

Abstract
Bacterial biofilms are communities of bacteria that are enclosed in an extracellular matrix. Within a biofilm the bacteria are protected from antimicrobials, environmental stresses, and immune responses from the host. Biofilms are often believed to have a highly developed organization that is derived from differential regulation of the genes that direct the synthesis of the extracellular matrix and the attachment to surfaces. The mycoplasmas have the smallest of the prokaryotic genomes and apparently lack complex gene-regulatory systems. We examined biofilm formation by Mycoplasma pulmonis and found it to be dependent on the length of the tandem repeat region of the variable surface antigen (Vsa) protein. Mycoplasmas that produced a short Vsa protein with few tandem repeats formed biofilms that attached to polystyrene and glass. Mycoplasmas that produced a long Vsa protein with many tandem repeats formed microcolonies that floated freely in the medium. The biofilms and the microcolonies contained an extracellular matrix which contained Vsa protein, lipid, DNA, and saccharide. As variation in the number of Vsa tandem repeats occurs by slipped-strand mispairing, the ability of the mycoplasmas to form a biofilm switches stochastically.
Biofilm formation can enhance the resistance of pathogens to antimicrobial agents (6) and immune surveillance (36). This has been attributed in part to the encasement within an extracellular matrix of the biofilm that isolates and protects the bacteria from the host immune response. The extracellular matrix of a biofilm is complex and usually contains lipid, protein, DNA, and exopolysaccharide (1, 2, 4, 38). Most studies have focused on the complexity of the molecular systems involved in the formation of the matrix constituents. The formation of biofilms by some bacteria is dependent on two-component and quorum-sensing systems that regulate macromolecules necessary for attachment and matrix synthesis (24). A recent trend is to describe biofilm formation in the context of a multicellular structure that develops as a result of programmed gene expression (7, 21).
Few studies address the formation of biofilms from the viewpoint of simplicity. The recent finding that mycoplasmas can form biofilms (19) provides us with a model in which microorganisms using minimally regulated genetic systems, or perhaps stochastically modulated genetic systems, can form biofilms. The mycoplasmas are the smallest and simplest of self-replicating cells (11, 22). Although some mycoplasmas can produce complex structures such as an attachment organelle, their genomes encode only a few hundred proteins. There are no known two-component regulatory systems or other recognizable global regulators in most species of mycoplasmas, including Mycoplasma pulmonis. The lack of such regulators can be attributed to the survival of the mycoplasmas in only one environmental niche: the animal host.
The variable surface antigen (Vsa) proteins of the murine respiratory pathogen M. pulmonis are associated with virulence (34) and mediate the susceptibility of the mycoplasma cells to complement (26, 27). A Vsa protein contains a conserved N-terminal region attached to one of several C-terminal tandem repeat regions (3, 28). Vsa phase variation results when DNA inversions recombine an alternate tandem repeat region with the vsa expression site (25, 29). Size variation results when slipped-strand DNA replication alters the length of the vsa tandem repeat region through the gain or loss of one or more repeat units. As both Vsa phase and size variations occur stochastically, cultures of mycoplasmas consist of mixed populations of cells that produce varied Vsa proteins.
Variation of the Vsa protein affects the surface characteristics of the mycoplasma cell (9, 37). Independent of the particular vsa gene that occupies the vsa expression site, mycoplasmas that produce a long form of the Vsa protein of about 40 tandem repeats do not adhere to polystyrene or erythrocytes. These mycoplasmas are resistant to killing by complement (26, 27). Mycoplasmas that produce a short Vsa protein with only a few tandem repeats (e.g., five repeats or less) adhere to polystyrene, adsorb erythrocytes, and are efficiently killed by complement.
We present here evidence that the length of the Vsa tandem repeat region affects the formation of a structured biofilm and an extracellular matrix. Cultures that produced the short Vsa protein formed biofilms attached to glass and polystyrene. Like biofilms that have been described for other bacteria, the matrix contained saccharide, lipid, protein, and DNA. Cultures of mycoplasmas that produced a long Vsa protein with many tandem repeats did not form a biofilm but did form free-floating microcolonies that contained an abundant extracellular matrix. These results demonstrate that the ability of the mycoplasmas to form a biofilm is modulated by stochastic events (slipped-strand mispairing within the vsa gene) in the absence of known, global gene regulatory systems.
MATERIALS AND METHODS
Mycoplasma strains.
Table 1 summarizes the characteristics of the strains of M. pulmonis used in this study. The R suffix on the strain and protein designations refers to the number of tandem repeats on the Vsa protein. All of the strains used in this study were derived from M. pulmonis strain CT and were grown in mycoplasma broth supplemented with 20% heat-inactivated (HIA) whole horse serum (HyClone) as described previously (12). Strains CT182-R3, CT182-R40, CTG-R5, CTG-R40, and CT228 are previously described mutants (26, 27, 29) that contain transposon Tn4001T (10) inserted in their genomes. Strains CT182-R3 and CT182-R40 produce a VsaA protein with 3 tandem repeats or about 40 tandem repeats, respectively. In strains CT228, CTG-R5, and CTG-R40, Tn4001T disrupts the gene encoding the HvsR recombinase that catalyzes vsa gene rearrangements, resulting in phase-locked strains that produce VsaA with 40 tandem repeats and VsaG with 5 or about 40 tandem repeats, respectively. Strain CTp12, which produces VsaA-R40, and strains CT39-6-2 and CTH.8, which produce VsaH, were previously described (26, 27) and do not contain transposon Tn4001T.
