

Abstract
Amprenavir is a protease inhibitor that has been shown to have secondary peaks postulated to be due to enterohepatic recycling. We propose a model to describe the pharmacokinetics of amprenavir which accommodates the secondary peak(s). A total of 82 healthy human immunodeficiency virus (HIV)-seronegative subjects were administered a single 600-mg dose of amprenavir as part of adult AIDS Clinical Trials Group protocol A5043. Serial blood samples were obtained over 24 h. Samples were analyzed for amprenavir and fit to a compartmental model using ADAPT II software, with all relevant parameters conditional with respect to bioavailability. The model accommodated secondary peaks by incorporating clearance out of the central compartment with delayed instantaneous release back into the gut compartment. The data were weighted by the inverse of the estimated measurement error variance; model discrimination was determined using Akaike's Information Criteria. A total of 76 subjects were evaluable in the study analysis. The data were best fit by a two-compartment model, with 98.7% of the subjects demonstrating a secondary peak. Amprenavir had a mean total clearance of 1.163 liters/h/kg of body weight (0.7), a central volume of distribution of 1.208 liters/kg (0.8), a peripheral volume of distribution of 8.2 liters/kg (0.81), and distributional clearance of 0.04 liters/h/kg (0.81). The time to the secondary peak was 7.86 h (0.17), and clearance into a recycling compartment was 0.111 liters/kg/h (0.74). Amprenavir pharmacokinetics has been well described using a two-compartment model with clearance to a recycling compartment and release back into the gut. The nature of the secondary peaks may be an important consideration for the interpretation of amprenavir plasma concentrations during therapeutic drug monitoring.
Many potent antiretroviral therapy regimens utilize human immunodeficiency virus type 1 (HIV-1) protease inhibitors as a backbone of HIV therapy. Amprenavir (APV) is a protease inhibitor approved in 2001 for the treatment of HIV-infected patients in combination with other antiretroviral therapies. With the increasing incidence of resistance, many steps have been taken to optimize antiretroviral systemic exposure such as adaptive feedback control (also known as therapeutic drug monitoring) for high-risk patients as well as patients who fail therapy, the study of relationships between plasma concentrations and intracellular concentrations in order to relate these to pharmacodynamic effects, and the study of drug-drug, drug-food, or drug-disease interactions. These strategies have in common a quest for better ways to optimize drug exposure by understanding the pharmacokinetics (PK) of individual antiretrovirals within combination regimens.
Published PK characterizations of amprenavir have mainly used noncompartmental methods. In addition, valid pharmacokinetic models and parameters are required to design PK-pharmacodynamic trials using tools such as optimal sampling theory and the development of maximum a posteriori Bayesian estimators or for Monte Carlo simulations. Amprenavir was also reported by Sadler et al. as exhibiting secondary peaks approximately 6 to 12 h after dosing (14), and these peaks have not been subsequently analyzed or characterized using a pharmacokinetic model. The phenomenon known as enterohepatic recycling has been observed with other drugs, and multiple approaches to pharmacokinetic analysis have been reported previously (2, 8, 11-13, 19, 22).
The purpose of this study was to use compartmental models to describe amprenavir data obtained from healthy volunteers, and among those models, one in particular that will accommodate secondary peaks, characterize the pharmacokinetic parameters associated with the drug, and determine the apparent amount of drug responsible for the secondary peaks.
MATERIALS AND METHODS
The AIDS Clinical Trials Group (ACTG) A5043 protocol was used for an open-label pharmacokinetic study that involved, in part, the administration of a single oral dose of APV (600 mg). An intravenous catheter was placed, and blood samples were collected prior to dosing and at 1, 2, 3, 4, 5, 6, 8, 10, 12, and 24 h after dosing.
Study subjects.
Healthy HIV-1-seronegative adults who met the inclusion criteria were enrolled in the study after signing an informed consent form. Subjects were admitted to a General Clinical Research Center on the morning of the PK day in a state of having fasted since midnight of the prior evening. A standard protocol-specified breakfast was given 1/2 h before the dose was administered at 8:00 a.m. Subjects were then given a single dose of APV (600 mg) in the clinic followed by a 24-h sampling period. Additional meals were scheduled at noon and 6 p.m. but were not standardized among the subjects.
APV assay.
