

Abstract
Host vesicles composed of amphiphilic β-cyclodextrin CD1 recognize metal-coordination complexes of the adamantyl-functionalized ethylenediamine ligand L via hydrophobic inclusion in the β-cyclodextrin cavities at the vesicle surface. In the case of Cu(II) and L, the resulting coordination complex was exclusively CuL2, and the interaction with the host vesicles was intravesicular, unless the concentration of metal complex and vesicles was high (>0.1 mM). In the case of Ni(II) and L, a mixture was formed consisting of mainly NiL and NiL2, the interaction with the host vesicles was effectively intervesicular, and addition of the guest–metal complex resulted in aggregation of the vesicles into dense, multilamellar clusters even in dilute solution [1 μM Ni(II)]. The metal–L complex could be eliminated by a strong chelator such as EDTA, and the intervesicular interaction could be suppressed by a competitor such as unmodified β-cyclodextrin. The result from this investigation is that the strongest metal-coordination complex [Cu(II) with L] binds exclusively intravesicularly, whereas the weakest metal-coordination complex [Ni(II) with L] binds predominantly intervesicularly and is the strongest interfacial binder. These experimental observations are confirmed by a thermodynamic model that describes multivalent orthogonal interactions at interfaces.
Keywords: self-assembly, vesicles, cyclodextrins
Multivalent, noncovalent interactions at the interface of cell membranes are involved in a variety of biological processes such as cell–cell signaling, pathogen identification, and inflammatory response (1). Multivalent binding events have collective properties that are qualitatively and quantitatively different from the contributing monovalent interactions. For example, multivalent interactions lead to higher binding affinities and can afford larger contact areas between surfaces (1–3). Multivalency can be conveniently described by an effective concentration (Ceff) term that represents a probability of interaction between two interlinked reactive or complementary entities and symbolizes the concentration of one of the reacting or interacting functionalities as experienced by its counterpart (4, 5). Versatile model systems to investigate multivalent noncovalent interactions at the dynamic interface between cell membranes and the surrounding aqueous solution include self-assembled monolayers (SAMs) (6–11), nanoparticles (12–14), solid-supported lipid bilayers (15, 16), and bilayer vesicles (17–19).
Metal–ligand coordination has been exploited to generate complex molecular architectures with specific topology, high stability, and original properties in aqueous solution (16, 20, 21). The N-nitrilotriacetic acid–histidine interaction is particularly interesting in a biological context. N-nitrilotriacetic acid-functionalized lipids (22, 23) and SAMs (24, 25) have been used to immobilize proteins through multivalent interactions. In a comparable approach, the multivalent binding of Cu(II) ions to a membrane-bound dansyl-ethylenediamine conjugate (26) and of Zn(II) to a membrane-embedded dipicolylamine receptor (27) has been reported.
Orthogonal recognition motifs are intermolecular interactions that operate independently of each other so that no crossover or interference occurs (28, 29). Orthogonal interactions can lead to higher stoichiometries, increased specificity, and more complex architectures than when only a single interaction motif is used (30, 31). The ultimate example of orthogonal multivalency is DNA, for which every pair of matching single strands is orthogonal to other pairs. This approach has been used to obtain DNA nanostructures (32–35).
The interactions between biological membranes are mediated by proteins and carbohydrates, and (at least conceptually) cell–cell interactions can be mimicked by the interaction of liposomes and/or bilayer vesicles. Intervesicular interaction such as adhesion, aggregation, and fusion can be induced by changes of pH, temperature, ionic strength, etc. but also by ion binding and specific molecular recognition (36). For example, it has been shown that liposomes equipped with terpyridine ligands bind Fe(II) (37), whereas liposomes equipped with bipyridine ligands bind Ni(II) and Co(II) (38), and that such liposomes aggregate and fuse through intervesicular metal coordination. Liposomes can also interact via complementary hydrogen-bonding groups (39). Very recently, the lateral phase separation and concomitant aggregation of liposomes mediated by intervesicular Cu(II)–histidine coordination was also reported (40).
In this article, we demonstrate that the interactions of vesicles can be controlled by orthogonal multivalent interaction between a metal–ligand complex and an artificial receptor in the vesicle membrane. Thus, two orthogonal interaction motifs (metal complexation and host–guest inclusion) operate simultaneously. This study represents a step forward in complexity compared with the studies cited above (36–40), which invariably are based on a single recognition motif only. Our model system is based on the multivalent inclusion binding of a coordination complex at the surface of β-cyclodextrin (βCD) host vesicles. Previously we reported the preparation of amphiphilic α-, β-, and γ-CDs and the corresponding bilayer vesicles, which have the ability to recognize and bind guests by size-selective inclusion in the CD host cavities at the vesicle surface (41–45). We also recently described the multivalent interaction of the Cu(II) and Ni(II) complexes of adamantyl (Ad)-functionalized ethylenediamine (en) L to a SAM of βCD molecules (46). In the present study we demonstrate that divalent binding of the metal-coordination complex formed between L and Cu(II) leads to exclusive intravesicular binding on the surface of the host vesicles. Binding of the metal complex formed between L and Ni(II) leads to intervesicular binding and aggregation of the host vesicles.