TABLE 1.
Summary of the characteristics of the M. pulmonis strains used in this study
| Strain | Vsa type | No. of tandem repeats | Ability to form a biofilma | Comments and/or reference |
|---|---|---|---|---|
| CTp12 | VsaA | 40 | − | 27 |
| CT182-R3-1 | VsaA | 3 | + | Isogenic sibling of CT182-R40 (27) |
| CT182-R3-2 | VsaA | 3 | + | |
| CT182-R40 | VsaA | 40 | − | Isogenic sibling of CT182-R3 (27) |
| CT228 | VsaA | 40 | − | Phase-locked (27) |
| CTG-R5 | VsaG | 5 | + | Phase-locked; isogenic sibling of CTG-R40 (26) |
| CTG-R40 | VsaG | 40 | − | Phase-locked; isogenic sibling of CTG-R5 (26) |
| CT39-6-2 | VsaH | 0 | + | 27 |
| CTH.8 | VsaH | 0 | + | 26 |
As determined by both macroscopic and microscopic observation.
Mycoplasmas were cultured in mycoplasma broth in polypropylene tubes (Fisher Scientific), in polystyrene flasks, or on glass coverslips at 37°C. To extend the time of growth of a culture, beginning on the second day of culture, the medium was changed daily.
Quantitative Vsa analysis.
The amount of VsaA protein, as a percentage of the total protein produced by a mycoplasma culture, was estimated by densitometric analysis of the VsaA protein bands in sodium dodecyl sulfate (SDS)-polyacrylamide gels that were stained with Coomassie blue. Cultures of M. pulmonis strain CT182-R3 were washed three times with phosphate-buffered saline (PBS), and the protein concentration was determined by the method of Bradford (Bio-Rad). Ten, 5, 2.5, and 1.25 micrograms of total mycoplasma proteins was resolved in 7.5% SDS-polyacrylamide gels and stained with Coomassie blue as previously described (26). The gels included lanes that contained 2.5, 1.25, 0.625, and 0.313 μg of bovine serum albumin (fraction V; Sigma) as standards. The bands containing Vsa protein were identified by Western analysis of parallel gels using Vsa-specific monoclonal antibody as described previously (26).The amount of protein in the VsaA major band was determined, and the percentage of VsaA protein produced was calculated as (amount of VsaA protein/amount of total protein) × 100. Although there are other minor bands of VsaA protein in a lane, this method provides a reasonable lower-limit estimate of amount of VsaA protein produced as a percentage of total mycoplasma protein production.
Scanning electron microscopy.
Mycoplasmas were grown in mycoplasma broth on plastic coverslips. The biofilms were washed three times in PBS, immersed in 2.5% glutaraldehyde in cacodylate buffer (0.1 M; pH 7.3) for 30 min, and rinsed three times in cacodylate buffer. The biofilms were immersed in 1.0% osmium tetroxide in cacodylate buffer for 1 h, followed by a brief rinse in distilled water. The biofilms were dehydrated through a 50%, 75%, 95%, and 100% series of washes in ethanol. This was followed by dehydration in a 1:1 (100% ethanol-hexamethyldisilizane) solution for 4 min and two final dehydrations in 100% hexamethyldisilizane for 4 min. The biofilms were dried overnight and sputter coated with gold palladium. The samples were observed with an International Scientific Instruments ISI-SX-40 scanning electron microscope.
Fluorescence microscopy.
Twenty-microliter samples were taken from cultures of mycoplasmas that produced the long R40 form of the Vsa protein, diluted with 50 μl of PBS, and placed on glass slides or nitrocellulose discs (Bio-Rad), and allowed to dry. Mycoplasmas that produced the short form of the Vsa protein were grown in mycoplasma broth on glass coverslips (Fisher Scientific). The coverslips were removed from the medium, washed once gently by being submerged in PBS, and allowed to dry. The dried cells were fixed in 10% neutral buffered formalin (4.0% formaldehyde, 0.4% sodium dihydrogen orthophosphate, 0.65% disodium hydrogen orthophosphate [pH 7.0]) for 15 min at room temperature, washed three times in PBS, and blocked for 30 min with PBS containing 5% HIA horse serum (HyClone). The cells were incubated with rabbit anti-VsaA immune serum diluted 200-fold in PBS containing 5% HIA horse serum, washed three times with PBS, incubated with donkey anti-rabbit antibodies (3) conjugated to either Alexa Fluor 488 or Alexa Fluor 647 (Molecular Probes, Eugene, OR; 10 μg/ml in PBS containing 5% HIA horse serum), counterstained with Hoechst 33342 at 20 μg/ml in water, and mounted to the slides with Pro Long Gold mounting medium (Molecular Probes).
For fluorescence microscopy with lectins, wheat germ agglutinin (WGA), Griffonia simplicifolia lectin-I (GS-I), and Griffonia simplicifolia lectin-II (GS-II) that were conjugated to fluorescein isothiocyanate (EY Laboratories) were used. Canavalia ensiformis lectin (ConA), which requires 4 mM Ca2+ in a Tris-based buffer for binding, and Arachis hypogaea lectin (PNA), which binds optimally in a bicarbonate-based buffer with a pH of 9.0, were diluted in PBS buffer without Ca2+ (pH 7.3) and used as controls for nonspecific binding. Mycoplasmas that were grown and fixed with formalin as described above were incubated with 50 μg/ml of the lectins in PBS at room temperature for 45 min, washed three times in PBS, and mounted as described above. PBS alone was also incubated with the mycoplasmas as a control for background.
