

Summary
Flp provides a unique opportunity to apply the tools of chemical biology to phosphoryl transfer reactions. Flp and other tyrosine recombinases catalyze site-specific DNA rearrangements via a phosphotyrosine intermediate, similar to the mechanism of type Ib topoisomerases [1]. Unlike most related enzymes, Flp’s nucleophilic tyrosine derives from a different protomer than the remainder of its active site [2, 3]. Because the tyrosine can be supplied exogenously, non-natural synthetic analogs can be used. Here we examine the catalytic role of Flp’s conserved H305. DNA cleavage was studied using a peptide containing either tyrosine (pKa≅10), or 3-fluoro-tyrosine (pKa≅8.4). Religation was studied using DNA substrates with 3’-phospho-cresol (pKa≅10) or 3’-para-nitro-phenol (pKa≅7.1) mimicking the covalent protein-DNA intermediate. In both cases, the tyrosine analog with the lower pKa specifically restored the activity of an H305 mutant. These results provide the first experimental evidence that this conserved histidine functions as a general acid/base catalyst in tyrosine recombinases.
Introduction
Site-specific DNA recombinases catalyze inversions, deletions and insertions of DNA segments. Far more common in bacteria than eukaryotes, these reactions have a broad variety of biological consequences, including the resolution of replicon dimers, the insertion of phage DNA, and the transfer of antibiotic resistance genes [1]. One of the few eukaryotic examples, Flp from S cerevisiae, is responsible for maintaining the copy number of the 2μM plasmid that encodes it. It does so by inverting a segment of DNA surrounding the plasmid’s origin of replication, resulting in rolling circle replication and, ultimately, the production of multiple copies of the plasmid with only one firing of the origin [4]. Flp is a member of the tyrosine recombinase family, which includes, among others, Cre and phage λ integrase[1].
Flp-catalyzed DNA recombination proceeds through a series of phosphotransfer reactions (figure 1a, b). In the cleavage steps, the DNA’s 5’ hydroxyl is displaced by the enzyme’s active site tyrosine, whereas in the religation steps, the tyrosine is displaced from the resulting covalent intermediate by attack of a different 5’ hydroxyl. Although recombination is normally orchestrated by a tetramer of Flp, the individual chemical steps do not require the complete oligomer [5]. Structural and biochemical studies have shown that tyrosine recombinases and the related type Ib topoisomerases surround the scissile phosphate with a constellation of conserved residues (in addition to the tyrosine), but, unlike many other phosphotransferases, do not require divalent metal ions (Figure 1)[1].
Fig. 1. Site-specific recombination mediated by Flp.
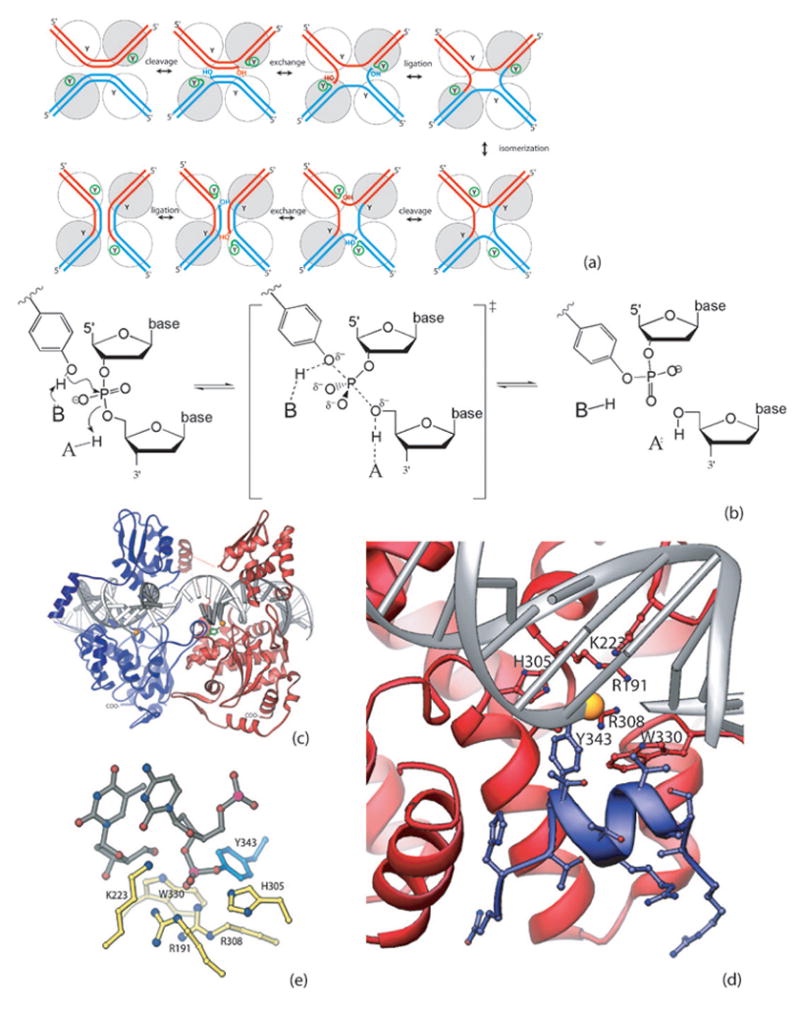
