

Previous studies have shown an association between chronic pancreatitis (CP) and mutations, especially the N34S mutation, in the serine protease inhibitor Kazal type 1 (SPINK1) gene.1,2 The human SPINK1 gene is approximately 7.5 kb long and consists of four exons.3 The gene product consists of 79 amino acids, including a 23 amino acid signal peptide. In exon 3, SPINK1 possesses a reactive site that serves as a specific target substrate for trypsin.4 It has been suggested that SPINK1 mutations might result in altered interaction between SPINK1 and trypsin, thus affecting the protease/antiprotease balance within the pancreas.1,2 But the underlying molecular mechanisms remain unclear. Splicing defects are estimated to account for approximately 10–15% of disease causing mutations in humans.5 Changes in the splicing patterns and in levels of normal transcripts lead to phenotypic differences. The prevalence of splicing mutations in the SPINK1 gene is unknown. Most reported mutations have only been described at the DNA level and have not been studied at the mRNA level, mainly due to unavailability of SPINK1 mRNA from patients.
We have recently shown that the [−215G>A; IVS3+2T>C] mutation is associated with familial and idiopathic CP in Japan.6,7 Because the IVS3+2T>C mutation affects the consensus splicing donor site,8 we hypothesised that this mutation leads to alternative splicing, resulting in decreased SPINK1 function. To overcome the difficulties in obtaining human pancreas samples, SPINK1 mRNA was harvested from the stomach, where SPINK1 is also abundantly expressed,9 of (1) a 70 year old male A with alcoholic CP carrying the homozygous [−215G>A; IVS3+2T>C] mutation, (2) his 40 year old daughter B, a heterozygote who had no abdominal complaints to date, and (3) a 54 year old female C with idiopathic CP, also heterozygous for the [−215G>A; IVS3+2T>C] mutation. Total RNA was isolated from biopsy specimen of the stomach. The entire coding region of the SPINK1 gene was amplified by reverse transcription‐polymerase chain reaction (PCR) and sequenced. Electrophoresis of the reverse transcription PCR products from subjects carrying the [−215G>A; IVS3+2T>C] mutation revealed two bands: a fragment corresponding to a normal (“F1”) and a truncated (“F2”) band (fig 1). Sequencing of the truncated fragment revealed complete deletion of exon 3. This mutated protein was predicted to consist of 63 amino acids: deletion of amino acid sequence from residues 30–64 and shifting of the reading frame at amino acid 65.
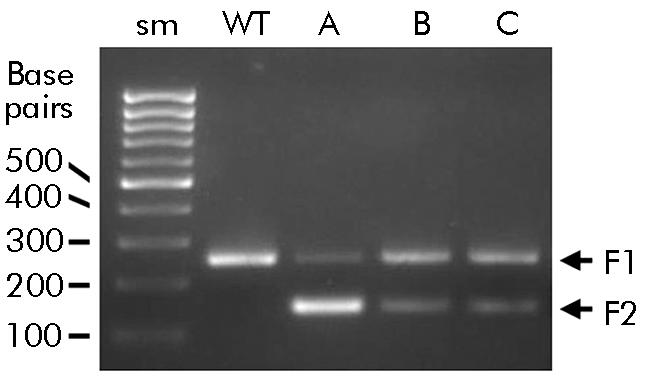
Figure 1 [−215G>A; IVS3+2] mutation produced a truncated transcript. Total RNA was isolated from the biopsy specimen of the stomach, and the entire coding region of the serine protease inhibitor Kazal type 1 (SPINK1) gene was amplified by reverse transcription polymerase chain reaction, followed by 2 % agarose gel electrophoresis. Sm, size marker (100 base pair ladders), WT, healthy control. Patient A with alcoholic chronic pancreatitis (CP) was homozygous for the [−215G>A; IVS3+2T>C] mutation. His daughter B and patient C with idiopathic CP were heterozygous. In subjects carrying the [−215G>A; IVS3+2T>C] mutation, two bands were observed: a fragment corresponding to a normal (“F1”) and a truncated (“F2”) band.
To our knowledge, this is the first study showing the splicing problem in the SPINK1 gene at the mRNA level. Northern blot analysis revealed that the size of the SPINK1 transcript was identical both in the pancreas and stomach,9 suggesting that exon 3 skipping is also likely to occur in the pancreas. It is logical to assume that skipping of exon 3 would result in functional loss of SPINK1, thus affecting the protease/antiprotease balance within the pancreas. Of note, the daughter of patient A carrying the heterozygous [−215G>A; IVS3+2T>C] mutation has not yet developed CP. Because this mutation has not been found in healthy controls,7 it is of interest to see whether she will develop CP in the future. Recently, Le Marechal and colleagues10 reported the IVS2+1G>A mutation in a CP patient carrying the P55S mutation in France. The IVS2+1G>A mutation affects the consensus splicing donor site of intron 2, implying a role of another splicing variation. Further studies using larger numbers of patients and different types of mutations will establish the role of splicing mutations in SPINK1 related CP.
Acknowledgements
This work was supported in part by a grant‐in‐aid from the Japan Society for the Promotion of Science (to AM and to TS).
Footnotes
*K Kume and A Masamune contributed equally to this work.
Conflict of interest: None declared
References
- 1.Witt H, Luck W, Hennies H C.et al Mutations in the gene encoding the serine protease inhibitor, Kazal type 1, are associated with chronic pancreatitis. Nat Genet 200025213–216. [DOI] [PubMed] [Google Scholar]
- 2.Pfutzer R H, Barmada M M, Brunskill A P.et al SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 2000119615–623. [DOI] [PubMed] [Google Scholar]
- 3.Horii A, Kobayashi T, Tomita N.et al Primary structure of human pancreatic secretory trypsin inhibitor (PSTI) gene. Biochem Biophys Res Commun 1987149635–641. [DOI] [PubMed] [Google Scholar]
- 4.Bartelt D C, Shapanka R, Greene L J. The primary structure of the human pancreatic secretory trypsin inhibitor: amino acid sequence of the reduced S‐aminoethylated protein. Arch Biochem Biophys 1977179189–199. [DOI] [PubMed] [Google Scholar]
- 5.Stenson P D, Ball E V, Mort M.et al Human gene mutation database (HGMD): 2003 update. Hum Mutat 200321577–581. [DOI] [PubMed] [Google Scholar]
- 6.Kaneko K, Nagasaki Y, Furukawa T.et al Analysis of the human pancreatic secretory trypsin inhibitor (PSTI) gene mutations in Japanese patients with chronic pancreatitis. J Hum Genet 200146293–297. [DOI] [PubMed] [Google Scholar]
- 7.Kume K, Masamune A, Mizutamari H.et al Mutations in the serine protease inhibitor Kazal type 1 (SPINK1) gene in Japanese patients with pancreatitis. Pancreatology 20055354–360. [DOI] [PubMed] [Google Scholar]
- 8.Faustino N A, Cooper T A. Pre‐mRNA splicing and human disease. Genes Dev 200317419–437. [DOI] [PubMed] [Google Scholar]
- 9.Marchbank T, Chinery R, Hanby A M.et al Distribution and expression of pancreatic secretory trypsin inhibitor and its possible role in epithelial restitution. Am J Pathol 1996148715–722. [PMC free article] [PubMed] [Google Scholar]
- 10.Le Marechal C, Chen J M, Le Gall C.et al Two novel severe mutations in the pancreatic secretory trypsin inhibitor gene (SPINK1) cause familial and/or hereditary pancreatitis. Hum Mutat 200423205. [DOI] [PubMed] [Google Scholar]