To determine the amount of nonspecific binding of WGA to the mycoplasmas, 50 μg/ml of WGA was incubated with 1.0 μg/ml or 0.1 μg/ml of N,N′,N"-triacetylchitotriose (a trimer of N-acetylglucosamine) in PBS for 30 min at room temperature. The lectins were then incubated with the mycoplasmas, and fluorescence microscopy was performed as described above. As a control, WGA was incubated with PBS alone.
For staining with Congo red, the mycoplasmas were fixed as described above and incubated in an aqueous solution containing 0.1% Congo red for 30 min at room temperature (30). Red fluorescence was observed using 350-nm excitation and 715-nm emission filters.
To detect the presence of nonviable mycoplasma cells in a culture, Hoechst 33342 and propidium iodide (PI) were added to unfixed cultures of mycoplasmas to give a final concentration of 20 μg/ml. Fluorescence from PI was observed using 590-nm excitation and 617-nm emission filters. As PI does not enter cells with intact cell membranes, while Hoechst stain does diffuse through cell membranes, we interpreted mycoplasmas that stained with both PI and Hoechst stain as cells that were nonviable.
Mycoplasma cell membrane was detected by fluorescence microscopy using the membrane dye FM4-64FX (Molecular Probes). Mycoplasma cells were incubated with 20 μg/ml of FM4-64FX for 30 min at room temperature prior to fixation with formalin as described above. Fluorescence was observed using 555-nm excitation and 617-nm emission filters.
The cell-to-cell spacing was determined by digitally measuring the distances between mycoplasma cells and their three closest neighboring cells. The median cell-to-cell spacing of cells in the microcolonies or the biofilms was analyzed by the Mann-Whitney rank sum test (SigmaStat version 2.03).
Digital images were acquired at a magnification of ×1,600 with a Leica HC fluorescence microscope fitted with the Chroma 86012v2 filter set. Hoechst fluorescence was observed with 350-nm excitation and 475-nm emission filters. Alexa Fluor 488 or the fluorescein isothiocyanate-labeled lectins were observed with 495-nm excitation and 535-nm emission filters. Digital images were also acquired with an Olympus B16 laser scanning confocal microscope. Images were analyzed using the ImageJ image analysis software (version 1.33u; National Institutes of Health; http://rsb.info.nih.gov/ij/), the GNU Image Manipulation Program (GIMP version 2.2.8; http://www.gimp.org), or the MetaMorph Imaging System (version 7.0; Molecular Devices Corporation, Sunnyvale, CA).
RESULTS
Vsa production by M. pulmonis.
The positions of VsaA protein bands on SDS-polyacrylamide gels were identified by comparing Western blots reacted with Vsa-specific antibody to gels stained with Coomassie blue (Fig. 1). The upper major protein band of the VsaA protein from M. pulmonis strain CT182-R3 was in a region of the gel that had a low background of other proteins. Strain CT182-R40 had a minor protein band at the same position as the major VsaA-R3 protein band. However, Western blot analysis indicated this band was a minor VsaA protein band that is normally observed in the Vsa ladder pattern. The amount of protein in the VsaA-R3 band of CT182-R3 was estimated by comparison to bovine serum albumin standards. From several preparations of CT182-R3, the amount of VsaA-R3 was estimated to range from 7% to 10% of the total protein produced by the mycoplasma cultures.
FIG. 1.
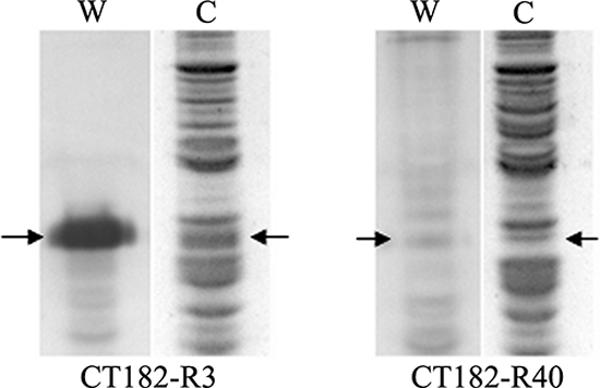
Estimation of VsaA production by M. pulmonis strain CT182-R3. Western blot analysis (lanes W) and SDS-polyacrylamide gels stained with Coomassie blue (lanes C) of the isogenic siblings M. pulmonis strains CT182-R3 and CT182-R40 are shown. The arrows point to the VsaA-R3 protein bands. For strain CT182-R3 this is the uppermost major VsaA band. For strain CT182-R40 the VsaA-R3 protein band is one of the lower minor VsaA bands. Densitometric analysis was performed on the major VsaA-R3 protein band of strain CT182-R40 in SDS-polyacrylamide gels that were stained with Coomassie blue.
Biofilm formation by mycoplasma cells is affected by the length of the Vsa tandem repeat.