Results and Discussion
Three complementary building blocks were used to study multivalent, orthogonal complexes at the surface of vesicle membranes. Vesicles of an amphiphilic CD1 were selected as model membranes. The preparation of amphiphilic CDs (Fig. 1) and their corresponding bilayer vesicles, as well as their size-selective inclusion interaction with suitable guests, have been described in detail (41, 44, 45). The Ad-functionalized en derivative L (Fig. 1) was used as a bifunctional ligand that can bind to βCD via hydrophobic inclusion of the Ad moiety and to suitable metal ions via bidentate coordination of the en moiety (47). The synthesis of L and its interaction with unmodified βCD and with βCD SAMs has been described (46). As the metal–ligand coordination motif, the Cu(II) and Ni(II) ions were used, with Cu(II) as a divalent coordinator and Ni(II) as a (potentially) trivalent coordinator. The basicity of the amino groups makes the complexation of L to metal cations pH-dependent. It is assumed that all L species present (protonated, unprotonated, or metal-complexed) are able to bind βCD. The oligo(ethylene glycol) chain provides enough length and flexibility to bind host vesicles of CD1 in a multivalent fashion while retaining water solubility and preventing nonspecific interactions.
Fig. 1.

Ad en conjugate L (Left), βCD (Center), and vesicle-forming amphiphilic βCD (Right) (CD1, n = 1–3).
Cu(II) forms divalent coordination complexes with en and en derivatives with a square-planar geometry (47). The cis and trans configurations (Fig. 2) are likely to behave similarly in this case, because the length of the linker between the Ad groups (≈3 nm) is significantly larger than the average distance between the cavities of CD1 in the vesicle membrane (2.2 nm) (44). The metal complex was prepared by mixing a solution of L and CuCl2 in a 2:1 molar ratio.
Fig. 2.

Distance between the Ad moieties in the most extended cis (Lower) and trans (Upper) configurations of CuL2 derived from Corey–Pauling–Koltun models.
The coordination of Cu(II) and L was not studied in detail. It is known that N-alkyl substitution of the en moiety hardly influences the protonation constants (48) but strongly reduces the metal-complex formation constants (49). Therefore, protonation (KHL = 2.00 × 1010 M−1 and KH2L = 3.39 × 107 M−1) (48) and metal-complex formation (KCuL = 8.71 × 109 M−1 and KCuL2 = 1.89 × 108 M−1) (49) constants corresponding to N-n-butylethylenediamine were used for L. These values lead to a pH dependence of the speciation of L in the absence and presence of Cu(II), as shown in Fig. 3. A pH of >6.5 favors the formation of CuL2.
Fig. 3.

Speciation of L in aqueous solution as a function of pH in the absence of metal(II) (Left), in the presence of Cu(II) (Center), and in the presence of Ni(II) (Right). [L]total = 1 mM; [Cu(II)]total = 0.5 mM; and [Ni(II)]total = 0.33 mM. In the presence of metal(II), solid lines represent metal(II) complexes, and dashed lines represent species without metal(II). Charges are omitted for clarity.
The interaction of L and CuL2 with unmodified βCD has been examined in detail by using isothermal titration calorimetry (ITC) (46). No significant pH effect on the host–guest complexation was observed. The average binding constant (Ka = 7.9 × 104 M−1) and the enthalpy of binding [ΔH° = −5.4 kcal·mol−1 (1 kcal = 4.18 kJ)] for L and βCD in the presence of Cu(II) are similar to the thermodynamic parameters obtained for the complexation of L with βCD in the absence of Cu(II). These observations led to the conclusion that the host–guest inclusion and the metal-coordination motifs are fully orthogonal and that there is no influence of pH on the host–guest complexation (46).
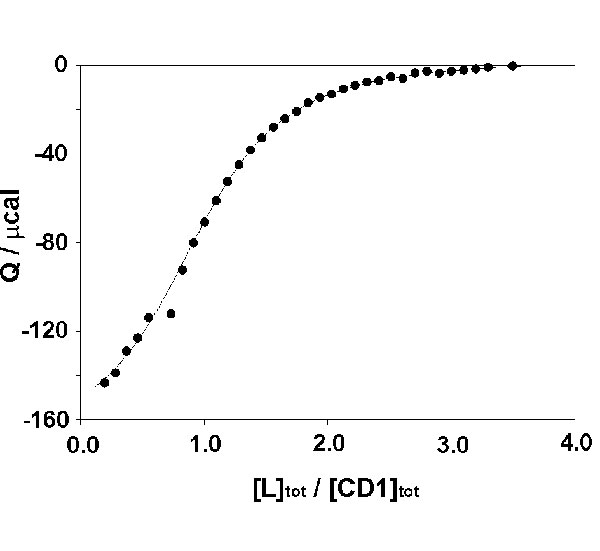
ITC was also used to study the interaction of L and vesicles of CD1. The titration was carried out by adding aliquots of guest L to a solution of host vesicles of CD1 at pH 9 (see supporting information (SI)). The concentration of CD1 was set at 50% of the total concentration, because ≈50% of CD1 reside at the inside of the unilamellar vesicles and are not accessible to guest L. The titration curve showed an inflection point at ≈1 equivalent of L added, which confirms the 1:1 stoichiometry of the interaction between CD1 and L. The titration data were fitted to a 1:1 binding model by using a nonlinear least-squares fitting procedure. The binding constant (Ka = 1.4 × 104 M−1) and the enthalpy of binding (ΔH° = −3.6 kcal·mol−1) for L and CD1 are similar to the thermodynamic parameters obtained for the complexation of other Ad derivatives with the CD1 (44). The decrease of Ka for CD1 (Ka = 1.4 × 104 M−1) compared with unmodified βCD (Ka = 7.9 × 104 M−1) is attributed to the presence of the ethylene glycol units on CD1 and is consistent with earlier findings (44). The ITC experiment demonstrates that the inclusion of the Ad group of L into the cavity of CD1 is not affected by the presence of the en group on L or the alkyl chains on CD1.