Plasma amprenavir concentrations were measured in the University at Buffalo ACTG Pharmacology Specialty Laboratory in an assay that also detects efavirenz, nelfinavir, M8, indinavir, ritonavir, and saquinavir with a validated liquid chromatography/mass spectrometry/mass spectrometry assay method (5, 10). Limits of detection and interassay variation were determined during method validation and were 16.3 ng/ml for amprenavir, with interassay variations of 12%, 12%, 10%, and 8% at 48 ng/ml, 240 ng/ml, 1,200 ng/ml, and 6,000 ng/ml, respectively.
Pharmacokinetic model.
The individual plasma concentrations were initially fit to candidate pharmacokinetic models using the maximum likelihood procedure available in ADAPT II software (3, 4). The maximum likelihood results were then used to compute maximum a posteriori Bayesian priors. The plasma concentrations were then fit by iterative two-stage analysis, a population analysis technique based on the methods described by Steimer et al. (20), and developed using the maximum a posteriori-Bayesian value estimator in ADAPT II.
Weighting was by the fitted inverse of the residual (error) variance. The observed standard deviation (SD) was described as linear with the fitted value (∨y) as follows: SD = SDslope · ∨y + SDintercept, where SDslope and SDintercept are the variance parameters initially empirically estimated based on assay error patterns and later fitted based on the data. The amount of drug in individual compartments and the area under the concentration-time curve were computed by numerically integrating the fitted model. Clearances and volumes were conditional based on systemic bioavailability (F) and were normalized by weight. Model discrimination was consistent with the rule of parsimony (9) based on Akaike's Information Criterion (AIC) (1).
RESULTS
A total of 82 subjects were enrolled, and 76 were evaluable for this analysis. Demographics for these subjects are presented in Table 1. The median age was 29. Nine subjects were excluded from the pharmacokinetic analysis due to dosing administration inconsistencies. The study medication was generally well tolerated. The final pharmacokinetic model was a linear, two-compartment model with a first-order input following a fitted lag time (Fig. 1). The final model accommodated secondary peaks with clearance into a recycling compartment and instantaneous release back into the gut after a fitted recycling time as shown in the following series of differential equations:
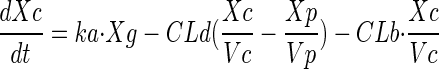 |
(1) |
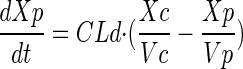 |
(2) |
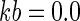 |
(3) |
If time > recycle time, then
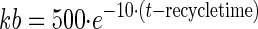 |
(4) |
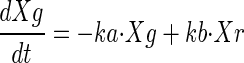 |
(5) |
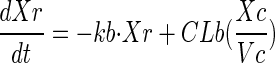 |
(6) |
A secondary peak was required by AIC for 98.7% of the subjects studied. The plasma concentration versus time curve data for a typical subject are shown in Fig. 2. The fit of the model to the data was excellent, with an overall r2 of 0.989 (observed = 0.989 · fitted + 0.00) and with the line of best fit not different from the line of identity (Fig. 3). The fitted SDslope and SDintercept terms were 0.1123 and 0.0124, respectively. The results of the pharmacokinetic analysis are shown in Table 2. The fraction of drug recycled was computed by obtaining the fraction of drug in the recycling compartment, before input back into the gut, relative to the administered dose. The “recycling time” was the fitted time (postdose) in which the recycling compartment emptied into the apparent absorptive site.
TABLE 1.
Enrollment and demographics
| Characteristic | Value for treatment arm (% value for data from all arms combined) |
|---|---|
| Age at baseline (yr) | |
| Median | 29 |
| 18 to 29 | 44 (54) |
| 30 to 39 | 20 (24) |
| 40 to 49 | 15 (18) |
| ≥50 | 3 (4) |
| Gender | |
| Male | 79 (96) |
| Female | 3 (4) |
| Race/ethnicity | |
| White non-Hispanic | 61 (74) |
| Black non-Hispanic | 16 (20) |
| Hispanic (regardless of race) | 2 (2) |
| Asian (Pacific Islander) | 3 (4) |
| Intravenous drug use at baseline | |
| Never | 82 (100) |
FIG. 1.

Pharmacokinetic model. F, systemic bioavailability; Xc, Xp, Xr, and Xg, amounts in the central, peripheral, “recycling,” and “gut” compartments (Cmpt), respectively; Vc, apparent volume of central compartment; Vp, apparent volume of the peripheral compartment; CLd, distributional clearance; CLb, clearance to the recycling compartment; CLt, total APV clearance; Kb, first-order rate constant for dispersion of the drug into the gut compartment at the time of recycling; TLag, lag time.
FIG. 2.

Representative amprenavir plasma concentrations (in milligrams per liter) versus time (in hours).
