. Regardless of the absolute values obtained, our estimates of TMRCA for each dataset have less overlap than do those reported for each study separately (Table 1). A first conclusion is that, when both datasets are analyzed separately using the same method, the results differ, probably because of differences in the geographical origin of the samples. Nevertheless, a pattern is consistent in both datasets: the Asian populations seem to be older and more diverse. The Americas consistently have the shortest TMRCA, as expected assuming a recent introduction of malaria to this continent, but there is a considerable difference in the TMRCA estimates for India. Interesting results emerge when the samples from the studies are combined. The older, and more diverse, population is still in Asia but the TMRCA estimates are between 280 000 and 360 000 years old, assuming two different mutation rates (Table 1). Estimates that are clearly older than that obtained from the data of each study are analyzed separately. The combined sample leads to estimates as old as 460 000 years for the extant P. vivax populations worldwide, which is longer than the TMRCA obtained from each study separately (Table 1). Given the lack of other information that enables the subpopulations to be defined better, these results indicate that the geographic structure in the dataset was poorly represented by the proposed subpopulations or regions. Thus, this analysis highlights the need for spatially explicit samples with specific populations that can be assumed to be evolving neutrally with random mating. The lack of a spatially well-defined sample could markedly affect TMRCA estimates.
Table 1.
| Scenarios | Africac | The Americas | Asia | India | Melanesiad | Entire sample | |
|---|---|---|---|---|---|---|---|
| Mu et al. | |||||||
| 4.31 × 10−9 | 50 000 (13 500) | 3800 (1300) | 275 500 (76 500) | 122 700 (30 400) | 139 000 (25 600) | 258 200 (32 400) | |
| 3.21 × 10−9 | 67 100 (18 100) | 5100 (1700) | 370 000 (102 700) | 164 900 (40 800) | 186 700 (34 400) | 346 800 (43 500) | |
| Jongwutiwes et al. | |||||||
| 4.31 × 10−9 | – | 5200 (2000) | 59 600 (20 600) | 1300 (600) | – | 162 400 (78 000) | |
| 3.21 × 10−9 | – | 6900 (2700) | 80 000 (27 700) | 1700 (800) | – | 218 000 (104 800) | |
| Joint data | |||||||
| 4.31 × 10−9 | 40 600 (10 800) | 4500 (1300) | 281 500 (85 700) | 143 400 (29 300) | 182 200 (35 500) | 346 000 (23 500) | |
| 3.21 × 10−9 | 54 500 (14 500) | 6000 (1700) | 378 000 (115 100) | 192 600 (39 400) | 244 700 (47 600) | 464 600 (31 500) |
Estimates (in years) were made using two mutation rates and the data from Mu et al. [11] and Jongwutiwes et al. [12].
Standard deviations estimated over the coalescent TMRCA are in parentheses.
No sequences were available from Africa for the Jongwutiwes et al. [12] dataset.
Melanesia was excluded from the study by Jongwutiwes et al. [12] because of the small sample size and highly divergent sequences.
Beyond geographic structure
Although we focused on population structure, demographic processes such as population expansion can also affect TMRCA estimates. Mu et al. [11] considered exponential growth and found no substantial differences from the model of constant population size in their sample. However, given that they did not consider geographic population structure when these models (constant population size and exponential growth) were compared, it is unclear how the underlying geographic structure could affect their ability to detect departures from the model of constant population size. Indeed, different regions could have different demographic histories. As an example, we looked for an excess of rare mutations, which is a marker of population expansion, by using Tajima’s D test [24]. In the Melanesian sample, we found a significant and negative D of −1.920, indicating an excess of rare mutations, whereas in Asia D was nonsignificant (Tajima’s D: −1.729; nonsignificant = 0.10 > P > 0.05). Regardless of whether these results indicate a strong population expansion within subpopulations, this could be the consequence of several subpopulations within a specific region that are poorly sampled and pooled together [25]. Finally, the mitochondrial genome is only one locus, in which all the proteins are linked and there is no recombination. Thus, these analyses reflect only the history of this genome. Other markers from the nucleus, such as microsatellites [26], are needed to understand the evolutionary history of P. vivax as a species better.