Transmission electron microscopy (TEM), dynamic light scattering (DLS), and UV-vis and fluorescence spectroscopy were used to investigate the interaction of CuL2 (added as CuCl2 and L in a 1:2 molar ratio) with CD1 vesicles. The addition of as much as 0.25 mM of CuL2 to a solution of vesicles of CD1 (60 μM at pH 9) did not result in significant changes in the average size distribution (measured by DLS; see SI) or the optical density of the solution (measured by UV-vis spectroscopy; see SI). At this pH and concentration, the CuL2 solution is expected to contain CuL2 as the major (if not exclusive) species. The vesicles in the presence of CuL2 were readily detected by using TEM (Fig. 4a). Interestingly, CuL2 functions as a positive staining agent that selectively binds to the vesicle surface, in contrast to conventional negative stains such as uranyl acetate (41, 44). From the TEM images it also seems that the vesicles do not tend to cluster in the presence of CuL2. We conclude that CuL2 adsorbs at the surface of the vesicles as a result of supramolecular divalent inclusion of CuL2 with CD1. DLS, UV-vis spectroscopy, and TEM consistently indicate that the interaction is exclusively intravesicular: both Ad anchors of CuL2 bind to the surface of one vesicle, and no interaction between vesicles is observed.
Fig. 4.

TEM of vesicles of CD1 (60 μM) in the presence of [CuL2] (60 μM) (a) and [NiL3] (6.0 μM) (b). pH = 9.
To quantify the interaction of CuL2 with CD1, a fluorescence titration of CuL2 to vesicles of CD1 (10 μM) containing 0.1% 7-nitrobenz-2-oxa-1,3-diazole (NBD) cholesterol was carried out in 10 mM carbonate buffer at pH 9 (45). The fluorescence intensity of NBD cholesterol embedded in the membrane of vesicles of CD1 is sensitive to the presence of Cu(II) within the Förster distance (i.e., at the vesicle surface). The fluorescence intensity of NBD cholesterol was efficiently quenched after adding CuL2 at a concentration as low as 10 μM (Fig. 5). The NBD fluorescence was recovered by addition of excess EDTA (data not shown). In addition, it was observed that when Cu(en)2 (a coordination complex without Ad anchors) was titrated to vesicles of CD1, no change in fluorescence occurred (Fig. 5). These observations strongly suggest that NBD fluorescence quenching resulted from a specific recognition of the divalent guest CuL2 at the host vesicle surface, which is not observed with Cu(en)2 because it lacks the proper recognition sites. The quenching of NBD fluorescence by CuL2 is relieved by the strong metal chelator EDTA, which scavenges the metal ion from the vesicle surface. From the Stern–Volmer plot, the apparent affinity constant Ka for the interaction of CuL2 and CD1 is estimated to be ≈2.4 × 105 M−1. The magnitude of this association constant is diagnostic for a divalent interaction of one guest complex CuL2 with two host molecules CD1. This observation is consistent with the divalent interaction of CuL2 with CD SAMs (46).
Fig. 5.

Quenching of NBD cholesterol fluorescence by Cu(II) with CuL2 (●) and with Cu(en)2 (○). [CD1] = 10 μM with embedded NBD cholesterol (1 mol%); pH = 9.
To increase our understanding of orthogonal multivalency at the host vesicle surface, we also prepared a metal complex of L by using Ni(II) as the metal ion. This divalent cation has a coordination number of six and tends to form complexes with an octahedral geometry. The protonation (KHL = 2.00 × 1010 M−1 and KH2L = 3.39 × 107 M−1) (48) and metal-complex formation (KNiL = 5.37 × 106 M−1, KNiL2 = 3.63 × 105 M−1, KNiL3 = 1.58 × 102 M−1) (49) constants of N-n-butylethylenediamine were used for the complexation of Ni(II) and L. It is evident that the metal-complexation constants for Ni(II) are many orders of magnitude lower than the metal-complexation constants for Cu(II). The stability constants lead to an expected pH dependence of the speciation of L in the presence of Ni(II), as shown in Fig. 3. Clearly, in aqueous solution at pH 9, divalent NiL2 dominates. Similar to the case of Cu(II), when complete orthogonality is assumed, all intrinsic stability constants for βCD complexation of any species of L are equal.