FIG. 3.

Observed amprenavir concentrations in plasma versus fitted concentrations in plasma. The diagonal is the line of best fit, which did not differ from the line of identity (r2 = 0.989).
TABLE 2.
Fitted amprenavir pharmacokinetic parameters with summary statistics
| Parametera | Mean | Median | Interquartile range |
|---|---|---|---|
| Vc/F (liters/kg) | 1.07 | 0.987 | 0.704-1.35 |
| Vp/F (liters/kg) | 7.65 | 5.61 | 2.39-11.8 |
| CLd (liters/h/kg) | 0.426 | 0.320 | 0.225-0.484 |
| CLt/F (liters/h/kg) | 1.17 | 0.917 | 0.680-1.40 |
| CLb/F (liters/h/kg) | 0.106 | 0.090 | 0.051-0.144 |
| Fraction recycled | 0.079 | 0.065 | 0.042-0.103 |
| Recycling time (h) | 7.88 | 7.89 | 7.42-8.28 |
| TLag (h) | 0.354 | 0.013 | 0.006-0.799 |
| ka (h−1) | 0.986 | 0.995 | 0.823-1.08 |
| kel (h−1) | 1.209 | 0.963 | 0.691-1.39 |
| kcp (h−1) | 0.496 | 0.363 | 0.203-0.546 |
| kpc (h−1) | 0.124 | 0.059 | 0.036-0.133 |
| α t1/2 (h) | 0.547 | 0.520 | 0.341-0.724 |
| β t1/2 (h) | 15.8 | 21.0 | 7.54-27.5 |
Vc, apparent volume of central compartment; F, systemic bioavailability; Vp, apparent volume of the peripheral compartment; CLd, distributional clearance; CLt, total APV clearance; CLb, clearance to the recycling compartment; TLag, lag time; ka, first-order rate constant of absorption; kel, elimination rate constant; kcp, elimination rate constant from central compartment to peripheral; kpc, elimination rate constant from peripheral compartment to central; α t1/2, distribution half-life; β t1/2, elimination half-life.
DISCUSSION
A number of recent reports have identified pharmacokinetic approaches to analyzing enterohepatic recycling in human and animal models (2, 8, 11-13, 19, 22). In this report, we utilized compartmental analysis to characterize the pharmacokinetics of amprenavir, in contrast to most published studies, which have used noncompartmental approaches (7, 14, 21). The secondary peak was modeled as a “very fast” first-order rate constant (essentially a bolus dose) after a fitted recycling time followed by first-order absorption.
The resultant terminal half-life shown in this study differed from reported values by 2.6-fold in spite of accounting for the apparent recycling component of the drug. The difference in half-life values may be due to inherent errors due to the use of noncompartmental techniques, which are not precise and are subjective. The presence of a secondary peak, also reported by Sadler et al. for half of their individual profiles at 6 to 12 h after the drug administration (16), is consistent with our finding of a median (interquartile range) time to recycling of 7.89 h (range, 7.42 to 8.28 h). In considering our data, it would be interesting to compare the amprenavir half-life values with and without the recycling component in the model. However, because our compartmental model has identified a prolonged terminal elimination half-life (rather than a half-life calculated from a noncompartmental approach), a follow-up study will be required to determine the influence of recycling on the terminal half-life we have identified using a compartmental modeling approach.
The relative size of the secondary peak for patients with HIV on chronic therapy is still unknown, but the presence of the peak was detected by Sadler et al. (15, 17, 18, 23, 24). Additional studies will have to be done to elucidate the cause of the secondary peak, particularly since this phenomenon might also be seen with fosamprenavir, as it is quickly hydrolyzed to amprenavir in the gut (6, 17, 18, 23, 24).
The apparent percentage of the dose that was recycled was 7.9% (4.2% to 10.3%), and the recycled amount was sufficient to show a secondary peak for 98.7% of the subjects studied. The clinical relevance of the secondary peak in patient data is unclear with regard to therapy; however, with the increasing utility of adaptive feedback control, the secondary peak may have clinical implications, since some patients are now participating in therapeutic drug monitoring studies and the second peak may be needed as a part of the overall interpretation of these plasma concentrations.
In summary, the development of a compartmental pharmacokinetic model for amprenavir accommodating secondary peaks, although the data were obtained with healthy volunteers, is a first step that will facilitate the use of novel study designs utilizing sparse drug sampling strategies and the development of adaptive feedback control algorithms for therapeutic drug monitoring.