Mycoplasmas that produced a short form of the VsaA, VsaG, or VsaH protein, i.e., strain CT182-R3, CT39-6-2, CT-H.8, or CTG-R5, grew adhered to untreated glass and formed a biofilm (Fig. 2A to E). In cultures of mycoplasmas that were grown overnight, individual cells or small clusters of cells were observed attached to the glass (Fig. 2B). In cultures that were grown for 1 to 2 days, the mycoplasmas formed a network of cells (Fig. 2C). Analysis of a series of images that represented different focal planes of the biofilms indicated that mycoplasma cells formed a honeycombed layer that contained numerous cavities (Fig. 2C). The cavities appeared to be continuous with the mycoplasma growth medium. Outgrowths of mycoplasma cells (Fig. 2A, D, and E), which resembled towers, extended from the honeycombed regions of all the biofilms that were observed. The diameters of the towers ranged from about 10 μm to greater than 50 μm, and channels were observed within the towers. After 3 days of growth, as the biofilms became increasingly dense, the prominence of the cavities was reduced and the diameter of the towers increased.
FIG. 2.

Immunofluorescent images of biofilms and microcolonies of M. pulmonis. VsaA epitopes are in green and DNA (Hoechst 33342) is in blue. The scale bars are in red. (A) Three-dimensional reconstruction of a biofilm formed by strain CT183-R3 after 3 days of growth on glass coverslips. The upper image shows the biofilm observed from an angle of elevation of 25 degrees, while the lower image is a horizontal reconstruction of the biofilm. The red arrows point to the tower structures. (B) Laser scanning confocal microscopy image of a small cluster of biofilm-forming mycoplasma cells grown overnight on glass coverslips. (C) Digital image of the honeycombed region. The red arrow points to the cavities in the honeycombs. (D) Scanning electron microscopic image of a tower structure. (E) Cross-sectional image of a biofilm tower acquired by laser scanning confocal microscopy. The arrow points to a channel within the tower. (F) Microcolony of a strain that produces VsaA-R40. The arrow points to extracellular VsaA epitopes. (G) Laser scanning confocal microscopy image of a region of cells adjacent to a microcolony. The red arrow denotes the web-like structures that contain VsaA epitopes. (H) Immunofluorescent image of a biofilm of mycoplasmas that produce the short form of VsaG (control showing that antibody is specific for VsaA).
In contrast, mycoplasmas that produced a long form of the Vsa protein (VsaA-R40 or VsaG-R40) did not grow adhered to glass or to polystyrene. They formed microcolonies that floated freely in the medium (Fig. 2F; see Fig. 4A) and ranged in diameter from about 10 μm to greater than 50 μm. The mycoplasma cells were distributed throughout the mass of the microcolonies, and a large amount of a matrix material was observed between the cells. Microcinematographic analysis revealed that microcolonies incubated with the DNA stain (Hoechst 33342) were pliable (see Fig. S1 in the supplemental material). We consistently observed this flexing (or changes in morphology) of unfixed microcolonies from cultures of mycoplasmas that produced the long Vsa protein.
FIG. 4.

Representative images from microcinematographic analysis of a viable CTp12 microcolony (A) and fluorescence microscopy of a CTG-R5 microcolony (B). The mycoplasma cultures were grown in polyurethane tubes and stained with Hoechst 33342. The scale bar represents 10 μm.
When the viable biofilms that were produced by strains CT182-R3 and CTG-R5 were costained with the DNA stains Hoechst 33342 and PI, few of the cells in the honeycombed regions bound PI (Fig. 3A). However, PI bound to large amounts of an amorphous material in the channels within the towers of the biofilms. In the microcolonies produced by strain CTp12, PI bound to very few of the mycoplasma cells (Fig. 3B).
FIG. 3.

DNA staining of a viable (nonfixed) biofilm (A) and a microcolony (B) with Hoechst 33342 and propidium iodide. The biofilms and microcolonies were from cultures that were grown for 2 days. Panel A shows both the honeycombed region of the biofilm and a tower structure (on the right side of panel A). Hoechst staining is shown in blue, and propidium iodide staining is shown in red. The scale bar represents 10 μm.
The length of the Vsa tandem repeat affects the extracellular spaces of the biofilms and microcolonies.
The median cell-to-cell spacing in the biofilms (9.25 μm; n = 56) was greater (P < 0.001) than the cell-to-cell spacing in the microcolonies of mycoplasmas that produced the long form of the Vsa protein (21.3 μm; n = 52). Cell-to-cell measurements within the towers of the biofilms were impractical to obtain, as these cells were too densely associated. Strains CT182-R3 and CTG-R5 formed microcolonies but not biofilms when they were grown in polypropylene tubes (Fig. 4B). The cell-to-cell measurements within these microcolonies, like the measurements between the cells in the towers of the biofilms, were too dense to obtain.
VsaA epitopes, observed by indirect immunofluorescence microscopy, were detected on the cells of all of the cultures of mycoplasmas that produced VsaA (Fig. 2). Control strains of mycoplasmas that produced VsaG had no detectable VsaA epitopes (Fig. 2H). After 2 to 3 days of growth of CT182-R3, VsaA epitopes were observed on the cells comprising the honeycombed region, and the greatest intensity of labeling was on the external cell layers of the towers of the biofilms (Fig. 2C and E). VsaA epitopes were rarely observed between the cells in the biofilm. However, the antibody apparently did not penetrate all areas of the biofilm, and the signal diminished as the distance from the outer surface of the tower increased. Few epitopes were detected on the cells that were located inside the towers. Additionally, VsaA epitopes were not detected on the mycoplasmas on the surface of the channels inside the towers. VsaA epitopes were detected on the external surface but not inside of the smaller clusters of cells in the biofilms that were 1 to 2 days old.