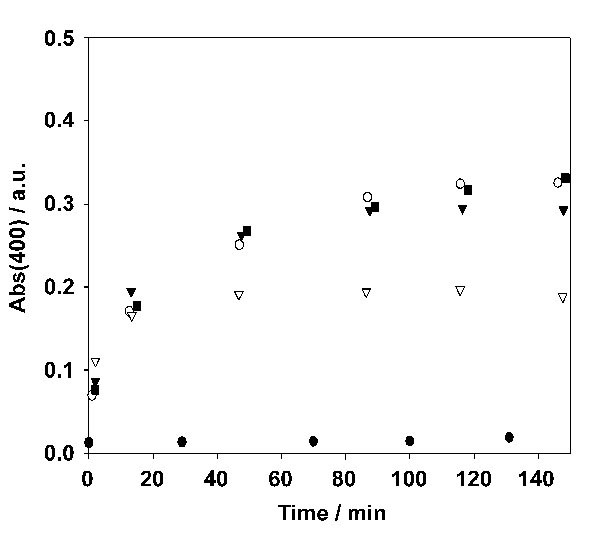
TEM, DLS, and UV-vis spectroscopy were used to investigate the interaction of NiL3 (added as NiCl3 and L in a 1:3 molar ratio) with vesicles of CD1. The addition of as little as 10 μM of NiL3 to a dilute solution of vesicles of CD1 (33 μM) resulted in a dramatic increase of the average size of the vesicles from ≈160 to >1,000 nm (see SI), accompanied by an increase in optical density from 0.01 to 0.3 (see SI). The original size distribution and the optical transparency of the vesicle solution were quickly recovered by addition of excess EDTA. Furthermore, the increase in optical density could be significantly suppressed by the addition of a large excess of unmodified βCD to the vesicle solution (see SI). In the presence of NiL3, the vesicles were readily observed by using TEM (Fig. 4b). Similar to CuL2, NiL3 also functions as a positive staining agent. From the TEM images, however, it is apparent also that the vesicles tend to cluster in the presence of NiL3. We conclude that NiL3 adsorbs at the surface of the vesicles because of supramolecular interaction of L with the cavities of CD1. DLS, UV-vis spectroscopy, and TEM consistently indicate that the interaction is significantly intervesicular: the Ad anchors bind to the surface of one vesicle but also between the surfaces of two vesicles, resulting in aggregation of vesicles. The intervesicular interaction can be suppressed by excess βCD in solution, and the interaction is quenched by the removal of Ni(II) by excess EDTA.
The host vesicles of CD1 were also investigated by using cryo-TEM (Fig. 6). For this methodology, the minimum concentration is ≈0.5 mM. The vesicles are clearly visible as spherical, mostly unilamellar vesicles with a diameter consistent with previous TEM observations and DLS measurements (41, 44). In the presence of either CuL2 or NiL3, the vesicles cluster and form extensive aggregates. Both metal–vesicle complexes give rise to a dense multilamellar arrangement with a bilayer spacing of ≈4.5 nm, which is consistent with the bilayer spacing observed with x-ray diffraction (44). In the presence of a large excess of EDTA, the metal–vesicle complexes dissociate, and vesicles are obtained again, although the fraction of multilamellar vesicles is higher than that in the original sample. From these observations we conclude that at the relatively high concentrations required for cryo-electron microscopy, both CuL2 and NiL3 adsorb at the surface of the vesicles and that the interaction is significantly intervesicular: the Ad anchors bind to the surface of one vesicle but also between the surfaces of two vesicles, resulting in aggregation and possibly also fusion of vesicles.
Fig. 6.

Cryo-TEM of vesicles of CD1 (Left) in the presence of CuL2 (Center) and NiL3 (Right). [CD1] ≈ 0.5 mM; and [CuL2] and [NiL3] ≈ 0.4 mM. The dark line in Left is the edge of the holey carbon film. (Scale bars, 200 nm.)
Finally, the interaction of vesicles of CD1 and βCD SAMs mediated by Cu(II) or Ni(II) and L was examined by surface plasmon resonance (SPR) spectroscopy (Fig. 7). To this end, a solution of vesicles of CD1 (10 μM) was flowed over a βCD SAM on gold (46). In the absence of Cu(II), Ni(II), and L, only a weak increase in reflectivity was observed after exposure of the βCD SAM to the vesicles, which instantly disappeared after rinsing with buffer. The small and rapid change in reflectivity likely results from some nonspecific interaction between the vesicles and the βCD SAM, as well as a difference in refractive index between the vesicle solution and the NaHCO3 buffer. If the βCD SAM is first saturated with CuL2 (100 μM) (46), a similar, reversible increase of reflectivity was observed after exposure of the βCD SAM to the vesicles, indicating that the vesicles do not adhere to a βCD SAM saturated with CuL2. In the case of NiL3 (10 μM), a strong and irreversible increase of reflectivity was observed, indicating that the vesicles adhere to a βCD SAM saturated with NiL3. Thus, these SPR measurements are fully consistent with the observations made above, that is, the interaction between vesicles of CD1, as well as between vesicles of CD1 and βCD SAMs, is mediated only by Ni(II) and L and not by Cu(II) and L.
Fig. 7.

SPR sensogram of vesicles of CD1 on βCD SAMs. Blue, vesicles of CD1 on βCD SAMs in the absence of metal(II) and L; red, vesicles of CD1 on βCD SAMs in the presence of Cu(II) and L; green, vesicles of CD1 on βCD SAMs in the presence of Ni(II) and L. Injections: □, 10 μM CD1 vesicles; ▾, 0.1 mM CuL2 or 10 μM NiL3; ●, 1 mM NaHCO3 buffer (pH 9). r.u., relative units.
The striking difference in the orthogonal multivalent interaction of Cu(II) and L versus Ni(II) and L with vesicles of CD1 can be interpreted conveniently in terms of the model for multivalent interactions on interfaces developed originally for multivalent guest molecules on SAMs of host molecules (5) and recently extended for metal–guest coordination complexes on SAMs of host molecules (46).