Acknowledgments
The participation of the study volunteers is appreciated in helping to answer the clinical research questions in ACTG protocol A5043. The research staff at the following adult ACTG units and General Clinical Research Centers that participated in protocol A5043 were as follows: AIDS clinical trials units, Indiana University, Johns Hopkins University, Ohio State University, Stanford University, University of Colorado, University of Rochester, University of Washington, Vanderbilt University, and Washington University; adult ACTG pharmacology support laboratory, University at Buffalo School of Pharmacy and Pharmaceutical Sciences faculty and laboratory staff; ACTG operation center, Barbara Brizz; clinical trials specialist, statistical data analysis center, Yoninah Segal; data management center, Ann Walawander and Courtney Ashton; Division of AIDS, Elaine Ferguson; and pharmaceutical company representatives, Pascal J. de Caprariis (Roche), Alfred J. Saah (Merck), Mark Becker (Agouron), Mary Wire, and Mark Shelton (GlaxoSmithKline).
Support was provided by the following grants: National Institute of Allergy and Infectious Diseases U01-AI-25859, -25903, -25924, -27658, -27664, -27666, -27668, -32770, -38855, and -38858; and General Clinical Research Center support from the National Institutes of Health National Center of Research Resources M01-RR-00034, -00036, -00037, -00044, -00051, -00052, -00070, and -00750.
A.F., R.D., S.R., M.F.P., K.E.Y., and R.C.R. are members of the adult AIDS Clinical Trials Group.
Footnotes
Published ahead of print on 5 February 2007.
REFERENCES
- 1.Akaike, H. 1979. Bayesian extension of the minimum AIC procedure of autoregressive model fitting. Biometrika 66237-242. [Google Scholar]
- 2.Cremers, S., R. Schoemaker, E. Scholten, J. den Hartigh, J. Konig-Quartel, E. van Kan, L. Paul, and J. de Fijter. 2005. Characterizing the role of enterohepatic recycling in the interactions between mycophenolate mofetil and calcineurin inhibitors in renal transplant patients by pharmacokinetic modeling. Br. J. Clin. Pharmacol. 60249-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D'Argenio, D., and A. Schumitzky. 1997. ADAPT II user's guide: pharmacokinetic/pharmacodynamic system analysis software. Biomedical Simulations Resource, Los Angeles, CA.
- 4.D'Argenio, D. Z., and A. Schumitzky. 1979. A program package for simulation and parameter estimation in pharmacokinetic systems. Comput. Programs Biomed. 9115-134. [DOI] [PubMed] [Google Scholar]
- 5.Frerichs, V. A., R. DiFrancesco, and G. D. Morse. 2003. Determination of protease inhibitors using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 787393-403. [DOI] [PubMed] [Google Scholar]
- 6.Furfine, E. S., C. T. Baker, M. R. Hale, D. J. Reynolds, J. A. Salisbury, A. D. Searle, S. D. Studenberg, D. Todd, R. D. Tung, and A. Spaltenstein. 2004. Preclinical pharmacology and pharmacokinetics of GW433908, a water-soluble prodrug of the human immunodeficiency virus protease inhibitor amprenavir. Antimicrob. Agents Chemother. 48791-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goujard, C., I. Vincent, J. L. Meynard, N. Choudet, D. Bollens, C. Rousseau, D. Demarles, C. Gillotin, R. Bidault, and A. M. Taburet. 2003. Steady-state pharmacokinetics of amprenavir coadministered with ritonavir in human immunodeficiency virus type 1-infected patients. Antimicrob. Agents Chemother. 47118-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeong, E. J., X. Liu, X. Jia, J. Chen, and M. Hu. 2005. Coupling of conjugating enzymes and efflux transporters: impact on bioavailability and drug interactions. Curr. Drug Metab. 6455-468. [DOI] [PubMed] [Google Scholar]
- 9.Jusko, W. J. 1992. Guidelines for collection and analysis of pharmacokinetic data, p. 21-43. In W. E. Evans, W. J. Jusko, and J. J. Schentag (ed.), Applied pharmacokinetics, 3rd ed. Applied Therapeutics, Washington, DC.