After 2 to 3 days of growth, VsaA epitopes were detected in between the cells of microcolonies of strains CT182-R40, CTp12, and CT228 (Fig. 2F and G). The extracellular Vsa epitopes appeared to be substantially more abundant in the microcolonies that produced VsaA-R40 (strains CTp12, CT228, and CT182-R40) (Fig. 2F) than in the biofilm produced by strain CT182-R3. Additionally, the extracellular epitopes were observed in fibrous, net-like structures in cultures of mycoplasmas that produced VsaA-R40 (Fig. 2G). These fibrous structures were not detected in the biofilms. In some areas of the microcolonies, DNA was present while VsaA epitopes were not detected.
Binding of lectins, Congo red, and membrane dyes to the mycoplasmas and matrix.
Amorphous material was detected in the extracellular spaces of the microcolonies and biofilms that were incubated with Congo red and observed by fluorescence microscopy. In the biofilms, large amounts of Congo red staining were observed throughout the mycoplasma towers of the biofilms and within the channels of the towers (Fig. 5A). Smaller amounts of the material were associated with the mycoplasmas in the honeycombed region of the biofilms. In the cultures of mycoplasmas that produced Vsa-R40, the amorphous material was abundant and was observed between the cells of the microcolonies (Fig. 5B). The amorphous material was not detected when mycoplasma broth alone was incubated with Congo red. Red fluorescence was not detected in mycoplasma biofilms or microcolonies that were incubated with Hoechst 33342 in the absence of Congo red.
FIG. 5.

Congo red fluorescence of a M. pulmonis tower (A) and a microcolony (B). Mycoplasma cells stained with Hoechst 33342 are shown in blue. The scale bar represents 10 μm.
All of the M. pulmonis strains tested bound the lectins WGA and GS-II (Fig. 6). The lectins ConA (Fig. 6C and F) and PNA (not shown) did not bind the mycoplasmas, and ConA was chosen as a negative control for subsequent experiments. GS-I, which was tested only with the biofilms, bound to strains CT182-R3, CTG-R5, CT39-6-2, and CTH.8. The towers of the biofilms bound the lectins to a greater extent than the honeycombed regions (Fig. 6B). The greatest intensity of binding by the lectins was observed in the channels within the towers. Analysis of microcolonies from the cultures that produced Vsa-R40 (including strains CTp12 [Fig. 6G] and CTG-R40 [Fig. 6H]) indicated that the lectins bound to copious amounts of saccharide between the cells. Whether the mycoplasmas produced a biofilm or a microcolony, the greatest degree of lectin binding was localized to regions between the mycoplasma cells.
FIG. 6.

Lectin binding to M. pulmonis biofilms that produce VsaA (strains CT182-R3 [A, B, and C] and CTp12 [G]) or VsaG (strains CTG-R5 [D, E, and F] and CTG-R40 [H]). The binding of GS-II (left panels), WGA (center panels), or ConA (right panels) is shown in green, and the DNA (Hoechst 33342) is shown in blue. The honeycombed region of the biofilm is shown in the optical cross-section in panel B (arrow). The red scale bars represent 10 μm.
The binding of WGA to the biofilms was inhibited when the lectin was incubated with as little as 0.1 μg/ml of N,N′,N"-triacetylchitotriose prior to its incubation with the biofilms (data not shown). The binding of WGA to the biofilms was not inhibited when the lectins were incubated with PBS prior to its reaction with the biofilms. This indicated that the binding of WGA to the biofilms was specific.
Staining of the mycoplasma membranes with the membrane dye FM4-64FX revealed an intense red fluorescent signal in the centers of the microcolonies and the towers of the biofilms (Fig. 7). The red fluorescence colocalized well with the DNA staining (Hoechst 33342). The channels within the towers contained an amorphous material that was intensely stained with the membrane dye and lightly stained with the Hoechst DNA stain (Fig. 7A and D). This same pattern of membrane staining was also observed in the microcolonies (Fig. 7B and E). No red fluorescent signal was detected when FM4-64FX was excluded from the protocol (Fig. 7F).
FIG. 7.

Detection of cell membranes in biofilms and microcolonies by using FM4-64FX. (A and D) Images of the same tower in a biofilm formed by M. pulmonis strain CT182-R3. (B and E) Images of the same microcolony formed by M. pulmonis strain CTp12. (C) Honeycombed region of the biofilm. The microcolony and the biofilm were stained with Hoechst 33342 (shown in blue) and FM4-46FX (shown in red). VsaA epitopes are in green. (F) Biofilm tower that was not stained with FM4-64FX (negative control). Red scale bars represent 10 μm. The yellow arrows indicate areas of intense membrane fluorescence and low DNA fluorescence.
DISCUSSION
The formation of biofilms by most microorganisms involves the regulation of genes that are essential for attachment to surfaces and the production of extracellular matrices (7, 20, 21, 31). Several mycoplasma species have recently been shown to form biofilms (19), but the macromolecules and the mechanisms that contribute to biofilm structure are unknown. The results presented here demonstrate that the length of the Vsa tandem repeats is associated with the formation of biofilms or microcolonies ands affect the interactions between the mycoplasma cells. As global regulatory systems are not known to exist in M. pulmonis and size variation of the Vsa protein occurs stochastically, the ability of M. pulmonis to form a biofilm is stochastic and may not involve any significant changes in gene regulation. Also, as the length of the Vsa protein and not the particular Vsa protein being produced (VsaA, VsaG, VsaH, or VsaI) affects the cell's susceptibility to complement and adsorption to erythrocytes (26), it suggests that the Vsa tandem repeat region nonspecifically modulates interactions at the mycoplasma surface. It is hypothesized that the long Vsa proteins sterically hinder interactions between the mycoplasma cell surface and the environment. As the Vsa protein constitutes a significant proportion (7% to 10%) of the total protein produced by the mycoplasmas, this would be consistent with the model in which Vsa can function as a steric shield.