Concluding remarks
In summary, we analyzed the available data regarding complete mitochondrial genomes to study the phylogeny of Southeast Asian malarial parasites of primates and the history of P. vivax populations. The phylogeny estimated from complete mitochondrial genomes confirms previous results [9,10] indicating that P. vivax originated from a malarial parasite of non-human primates that is related to the species currently found in Asian macaques. We also found that the Asian populations of P. vivax consistently seem to be older than other populations in our analyses. These lines of evidence (phylogenetic and population genetics) are most parsimoniously explained by an Asian origin of P. vivax. However, our analyses illustrate that geographic population structure is an important factor that must be taken into account to estimate TMRCA properly. We have shown that TMRCA estimates are affected by the samples considered in the analysis. We also found an excess of rare haplotypes, indicating that demographic processes such as population expansion could affect TMRCA estimates. The relevance of demography versus population structure when estimating TMRCA depends on the goal and scale of each investigation. This situation resembles the one described for human populations, in which extensive geographic sampling is needed to understand the demographic history of the species better [25,27].
Mu et al. [11] and Jongwutiwes et al. [12] have enabled the evolutionary history of extant populations of P. vivax to be addressed. We suggest that an extensive and spatially defined sampling, together with additional independent markers [26], is needed to understand the history of this malarial parasite of humans better. Such investigations will improve the understanding of P. vivax genetic variation, enabling the identification of adaptive variation, the interpretation of linkage patterns, leading to the identification of genes potentially involved in drug resistance, and the development of molecular surveillance protocols to identify the source of parasites involved in outbreaks in areas in which malaria is imported [7,14–16].
Figure 2.
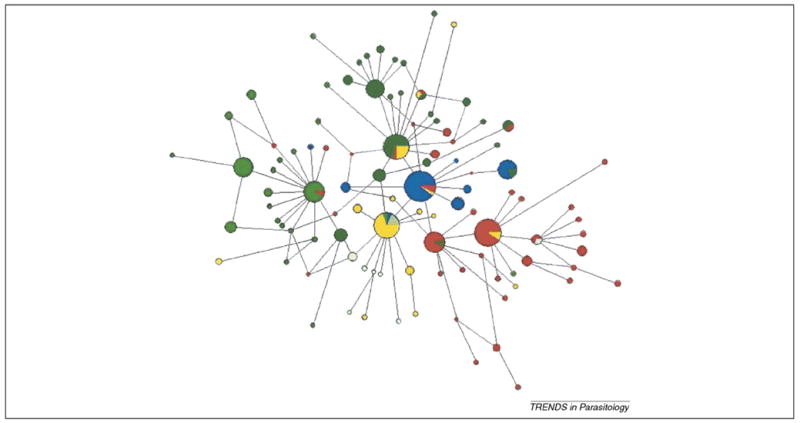
Haplotype network showing the relationships among Plasmodium vivax populations. Haplotype network of the 282 mitochondrial genomes (joint datasets) inferred under a median-joining algorithm with posterior pruning using maximum parsimony criteria as implemented in Network 4.1.1.2 [32]. The size of the circles is proportional to the haplotype frequency, with each color indicating the geographic origin of the sample. The Asian populations (dark green) include isolates from China, Thailand, Indonesia and Vietnam. There are 45 haplotypes in 114 sequences from this region, with a haplotype diversity of 0.948. The Melanesian sample (red) includes 32 haplotypes from 73 sequences, with a haplotype diversity of 0.889; the isolates are from Papua New Guinea, Salomon Islands and Vanuatu. The India–Middle-East sample (yellow) includes 16 haplotypes out of 35 sequenced and a haplotype diversity of 0.881. There is more haplotype diversity in Asia and Melanesia than in any other region considered in this analysis. The population from the Americas (blue) has a small number of haplotypes (seven out of 47 sequences and a haplotype diversity of 0.645), smaller, even, than the African population (light green) (eight haplotypes in 12 sequences, with a haplotype diversity of 0.894).