(a) Recombination pathway. Two DNA segments and four enzyme molecules (spheres, with Y representing the catalytic tyrosine) are required. Only two protomeres are active at each step, as indicated by the small circle around the Ys. The catalytic states of the individual protomers are switched in the isomerization step. The two DNA strands are sequentially cleaved, exchanged and religated. (b) General acid/base catalysis during the DNA cleavage and ligation reactions. In the DNA cleavage reaction, a general base presumably accepts a proton from the attacking tyrosyl hydroxyl group while a general acid donates a proton to the DNA’s 5’-bridging oxygen. This process is reversed in the ligation reaction. (c) Flp cleaves DNA in trans. The interface between two protomers is viewed from the center of the Holliday junction. Y343 of the blue protomer, shown in ball and stick mode, is donated to the red protomer and cleaves the DNA bound by that one. The scissile phosphate is shown as a gold sphere. (d) Flp Helix M (blue) packing into the active site of a neighboring Flp protomer (red). The segment shown corresponds to the peptide used in this study, and all side chains are shown. (e) Catalytic residues. The active state of the catalytic site, containing the covalent phosphotyrosine linkage, is shown. Six conserved residues, including Y343, surround the scissile phosphate. PDB code 1M6X
Enzymes can enhance phosphotransfer reaction rates in a number of ways. First, they can greatly enhance the reaction simply by binding the reactants such that they are appropriately oreinted for the ensuing reaction. For phosphodiesters such as DNA these reactions are thought to involve a pentacovalent transition state, in which bond formation to the nucleophile is at least to some degree simultaneous with breakage of the bond to the leaving group. The reaction could therefore be stimulated by both a general base to aid deprotonation of the nucleophile, and a general acid to protonate the leaving group. An enzyme could also preferentially stabilize the transition state, which has a slightly different geometry than the ground state and may also bear additional negative charge [6, 7].
How are these catalytic roles addressed by the conserved residues found in the active sites of Flp and related enzymes (figure 1e)? Since all the reactants are macromolecules, the job of simply localizing the players is accomplished by formation of the protein-DNA complex as a whole. This may at least in part explain why these enzymes do not need Mg++, which often coordinates the attacking water molecule in hydrolases. The two arginines (Flp R191 and R308) probably help position the scissile phosphate and may also stabilize the geometry and charge of the transition state. A combination of structural data and 5’ bridging sulfur substitution studies with the type Ib topoisomerase from vaccinia virus have implicated the lysine (K223 in Flp) and possibly the 1st of the two arginines in protonating the leaving 5’oxygen during DNA cleavage [8, 9]. The most important role of W330 in Flp appears to be structural [10]: in a full recombination assay, W330F was indistinguishable from WT, although in a cleavage assay it was ~5-fold slower. This also showed that the conformational changes required of both the DNA and the protein in a full recombination assay can mask subtle catalytic defects that are revealed in simpler (single-step) assays. In related proteins W330 is usually replaced by a histidine, which may also stabilize the transition state through a hydrogen bond to one of the non-bridging oxygens of the scissile phosphate [11–14].