In the case of Cu(II) and L interacting with vesicles of CD1, the explanation is straightforward: as a result of the very high metal-coordination constants for Cu(II) and L, this complex is present exclusively in the CuL2 form at pH 9 (Fig. 3). Indeed, it was found that CuL2 binds as a divalent guest to a βCD SAM (46). The results presented here indicate that CuL2 also binds as a divalent guest to a vesicle of CD1. The multivalency model predicts that the fraction of CuL or free L bound to the vesicle surface is negligible at all concentrations of Cu(II) between 10−8 and 10−2 M (Fig. 8a). This prediction implies that throughout this concentration range, no intervesicular metal–ligand coordination can be expected, because all CuL2 complexes are fully saturated. It also predicts that at concentrations of Cu(II) >10−4 M, a significant fraction of CuL2 will bind with one Ad only to accommodate a higher number of CuL2§. This prediction implies that at concentrations of CuL2 >10−4 M, one may expect intervesicular interaction because of the complexation of a small (but significant) fraction of CuL2 bound with one L only and a small (but significant) fraction of empty host molecules on the vesicle surface, which explains the aggregation observed in the cryo-TEM images at [CuL2] = [CD1] ≈ 0.5 mM.
Fig. 8.

Surface-adsorbed species according to the multivalency model. (a) Cu(II) and L. (b) Ni(II) and L.
In the case of Ni(II) and L interacting with CD vesicles, the explanation is more elaborate: as a result of the weak metal-coordination constants for Ni(II) and L, this complex is present mostly in the NiL and NiL2 forms at pH 9 (Fig. 3). The trivalent complex NiL3 is not present in a significant amount at any concentration. It was found that Ni(II) and L bind predominantly as a divalent guest to a βCD SAM (46). The multivalency model predicts that the fractions of NiL2 and NiL, as well as free L bound to the vesicle surface, are significant at all concentrations of Ni(II) >10−6 M (Fig. 8b). This prediction implies that above this concentration, intervesicular metal–ligand coordination can be expected, because multiple vacant coordination sites are available on NiL2 and NiL bound to the vesicles, and also a significant amount of free L is present on the surface of the vesicles. This nicely explains the onset of vesicle aggregation at [Ni(II)] and [L] in the micromolar range. It is plausible that the NiL on one vesicle interacting with L on another vesicle constitutes the main driving force for aggregation at this concentration and not the more abundant NiL2 because of the much lower stability constant KNiL3 = 1.58 × 102 M−1 compared with KNiL2 = 3.63 × 105 M−1 (49). Furthermore, the model predicts that, similar to the case for Cu(II) and L, at concentrations of Ni(II) of ≈10−4 M a significant fraction of NiL2 will bind with one Ad only to accommodate a higher number of NiL2. This prediction implies that at concentrations of NiL2 >10−4 M, one may, in addition, expect intervesicular interaction resulting from the complexation of a small (but significant) fraction of NiL2 bound with one L only and a small (but significant) fraction of empty host molecules on the vesicle surface.
The results of the multivalency model are illustrated in Fig. 9. In the case of Cu(II) and L, the surface of the vesicles is entirely covered by CuL2 (Fig. 9a). No coordination vacancies and no free ligand L are available. No intervesicular interaction occurs if [CuL2] < 0.1 mM. In the case of Ni(II) and L, the surface of the vesicles is covered by a mixture of NiL2, NiL, and L (Fig. 9b), which implies that multiple coordination vacancies, as well as free ligand L, are available. If two vesicles collide, they can form numerous interaction pairs in the contact area, even at [Ni(II)] = [L] = 10−6 M. Hence, the vesicles rapidly aggregate as a result of intervesicular interaction (Fig. 9c).
Fig. 9.

Orthogonal multivalent interactions on interfaces and between interfaces. (a) Vesicle surface saturated with Cu(II) and L. CuL2 is the only significant species. (b) Vesicle surface saturated with Ni(II) and L. L largely saturates the CD sites, but many vacant coordination sites on Ni(II) and L are available. (c) Two vesicles interacting via multiple coordination sites on Ni(II) and L.
Conclusions
Vesicles of amphiphilic cyclodextrin CD1 recognize metal-coordination complexes with Ad ligands via hydrophobic inclusion in the host cavities at the vesicle surface. In the case of divalent Cu(II) complexes, the interaction is predominantly intravesicular unless the concentration of metal complex and vesicles is high (>0.1 mM). In the case of Ni(II), the interaction is effectively intervesicular, and addition of the guest–metal complex results in aggregation of the vesicles into dense, multilamellar clusters even in dilute, micromolar solution. The valency of molecular recognition at the surface of vesicles and the balance between intravesicular and intervesicular interaction can be tuned by metal coordination of guest molecules. The paradoxical result from this investigation is that the strongest metal-coordination complex [Cu(II) with L] binds exclusively intravesicularly, whereas the weakest metal-coordination complex [Ni(II) with L] binds predominantly intervesicularly and is the strongest interfacial binder. These dynamic model systems are helpful to elucidate the interplay of molecular recognition and interfacial interactions that occur in cell–cell interactions and viral infection. In addition, the findings described here demonstrate the potential of multivalent, orthogonal supramolecular interactions to design materials at the nano scale by interfacial self-assembly.
Materials and Methods
Materials.
Chemicals were obtained from commercial sources and used as received. Solvents were purified according to standard laboratory methods. Millipore water with a resistivity >18 MΩ·cm was used in all our experiments. The synthesis of L was described previously (46). The synthesis of the amphiphilic CD1 and the preparation of unilamellar vesicles of CD1 have also been described (41, 42, 44). The preparation of βCD SAMs on gold substrates was described in detail previously (50).
Preparation of the Metal Complex of L with Cu(II) and Ni(II).