Acknowledgments
A.A.E. is supported by NIH grant R01 GM60740. O.E.C. is supported by the Graduate Division of Biological and Biomedical Sciences at Emory University. We thank Andrea McCollum, Janet Cox, Lisa Jones-Engel, Maria Pacheco and two anonymous reviewers for comments that improved the manuscript.
Glossary
- Bayesian phylogenetic methods
phylogenetic reconstruction method based on Bayesian statistics. This method estimates the phylogeny or set of phylogenies that is more likely to be explained by the data (aligned sequences), allowing for uncertainty in the phylogenetic inference
- Coalescent
under this theory, all alleles in a sample are assumed to derive from a common ancestor. Therefore, it is possible to estimate the coalescent time of two alleles into one common ancestor. After one coalescent event, a sample of N alleles is reduced to N − 1. Time is considered to move backwards in the model until the time to a common ancestor for the entire sample is estimated. By modeling this stochastic process, a distribution of genealogies emerges, enabling the estimation of parameters of interest, such as the TMRCA
- DNA substitution models
evolutionary models that estimate the probability of nucleotide substitutions per site (a site is a position in a group of aligned DNA sequences) correcting for different factors (for review, see Ref. [31])
- Effective population size
the size of an ideal population that has the same variation as the actual population in its allele frequencies by random genetic drift
- Infinite-sites model
assumes that the mutation rate per site is extremely low, so the probability of multiple mutations at the same position is considered negligible. It assumes that no recombination occurs
- Molecular clock
the neutral theory of molecular evolution claims that most of the observed substitutions at DNA and amino acid level are neutral that is these substitutions have no effect on the ability of the organism to reproduce (fitness). In such a scenario, it is possible to assume a constant rate of evolution, enabling estimation of the time to a common ancestor from when the sequences diverged
- Neighbor-joining tree
phylogenetic reconstruction method based on an algorithm that minimizes the total length of the tree. This method does not explore or compare possible topologies [31]
- Tajima’s D test
summarizes the spectrum of substitutions found in an alignment of DNA sequences. It can be used to make preliminary inferences about deviations from the neutral model of molecular evolution, such as population growth and natural selection
References
- 1.Snow RW, et al. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garnham PCC. Malaria Parasites and Other Haemosporidia. Blackwell Scientific Publications; 1966. [Google Scholar]
- 3.Coatney RG, et al. The Primate Malaria. US Government Printing Office; 1971. [Google Scholar]
- 4.Hartl DL. The origin of malaria: mixed messages from genetic diversity. Nat Rev Microbiol. 2004;2:15–22. doi: 10.1038/nrmicro795. [DOI] [PubMed] [Google Scholar]
- 5.Joy DA, et al. Early origin and recent expansion of Plasmodium falciparum. Science. 2003;300:318–321. doi: 10.1126/science.1081449. [DOI] [PubMed] [Google Scholar]
- 6.Hughes AL, Verra F. Very large long-term effective population size in the virulent human malaria parasite Plasmodium falciparum. Proc Biol Sci. 2001;268:1855–1860. doi: 10.1098/rspb.2001.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Escalante AA, et al. Assessing the effect of natural selection in malaria parasites. Trends Parasitol. 2004;20:388–395. doi: 10.1016/j.pt.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Mendis K, et al. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001;64:97–106. doi: 10.4269/ajtmh.2001.64.97. [DOI] [PubMed] [Google Scholar]