Cultures of mycoplasmas that produced a short VsaA, VsaH, or VsaG protein formed biofilms on glass surfaces. Regardless of the particular Vsa protein (VsaA, VsaH, or VsaG) being produced, the structures of the biofilms were similar and they contained honeycombed regions and towers. The formation of a biofilm by a strain (CTG-R5) that was phase locked and could produce only VsaG indicates that phase variation of the Vsa proteins is not required for the formation of a biofilm. That only few cells in the honeycombed region bound PI, compared to the towers, suggested that little cell death occurs in the honeycombed region. As with other biofilms, the cavities of the honeycombed region may facilitate the uptake of nutrients (17, 18).
Mycoplasma strains that produced long Vsa protein formed microcolonies that did not adhere to glass. However, the microcolonies did share a characteristic that is similar to that of biofilms that are attached to surfaces. They produced an abundant matrix material. The distance between the cells in the microcolonies was greater than the distance between the cells in the biofilms, and the unfixed microcolonies appeared to be flexible and moved en masse. The extracellular matrix, as revealed by the amount of space between the cells and the abundance of lectin and Congo red binding, appeared to be substantially more abundant in the microcolonies than in the biofilms. These observations are consistent with the presence of a matrix that functions to hold the cellular mass together. As such, the microcolonies could be thought of as free-floating biofilms. As few cells in the microcolonies bound PI, it is unlikely that the extracellular matrix impedes nutrients that are critical for mycoplasma growth.
Consistent with what has been described for the extracellular matrices formed by other bacteria, the matrix formed by the mycoplasmas contained protein (VsaA epitopes), lipid, DNA, and saccharide (2, 21, 23, 36, 38). Vsa is anchored to the cell surface via the acyl moiety of this lipoprotein (3, 28). The VsaA epitopes could have been released into the extracellular matrix by blebbing of the mycoplasma cell membrane and/or cell death, as have been extensively described for other prokaryotes (16). In support of either of these mechanisms, VsaA epitopes were associated with the membrane dye FM4-64FX, and VsaA epitopes were not detected in the supernatants of the culture medium (not shown). This suggests that the extracellular VsaA epitopes remained attached to membrane.
The binding of lectins and Congo red to the microcolonies and the biofilms indicates that saccharide, perhaps copious amounts of it, occupies the spaces between the mycoplasma cells. Although Congo red has been shown to bind to a number of substrates (13, 15, 30), it interacts strongly with various polysaccharides (35). The staining of bacterial cultures with Congo red may provide a rapid method of identifying pathogenic bacterial species that produce extracellular matrices. The strong binding of Congo red and the lectins to material between the cells suggests that saccharides are important for matrix function. The binding of WGA and GS-II indicates that N-acetylglucosamine (GlcNAc) is present in a poly- or oligosaccharide. As the ligands for GS-I include α-galactose, the binding of GS-I to the mycoplasmas suggests that the composition of the saccharides in the matrix is complex. These saccharides are likely synthesized by the mycoplasma, as several enzymes that could utilize GlcNAc for polysaccharide synthesis and a glycosyltransferase are predicted from the M. pulmonis genome sequence (5). It is unlikely that the Vsa proteins are associated with absorbing the saccharides from the medium. As the towers and the microcolonies appeared to retain similar amounts of saccharide and as strains CT39-6-2 and CTH.8, which produce a Vsa protein with no tandem repeats, bound WGA, GS-I, and GS-II, the tandem repeat region of the Vsa protein is not necessary for retaining saccharides in the matrix.
The mycoplasmas that produced the longer Vsa-R40 protein contained a larger volume of matrix than did the mycoplasmas that produced the short Vsa-R3 or Vsa-R5 protein. This result was true regardless of whether the cells producing the short form of the Vsa protein were grown as a biofilm or grown as detached microcolonies in polypropylene tubes. These data indicate that the cell-to-cell spacing is affected by the length of the Vsa tandem repeat region. If the Vsa proteins contribute to the matrix by a mechanism that is independent of the saccharides, perhaps the Vsa tandem repeat regions interact with each other or some other ligand or affect the hydration of the matrix.
As extracellular matrices have been shown to impart structure to biofilms (7), it is likely that the main function of the matrix in the mycoplasmal biofilm and microcolonies is to hold the mycoplasma cell mass together. Whether the mycoplasmas form an attached biofilm or a free-floating microcolony apparently depends on the Vsa protein's modulation of attachment to surfaces. This is plausible, as the formation of a biofilm can result from a two-step process, i.e., the initial attachment of bacteria to a surface and the subsequent accumulation of a matrix (39). As microcolonies do not attach to surfaces, this indicates that coaggregation is not sufficient for the formation of biofilms on a surface as has been described for other microorganisms (14). Although the possibility that regulatory events contribute to the maturation of the mycoplasmal biofilm cannot be excluded, these results indicate that the length of the Vsa protein is the dominant factor contributing to biofilm formation.