The complexes of Cu(II) and Ni(II) with L were prepared by adding aliquots of a concentrated solution of CuCl2 and NiCl2 in distilled water (Millipore) to a solution of L. The molar ratio of metal and L was maintained at exactly 1:2 [Cu(II)] and 1:3 [Ni(II)] to prevent the formation of metal hydroxides. After addition of the metal salts, the solutions were diluted into a 10 mM NaHCO3 buffer solution (pH = 9).
ITC.
ITC was performed at 25°C by using a Microcal (Amherst, MA) VP-ITC instrument with a cell volume of 1.4115 ml. Sample solutions were prepared in Millipore water. Five-microliter aliquots of a 5 mM solution of L were added to a 0.5 mM solution of CD1 in the calorimetric cell. The concentration of CD1 was set at 50% of the total concentration, because ≈50% of CD1 resides at the inside of the vesicles and is not accessible to L. (The available concentration of CD1 can also be used as a fitting parameter.)
TEM.
Samples for TEM were prepared on 200-mesh formvar-carbon-coated copper grids. A drop of vesicle solution with the appropriate concentration of metal(II) and L was left on the grid for 2 min and then gently blotted with filter paper. The samples were investigated in a JEOL 2000 transmission electron microscope operating at 80 kV. Samples for cryo-TEM were prepared by deposition of a few microliters of vesicle solution with the appropriate concentration of metal(II) and L on holey carbon-coated grids. After blotting the excess liquid, the grids were vitrified in liquid ethane and transferred to a Philips (Eindhoven, the Netherlands) CM 120 cryo-electron microscope equipped with a Gatan (Pleasanton, CA) model 626 cryostage operating at 120 kV. Micrographs were recorded under low-dose conditions with a slow-scan CCD camera.
DLS.
DLS measurements were carried out at room temperature by using Malvern (Worcestershire, U.K.) instrumentation. The vesicle solutions were filtered through 0.45-μm Gelman Acrodisk syringe filters before light-scattering measurements. Size distributions were obtained from a CONTIN analysis of the scattering data.
Fluorescence and UV-vis Spectroscopy.
Fluorescence spectroscopy was performed on an Edinburgh (Edinburgh, U.K.) FS900 fluorospectrophotometer in which a 450-W xenon arc lamp was used as an excitation source. Signals were detected by a Peltier (Bedford, NH) element cooled, red-sensitive, Hamamatsu (Middlesex, NJ) R928 photomultiplier system. The excitation wavelength is 480 nm for NBD cholesterol, and emission was recorded at 530 nm. Fluorescence titrations were carried out in 10 mM carbonate buffer at pH 9. The optical density at 400 nm was recorded on a Varian Cary 3E UV-vis spectrophotometer in a time interval. The sample solutions were prepared in 10 mM carbonate buffer at pH 9.
SPR Spectroscopy.
The SPR setup was obtained from Resonant Probes (Goslar, Germany) (51). The sample cell is mounted on top of a θ–2θ goniometer with the detector measuring the reflectivity changes as a function of the angle of incidence of the p-polarized incoming laser beam. The incoming s/p laser beam passes through a beam splitter, which splits the p- and the s-polarized light. The s-polarized light is conducted to a reference detector. The p-polarized light passes a beam-expanding unit (spatial filter) with a pinhole (25 μm) for spectral cleaning and control of the intensity of p-polarized light and is collected into a photodiode detector. Titrations were measured in real time by recording the changes in the reflectivity in the fixed-angle mode (55.2°). SPR spectroscopy was performed under continuous flow by using a peristaltic pump at 0.5 ml/min.
Modeling.
The speciation of L in solution (Fig. 3) was calculated as described (46) by using the parameters provided below. The thermodynamic model for multivalent interaction at surfaces has been described in detail for interactions on host SAMs (5, 46). For vesicles of CD1, the following parameters were used: [CD1] = 10−6 M; Ki = 1.4 × 104 M−1; and Ceff,max = 0.1 M. The solution pH was set at 9, and the solution concentration of βCD was set at 0 M. For the interaction with Cu(II) and L, the following parameters were used: KHL = 2.00 × 1010 M−1; KH2L = 3.39 × 107 M−1; KCuL = 8.71 × 109 M−1; and KCuL2 = 1.89 × 108 M−1 (48, 49). For the interaction with Ni(II) and L, the following parameters were additionally used: KNiL = 5.37 × 106 M−1; KNiL2 = 3.63 × 105 M−1; and KNiL3 = 1.58 × 102 M−1 (49). In the calculation, the speciation of L on the vesicle surface was calculated as a function of the concentration of Cu(II) and L and the concentration of Ni(II) and L, respectively. The ratio of Cu(II) to L was set at 1:2. The ratio of Ni(II) to L was set at 1:3. Substitution of the equilibrium constants, [CD1], the pH, Ki, and Ceff,max into the mass balances for L, Cu(II), and Ni(II) provides the speciation of L at the surface of the host vesicles (46). The surface coverage of the vacant recognition sites (Fig. 8) was calculated from a summation of the relevant surface species:
In the case of [Ad]vacant, the last two terms only apply to Ni(II) and L and were found to be negligible under all conditions described here. The vacant ML2 sites ([ML2]vacant) are relevant only for Ni(II) and L, because the CuL2 complex is coordinatively saturated. Fig. 8 provides the surface coverage of vacant recognition sites ΓX of any of the above-named sites X as:
Supplementary Material
Acknowledgments
We are grateful for financial support by the Council for Chemical Sciences of The Netherlands Organization for Scientific Research in the Young Chemists program Grant 700.50.522 (to J.H.).