- 9.Escalante AA, et al. The evolution of primate malaria parasites based on the gene encoding cytochrome b from the linear mitochondrial genome. Proc Natl Acad Sci U S A. 1998;95:8124–8129. doi: 10.1073/pnas.95.14.8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escalante AA, et al. A monkey’s tale: the origin of Plasmodium vivax as a human malaria parasite. Proc Natl Acad Sci U S A. 2005;102:1980–1985. doi: 10.1073/pnas.0409652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mu J, et al. Host switch leads to emergence of Plasmodium vivax malaria in humans. Mol Biol Evol. 2005;22:1686–1693. doi: 10.1093/molbev/msi160. [DOI] [PubMed] [Google Scholar]
- 12.Jongwutiwes S, et al. Mitochondrial genome sequences support ancient population expansion in Plasmodium vivax. Mol Biol Evol. 2005;22:1733–1739. doi: 10.1093/molbev/msi168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCutchan TF, et al. Evolutionary relatedness of Plasmodium species as determined by the structure of DNA. Science. 1984;225:808–811. doi: 10.1126/science.6382604. [DOI] [PubMed] [Google Scholar]
- 14.Severini C, et al. Risk of Plasmodium vivax malaria reintroduction in Uzbekistan: genetic characterization of parasites and status of potential malaria vectors in the Surkhandarya region. Trans R Soc Trop Med Hyg. 2004;98:585–592. doi: 10.1016/j.trstmh.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez-Morales AJ, et al. Impact of imported malaria on the burden of disease in northeastern Venezuela. J Travel Med. 2006;13:15–20. doi: 10.1111/j.1708-8305.2006.00006.x. [DOI] [PubMed] [Google Scholar]
- 16.Centers for Disease Control and Prevention. Multifocal autochthonous transmission of malaria – Florida. MMWR Morb Mortal Wkly Rep. 2004;53:412–413. [PubMed] [Google Scholar]
- 17.Roos C, et al. Molecular phylogeny of Mentawai macaques: taxonomic and biogeographic implications. Mol Phylogenet Evol. 2003;29:139–150. doi: 10.1016/s1055-7903(03)00076-9. [DOI] [PubMed] [Google Scholar]
- 18.Jablonski NG, et al. The influence of life history and diet on the distribution of catarrhine primates during the Pleistocene in eastern Asia. J Hum Evol. 2000;39:131–157. doi: 10.1006/jhev.2000.0405. [DOI] [PubMed] [Google Scholar]
- 19.Nordborg M, Krone S. Separation of time scales and convergence to the coalescent in structured populations. In: Slatkin M, Veuille M, editors. Modern Developments in Theoretical Population Genetics. Oxford University Press; 2002. pp. 194–232. [Google Scholar]
- 20.Sjödin P, et al. On the meaning and existence of an effective population size. Genetics. 2005;169:1061–1070. doi: 10.1534/genetics.104.026799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution Int J Org Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 22.Excoffier LG, et al. Arlequin ver. 30: an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 23.Bahlo M, Griffiths RC. Inference from gene trees in a subdivided population. Theor Popul Biol. 2000;57:79–95. doi: 10.1006/tpbi.1999.1447. [DOI] [PubMed] [Google Scholar]
- 24.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–589. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hammer MF, et al. Human population structure and its effects on sampling Y chromosome sequence variation. Genetics. 2003;164:1495–1509. doi: 10.1093/genetics/164.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imwong M, et al. Microsatellite variation, repeatarray length and population history of Plasmodium vivax. Mol Biol Evol. 2006;23:1016–1018. doi: 10.1093/molbev/msj116. [DOI] [PubMed] [Google Scholar]
- 27.Excoffier L. Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol. 2004;13:853–864. doi: 10.1046/j.1365-294x.2003.02004.x. [DOI] [PubMed] [Google Scholar]
- 28.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogeny. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 29.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 30.Kumar S, et al. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 31.Nei M, Kumar S. Molecular Evolution and Phylogenetics. Oxford University Press; 2000. [Google Scholar]
- 32.Bandelt HJ, et al. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]