The ability of M. pulmonis to form a biofilm may be a virulence factor. Biofilm formation may be an alternate mechanism to resist the host's immune system. M. pulmonis cells that produce the short VsaH protein are sensitive to complement lysis in vitro but survive in an immunocompetent host, indicating that the mycoplasma has an alternate mechanism to resist innate host defenses such as complement (8). Mycoplasma cells form aggregates and microcolonies on cell monolayers (33) and tracheal explants (32), suggesting that components of the biofilm, perhaps the towers, could form in the host. Our previous work demonstrating that mycoplasmas that produce the short Vsa protein are efficiently killed by complement was done with preparations of mycoplasma cells that were dispersed by gentle sonication. It remains to be determined whether the mycoplasmas in biofilms would be protected from the effects of complement or other immune components. It is plausible that the large amount of extracellular matrix material could impede interactions with complement or phagocytes.
Supplementary Material
Acknowledgments
We thank Portia Caldwell and Abha Soni for their expert technical assistance. Technical assistance for the scanning electron microscopy and laser scanning confocal microscopy was provided by Leigh Millican and Albert Tousson from the High-Resolution Imaging Facility at The University of Alabama at Birmingham.
This work was supported by Public Health Service grant AI64848 from the National Institutes of Health.
Footnotes
Published ahead of print on 1 December 2006.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Beveridge, T. 1999. Structures of gram-negative cell walls and their derived membrane vesicles. J. Bacteriol. 181:4725-4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beveridge, T. J., S. A. Makin, J. L. Kadurugamuwa, and Z. Li. 1997. Interactions between biofilms and the environment. FEMS Microbiol. Rev. 20:291-303. [DOI] [PubMed] [Google Scholar]
- 3.Bhugra, B., L. L. Voelker, N. Zou, H. Yu, and K. Dybvig. 1995. Mechanism of antigenic variation in Mycoplasma pulmonis: interwoven, site-specific DNA inversions. Mol. Microbiol. 18:703-714. [DOI] [PubMed] [Google Scholar]
- 4.Branda, S. S., A. Vik, L. Friedman, and R. Kolter. 2005. Biofilms: the matrix revisited. Trends Microbiol. 13:20-26. [DOI] [PubMed] [Google Scholar]
- 5.Chambaud, I., R. Heilig, S. Ferris, V. Barbe, D. Samson, F. Galisson, I. Moszer, K. Dybvig, H. Wroblewski, A. Viari, E. P. C. Rocha, and A. Blanchard. 2001. The complete genome sequence of the murine respiratory pathogen Mycoplasma pulmonis. Nucleic Acids Res. 29:2145-2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318-1322. [DOI] [PubMed] [Google Scholar]
- 7.Danese, P. N., L. A. Pratt, and R. Kolter. 2001. Biofilm formation as a developmental process. Methods Enzymol. 336:19-26. [DOI] [PubMed] [Google Scholar]
- 8.Denison, A. M., B. Clapper, and K. Dybvig. 2005. Avoidance of the host immune system through phase variation in Mycoplasma pulmonis. Infect. Immun. 73:2033-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dybvig, K., J. Alderete, H. L. Watson, and G. H. Cassell. 1988. Adsorption of mycoplasma virus P1 to host cells. J. Bacteriol. 170:4373-4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dybvig, K., C. T. French, and L. L. Voelker. 2000. Construction and use of derivatives of transposon Tn4001 that function in Mycoplasma pulmonis and Mycoplasma arthritidis. J. Bacteriol. 182:4343-4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dybvig, K., and L. L. Voelker. 1996. Molecular biology of mycoplasmas. Annu. Rev. Microbiol. 50:25-57. [DOI] [PubMed] [Google Scholar]
- 12.Gumulak-Smith, J., A. Teachman, A. H. Tu, J. W. Simecka, J. R. Lindsey, and K. Dybvig. 2001. Variations in the surface proteins and restriction enzyme systems of Mycoplasma pulmonis in the respiratory tract of infected rats. Mol. Microbiol. 40:1037-1044. [DOI] [PubMed] [Google Scholar]
- 13.Jin, L. W., K. A. Claborn, M. Kurimoto, M. A. Geday, I. Maezawa, F. Sohraby, M. Estrada, W. Kaminksy, and B. Kahr. 2003. Imaging linear birefringence and dichroism in cerebral amyloid pathologies. Proc. Natl. Acad. Sci. USA 100:15294-15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamaguchi, A., K. Nakayama, S. Ichiyama, R. Nakamura, T. Watanabe, M. Ohta, H. Baba, and T. Ohyama. 2003. Effect of Porphyromonas gingivalis vesicles on coaggregation of Staphylococcus aureus to oral microorganisms. Curr. Microbiol. 47:485-491. [DOI] [PubMed] [Google Scholar]
- 15.Kerstens, S., W. F. Decraemer, and J. P. Verbelen. 2001. Cell walls at the plant surface behave mechanically like fiber-reinforced composite materials. Plant Physiol. 127:381-385. [PMC free article] [PubMed] [Google Scholar]
- 16.Kuehn, M. J., and N. C. Kesty. 2005. Bacterial outermembrane vesicles and the host-pathogen interaction. Genes Dev. 19:2645-2655. [DOI] [PubMed] [Google Scholar]