Abbreviations
- SAM
self-assembled monolayer
- CD
cyclodextrin
- Ad
adamantyl
- en
ethylenediamine
- ITC
isothermal titration calorimetry
- TEM
transmission electron microscopy
- DLS
dynamic light scattering
- NBD
7-nitrobenz-2-oxa-1,3-diazole
- SPR
surface plasmon resonance.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0611123104/DC1.
When the concentration of CuL2 increases, intermolecular monovalent binding of a second CuL2 complex from solution to an empty cavity of CD1 becomes more likely relative to intramolecular divalent binding of CuL2.
References
- 1.Mammen M, Choi SK, Whitesides GM. Angew Chem Int Ed. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Lundquist JJ, Toone EJ. Chem Rev (Washington, DC) 2002;102:555–578. doi: 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]
- 3.Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulder A, Auletta T, Sartori A, Del Ciotto S, Casnati A, Ungaro R, Huskens J, Reinhoudt DN. J Am Chem Soc. 2004;126:6627–6636. doi: 10.1021/ja0317168. [DOI] [PubMed] [Google Scholar]
- 5.Huskens J, Mulder A, Auletta T, Nijhuis CA, Ludden MJW, Reinhoudt DN. J Am Chem Soc. 2004;126:6784–6797. doi: 10.1021/ja049085k. [DOI] [PubMed] [Google Scholar]
- 6.Mann DA, Kanai M, Maly DJ, Kiessling LL. J Am Chem Soc. 1998;120:10575–10582. [Google Scholar]
- 7.Rao JH, Yan L, Xu B, Whitesides GM. J Am Chem Soc. 1999;121:2629–2630. [Google Scholar]
- 8.Metallo SJ, Kane RS, Holmlin RE, Whitesides GM. J Am Chem Soc. 2003;125:4534–4540. doi: 10.1021/ja030045a. [DOI] [PubMed] [Google Scholar]
- 9.Smith EA, Thomas WD, Kiessling LL, Corn RM. J Am Chem Soc. 2003;125:6140–6148. doi: 10.1021/ja034165u. [DOI] [PubMed] [Google Scholar]
- 10.Crespo-Biel O, Péter M, Bruinink CM, Ravoo BJ, Reinhoudt DN, Huskens J. Chemistry. 2005;11:2426–2432. doi: 10.1002/chem.200400393. [DOI] [PubMed] [Google Scholar]
- 11.Nijhuis CA, Yu F, Knoll W, Huskens J, Reinhoudt DN. Langmuir. 2005;21:7866–7876. doi: 10.1021/la051156l. [DOI] [PubMed] [Google Scholar]
- 12.Fantuzzi G, Pengo P, Gomila R, Ballester P, Hunter CA, Pasquato L, Scrimin P. Chem Commun. 2003:1004–1005. doi: 10.1039/b211544a. [DOI] [PubMed] [Google Scholar]
- 13.Barrientos AG, De La Fuente JM, Rojas TC, Fernández A, Penadés S. Chemistry. 2003;9:1909–1921. doi: 10.1002/chem.200204544. [DOI] [PubMed] [Google Scholar]
- 14.Lin CC, Yeh YC, Yang CY, Chen GF, Chen YC, Wu YC, Chen CC. Chem Commun. 2003:2920–2921. doi: 10.1039/b308995a. [DOI] [PubMed] [Google Scholar]
- 15.Sackmann E. Science. 1996;271:43–48. doi: 10.1126/science.271.5245.43. [DOI] [PubMed] [Google Scholar]
- 16.Radler U, Mack J, Persike N, Jung G, Tampé R. Biophys J. 2000;79:3144–3152. doi: 10.1016/S0006-3495(00)76548-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thibault RJ, Galow TH, Turnberg EJ, Gray M, Hotchkiss PJ, Rotello VM. J Am Chem Soc. 2002;124:15249–15254. doi: 10.1021/ja026418+. [DOI] [PubMed] [Google Scholar]
- 18.Espuelas S, Haller P, Schuber F, Frisch B. Bioorg Med Chem Lett. 2003;13:2557–2560. doi: 10.1016/s0960-894x(03)00472-4. [DOI] [PubMed] [Google Scholar]
- 19.Lee HK, Park KM, Jeon YJ, Kim D, Oh DH, Kim HS, Park CK, Kim K. J Am Chem Soc. 2005;127:5006–5007. doi: 10.1021/ja042172s. [DOI] [PubMed] [Google Scholar]
- 20.Holliday BJ, Mirkin CA. Angew Chem Int Ed. 2001;40:2022–2043. [PubMed] [Google Scholar]
- 21.Cotton FA, Lin C, Murillo CA. Proc Natl Acad Sci USA. 2002;99:4810–4813. doi: 10.1073/pnas.012567599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorn IT, Eschrich R, Seemuller E, Guckenberger R, Tampé R. J Mol Biol. 1999;288:1027–1036. doi: 10.1006/jmbi.1999.2714. [DOI] [PubMed] [Google Scholar]
- 23.Thess A, Hutschenreiter S, Hofmann M, Tampé R, Baumeister W, Guckenberger R. J Biol Chem. 2002;277:36321–36328. doi: 10.1074/jbc.M202145200. [DOI] [PubMed] [Google Scholar]