- 10.Keil, K., V. A. Frerichs, R. DiFrancesco, and G. D. Morse. 2003. Reverse phase high performance liquid chromatography method for the analysis of amprenavir, efavirenz, indinavir, lopinavir, nelfinavir and its active metabolite, M8, ritonavir, and saquinavir in heparinized human plasma. Ther. Drug Monit. 25340-346. [DOI] [PubMed] [Google Scholar]
- 11.Kuchimanchi, K. R., C. Udata, T. P. Johnston, and A. K. Mitra. 2000. Pharmacokinetics, biliary excretion, and tissue distribution of novel anti-HIV agents, cosalane and dihydrocosalane, in Sprague-Dawley rats. Drug Metab. Dispos. 28403-408. [PubMed] [Google Scholar]
- 12.Liu, Y., Y. Liu, Y. Dai, L. Xun, and M. Hu. 2003. Enteric disposition and recycling of flavonoids and ginkgo flavonoids. J. Altern. Complement. Med. 9631-640. [DOI] [PubMed] [Google Scholar]
- 13.Roberts, M. S., B. M. Magnusson, F. J. Burczynski, and M. Weiss. 2002. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin. Pharmacokinet. 41751-790. [DOI] [PubMed] [Google Scholar]
- 14.Sadler, B. M., C. Gillotin, Y. Lou, J. J. Eron, W. Lang, R. Haubrich, and D. S. Stein. 2001. Pharmacokinetic study of human immunodeficiency virus protease inhibitors used in combination with amprenavir. Antimicrob. Agents Chemother. 453663-3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadler, B. M., C. Gillotin, Y. Lou, and D. S. Stein. 2001. Pharmacokinetic and pharmacodynamic study of the human immunodeficiency virus protease inhibitor amprenavir after multiple oral dosing. Antimicrob. Agents Chemother. 4530-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadler, B. M., C. D. Hanson, G. E. Chittick, W. T. Symonds, and N. S. Roskell. 1999. Safety and pharmacokinetics of amprenavir (141W94), a human immunodeficiency virus (HIV) type 1 protease inhibitor, following oral administration of single doses to HIV-infected adults. Antimicrob. Agents Chemother. 431686-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shelton, M. J., S. L. Ford, J. Borland, Y. Lou, M. B. Wire, S. S. Min, Z. G. Xue, and G. Yuen. 2006. Coadministration of esomeprazole with fosamprenavir has no impact on steady-state plasma amprenavir pharmacokinetics. J. Acquir. Immune Defic. Syndr. 4261-67. [DOI] [PubMed] [Google Scholar]
- 18.Shelton, M. J., M. B. Wire, Y. Lou, B. Adamkiewicz, and S. S. Min. 2006. Pharmacokinetic and safety evaluation of high-dose combinations of fosamprenavir and ritonavir. Antimicrob. Agents Chemother. 50928-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shou, M., W. Lu, P. H. Kari, C. Xiang, Y. Liang, P. Lu, D. Cui, W. B. Emary, K. B. Michel, J. K. Adelsberger, J. E. Brunner, and A. D. Rodrigues. 2005. Population pharmacokinetic modeling for enterohepatic recirculation in rhesus monkey. Eur. J. Pharm. Sci. 26151-161. [DOI] [PubMed] [Google Scholar]
- 20.Steimer, J. L., A. Mallet, J. L. Golmard, and J. F. Boisvieux. 1984. Alternative approaches to estimation of population pharmacokinetic parameters: comparison with the nonlinear mixed-effect model. Drug Metab. Rev. 15265-292. [DOI] [PubMed] [Google Scholar]
- 21.Veronese, L., J. Rautaureau, B. M. Sadler, C. Gillotin, J.-P. Petite, B. Pillegand, M. Delvaux, C. Masliah, S. Fosse, Y. Lou, and D. S. Stein. 2000. Single-dose pharmacokinetics of amprenavir, a human immunodeficiency virus type 1 protease inhibitor, in subjects with normal or impaired hepatic function. Antimicrob. Agents Chemother. 44821-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang, Y., A. Roy, L. Sun, and C. E. Lau. 1999. A double-peak phenomenon in the pharmacokinetics of alprazolam after oral administration. Drug Metab. Dispos. 27855-859. [PubMed] [Google Scholar]
- 23.Wire, M. B., K. L. Baker, L. S. Jones, M. J. Shelton, Y. Lou, G. J. Thomas, and M. M. Berrey. 2006. Ritonavir increases plasma amprenavir (APV) exposure to a similar extent when coadministered with either fosamprenavir or APV. Antimicrob. Agents Chemother. 501578-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wire, M. B., M. J. Shelton, and S. Studenberg. 2006. Fosamprenavir: clinical pharmacokinetics and drug interactions of the amprenavir prodrug. Clin. Pharmacokinet. 45137-168. [DOI] [PubMed] [Google Scholar]