- 17.Li, Y. H., P. C. Y. Lau, N. Tang, G. Svensater, R. P. Ellen, and D. G. Cvitkovitch. 2002. Novel two-component regulatory system involved in biofilm formation and acid resistance in Streptococcus mutans. J. Bacteriol. 184:6333-6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsh, E. J., H. Luo, and H. Wang. 2003. A three-tiered approach to differentiate Listeria monocytogenes biofilm-forming abilities. FEMS Microbiol. Lett. 228:203-210. [DOI] [PubMed] [Google Scholar]
- 19.McAuliffe, L., R. J. Ellis, K. Miles, R. D. Ayling, and R. A. J. Nicholas. 2006. Biofilm formation by mycoplasma species and its role in environmental persistence and survival. Microbiology 152:913-922. [DOI] [PubMed] [Google Scholar]
- 20.Miller, M. B., and B. L. Bassler. 2001. Quorum sensing in bacteria. Annu. Rev. Microbiol. 55:165-199. [DOI] [PubMed] [Google Scholar]
- 21.Parsek, M. R., and C. Fuqua. 2004. Biofilms 2003: emerging themes and challenges in studies of surface-associated microbial life. J. Bacteriol. 186:4427-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Razin, S., D. Yogev, and Y. Naot. 1998. Molecular biology and pathogenicity of mycoplasmas. Microbiol. Mol. Biol. Rev. 62:1094-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Renelli, M., V. Matias, R. Y. Lo, and T. J. Beveridge. 2004. DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiology 150:2161-2169. [DOI] [PubMed] [Google Scholar]
- 24.Schembri, M. A., K. Kjaergaard, and P. Klemm. 2003. Global gene expression in Escherichia coli biofilms. Mol. Microbiol. 48:253-267. [DOI] [PubMed] [Google Scholar]
- 25.Shen, X., J. Gumulak, H. Yu, C. T. French, N. Zou, and K. Dybvig. 2000. Gene rearrangements in the vsa locus of Mycoplasma pulmonis. J. Bacteriol. 182:2900-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simmons, W. L., A. M. Denison, and K. Dybvig. 2004. Resistance of Mycoplasma pulmonis to complement lysis is dependent on the number of Vsa tandem repeats: shield hypothesis. Infect. Immun. 72:6846-6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simmons, W. L., and K. Dybvig. 2003. The Vsa proteins modulate susceptibilty of Mycoplasma pulmonis to complement, hemadsorption and adherence to polystyrene. Infect. Immun. 71:5733-5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simmons, W. L., C. Zuhua, J. I. Glass, J. W. Simecka, G. H. Cassell, and H. L. Watson. 1996. Sequence analysis of the chromosomal region around and within the V-1-encoding gene of Mycoplasma pulmonis: evidence for DNA inversion as a mechanism for V-1 variation. Infect. Immun. 64:472-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sitaraman, R., A. M. Denison, and K. Dybvig. 2002. A unique, bifunctional site-specific DNA recombinase from Mycoplasma pulmonis. Mol. Microbiol. 46:1033-1040. [DOI] [PubMed] [Google Scholar]
- 30.Slifkin, M., and R. Cumbie. 1988. Congo red as a fluorochrome for the rapid dectection of fungi. J. Clin. Microbiol. 26:827-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Southey-Pillig, C. J., D. G. Davies, and K. Sauer. 2005. Characterization of temporal protein production in Pseudomonas aeruginosa biofilms. J. Bacteriol. 187:8114-8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stadtlander, C. T., H. L. Watson, J. W. Simecka, and G. H. Cassell. 1991. Cytopathic effects of Mycoplasma pulmonis in vivo and in vitro. Infect. Immun. 59:4201-4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svenstrup, H. F., P. K. Nielson, M. Drasbek, S. Birklund, and G. Christiansen. 2002. Adhesion and inhibition assay of Mycoplasma genitalium and M. pneumoniae by immunofluorescence microscopy. J. Med. Microbiol. 51:361-373. [DOI] [PubMed] [Google Scholar]
- 34.Talkington, D. F., M. T. Fallon, H. L. Watson, R. K. Thorp, and G. H. Cassell. 1989. Mycoplasma pulmonis V-1 surface protein variation: occurrence in vivo and association with lung lesions. Microb. Pathog. 7:429-436. [DOI] [PubMed] [Google Scholar]
- 35.Teather, R. M., and P. J. Wood. 1982. Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl. Environ. Microbiol. 43:777-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vuong, C., J. M. Voyich, E. R. Fischer, K. R. Braughton, A. R. Whitney, F. R. DeLeo, and M. Otto. 2004. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 6:269-275. [DOI] [PubMed] [Google Scholar]
- 37.Watson, H. L., D. K. Blalock, and G. H. Cassell. 1990. Relationship of hydrophobicity of the V-1 antigen of Mycoplasma pulmonis to adherence, p. 650-654. In G. Stanek, G. H. Cassell, J. G. Tully, and R. F. Whitcomb (ed.), Recent advances in mycoplasmology, vol. 20. Gustav Fischer Verlag, Stuttgart, Germany. [Google Scholar]
- 38.Whitchurch, C. B., T. Tolker-Nielsen, P. C. Ragas, and J. S. Mattick. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. [DOI] [PubMed] [Google Scholar]
- 39.Ziebuhr, W., C. Heilmann, F. Gotz, P. Meyer, K. Wilms, E. Straube, and J. Hacker. 1997. Detection of the intercellular adhesion gene cluster (ica) and phase variation in Staphylococcus epidermidis blood culture strains and mucosal isolates. Infect. Immun. 65:890-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.