- 24.Gamsjaeger R, Wimmer B, Kahr H, Tinazli A, Picuric S, Lata S, Tampé R, Maulet Y, Gruber HJ, Hinterdorfer P, et al. Langmuir. 2004;20:5885–5890. doi: 10.1021/la0498206. [DOI] [PubMed] [Google Scholar]
- 25.Tinazli A, Tang JL, Valiokas R, Picuric S, Lata S, Piehler J, Liedberg B, Tampé R. Chemistry. 2005;11:5249–5259. doi: 10.1002/chem.200500154. [DOI] [PubMed] [Google Scholar]
- 26.Doyle EL, Hunter CA, Phillips HC, Webb SJ, Williams NH. J Am Chem Soc. 2003;125:4593–4599. doi: 10.1021/ja021048a. [DOI] [PubMed] [Google Scholar]
- 27.Jiang H, Smith BD. Chem Commun. 2006:1407–1409. doi: 10.1039/b517940h. [DOI] [PubMed] [Google Scholar]
- 28.Fyfe MCT, Stoddart JF. Coord Chem Rev. 1999;183:139–155. [Google Scholar]
- 29.Hofmeier H, Schubert US. Chem Commun. 2005:2423–2432. doi: 10.1039/b419171d. [DOI] [PubMed] [Google Scholar]
- 30.Thalladi VR, Goud BS, Hoy VJ, Allen FH, Howard JAK, Desiraju GR. Chem Commun. 1996:401–402. [Google Scholar]
- 31.Funeriu DP, Lehn JM, Baum G, Fenske D. Chemistry. 1997;3:99–104. [Google Scholar]
- 32.Rosi NL, Mirkin CA. Chem Rev (Washington, DC) 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- 33.Samori B, Zuccheri G. Angew Chem Int Ed. 2005;44:1166–1181. doi: 10.1002/anie.200400652. [DOI] [PubMed] [Google Scholar]
- 34.Liao SP, Seeman NC. Science. 2004;306:2072–2074. doi: 10.1126/science.1104299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winfree E, Liu FR, Wenzler LE, Seeman NC. Nature. 1998;394:539–544. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 36.Paleos CM, Sideratou Z, Tsiorvas D. ChemBioChem. 2001;2:305–310. doi: 10.1002/1439-7633(20010504)2:5<305::AID-CBIC305>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 37.Constable EC, Meier W, Nardin C, Mundwiler S. Chem Commun. 1999:1483–1484. [Google Scholar]
- 38.Richard A, Marchi-Artzner V, Lalloz MN, Brienne MJ, Artzner F, Gulik-Krzywicki T, Guedeau-Boudeville MA, Lehn JM. Proc Natl Acad Sci USA. 2004;101:15279–15284. doi: 10.1073/pnas.0406625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marchi-Artzner V, Gulik-Krzywicki T, Guedeau-Boudeville MA, Gosse C, Sanderson JM, Dedieu JC, Lehn JM. ChemPhysChem. 2001;2:367–376. doi: 10.1002/1439-7641(20010618)2:6<367::AID-CPHC367>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 40.Mart RJ, Liem KP, Wang X, Webb SJ. J Am Chem Soc. 2006;128:14462–14463. doi: 10.1021/ja0657612. [DOI] [PubMed] [Google Scholar]
- 41.Ravoo BJ, Darcy R. Angew Chem Int Ed. 2000;39:4324–4326. doi: 10.1002/1521-3773(20001201)39:23<4324::AID-ANIE4324>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 42.Mazzaglia A, Donohue R, Ravoo BJ, Darcy R. Eur J Org Chem. 2001:1715–1721. [Google Scholar]
- 43.Ravoo BJ, Jacquier JC, Wenz G. Angew Chem Int Ed. 2003;42:2066–2070. doi: 10.1002/anie.200350923. [DOI] [PubMed] [Google Scholar]
- 44.Falvey P, Lim CW, Darcy R, Revermann T, Karst U, Giesbers M, Marcelis ATM, Lazar A, Coleman AW, Reinhoudt DN, et al. Chemistry. 2005;11:1171–1180. doi: 10.1002/chem.200400905. [DOI] [PubMed] [Google Scholar]
- 45.Lim CW, Ravoo BJ, Reinhoudt DN. Chem Commun. 2005:5627–5629. doi: 10.1039/b510540d. [DOI] [PubMed] [Google Scholar]
- 46.Crespo-Biel O, Lim CW, Ravoo BJ, Reinhoudt DN, Huskens J. J Am Chem Soc. 2007;129:17024–17032. doi: 10.1021/ja0637705. [DOI] [PubMed] [Google Scholar]
- 47.Mellor DP. Chem Rev (Washington, DC) 1943;33:137–183. [Google Scholar]
- 48.Basolo F, Murmann RK. J Am Chem Soc. 1952;74:2373–2374. [Google Scholar]
- 49.Basolo F, Murmann RK. J Am Chem Soc. 1952;74:5239–5246. [Google Scholar]
- 50.Beulen MJW, Bügler J, De Jong MR, Lammerink B, Huskens J, Schönherr H, Vancso GJ, Boukamp BA, Wieder H, Offenhäuser A, et al. Chemistry. 2000;6:1176–1183. doi: 10.1002/(sici)1521-3765(20000403)6:7<1176::aid-chem1176>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- 51.Aust EF, Ito S, Sawondny M, Knoll W. Trends Polym Sci. 1994;2:313–323. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}