Is the tyrosine assisted by a general acid/base? The active sites of the type Ib topoisomerases do not contain side chains that are good candidates for this role. Instead, a bound water molecule is thought to perform this duty [15]. No such appropriately positioned water has been noted in the tyrosine recombinases, but they do have a conserved histidine (H305 in Flp) that is well positioned to act as a general base in the cleavage reaction and conversely, general acid in the religation reaction [16–19]. Previous qualitative studies have observed that the H305A, Q and L mutants were defective in catalyzing the recombination reaction and tended to accumulate the DNA cleavage products [20, 21]. In Cre, the equivalent H289 has shown decreased activity as well. Both H289A and H289N mutants are ~4x less active than WT in a full recombination assay (in which the chemistry of cleavage is probably not normally rate-limiting). (Gelato, Baldwin et al 2005). Recently the Baldwin lab has shown that Cre H289N is 1200X slower than WT in a cleavage assay (personal communication). (We have found no published measurements of cleavage rates for mutations of the conserved active site histidine in any tyrosine recombinase). In the Flp-DNA complex model, the Nε2 atom of H305 is ~2.8 Å from the oxygen linking the tyrosine and DNA. Interestingly, this histidine lies across the DNA’s minor groove, with its Nδ1 atom relatively close to the phosphates of the opposite DNA backbone (figure 1d), making a Y-H-PO4 arrangement reminiscent of the S-H-D triad of the serine proteases. This hypothesis could not be tested by previous biochemical experiments, and the general acid/base that assists the tyrosine remained elusive.
We have taken advantage of Flp’s unique active site architecture to investigate this. In Flp, the conserved pentad of one protomer activates the scissile phosphate while Y343 is donated by a second protomer that inserts one helix into its neighbor’s active site (Figure 1). The active site is thus prepared to accept an exogenous tyrosine. Cleavage and ligation were studied separately using synthetic substrates bearing tyrosine analogs with lowered pKa’s that should mitigate the effects of mutating the general acid/base.
Results and Discussion
DNA cleavage and ligation reactions were investigated with three variants of Flp: Y343F, Y343F/H305Q and Y343F/K223R. All Flp proteins used also carry the four point mutations in the “Flpe” variant that enhance its thermostability [22]. We chose Q to replace H305 in order to maintain approximately the same volume and hydrogen bonding capacity while removing the labile proton As a control, we also included a mutant of K223, which is proposed to be the general acid/base that assists the DNA 5’oxygen in catalysis, but is not expected to interact with the tyrosine [8, 9]. The K223A mutation essentially abolishes the enzymatic activity, which is partially restored in the K223R mutant that we use here in order to ensure measurable amounts of product [10]. Except for the H305 – Y343 hydrogen bond, neither H305 nor K223 participates in any direct interactions with the catalytic tyrosine - containing “trans” helix (fig 1d). Mutations at these residues are therefore unlikely to affect the packing of this helix into the active site pocket.
Initial DNA cleavage
To study DNA cleavage, we supplied the tyrosine as part of a 13aa peptide spanning residues 334–346 of the Flp sequence (fig 1d). Previous studies have demonstrated that Flp Y343F, lacking its own nucleophile, could still facilitate the attack of alternative nucleophiles such as tyramine, glycerol and hydrogen peroxide on the scissile phosphate [5, 23], albeit quite inefficiently. We found that this cleavage reaction is much more robust when the nucleophile is supplied on the peptide, probably due to extensive interactions between other residues on the trans helix and the Flp protomer it docks into. Two versions of a peptide were synthesized, one with a native tyrosine (pKa≅10), and another with a 3-fluorotyrosine (pKa≅8.4). Fluorinated tyrosines have been used successfully in protein kinase studies [7, 24]. The nucleophile with the lower pKa should show less dependence on a general base during the cleavage reaction.
The DNA cleavage assay corresponds to the first chemical step in the recombination pathway (Figure 2). We used a suicide substrate containing a single Flp-binding site with only two nucleotides extending past the scissile phosphate on the cleaved strand [25]. After cleavage, this dinucleotide is diluted into solution, preventing the reverse reaction and leaving the peptide covalently linked to the DNA. Studying this first step of the reaction alone avoids the conformational changes that are known to be rate-limiting in a full recombination reaction [10, 26].
Fig. 2. Peptide cleavage of DNA catalyzed by Flp variants.
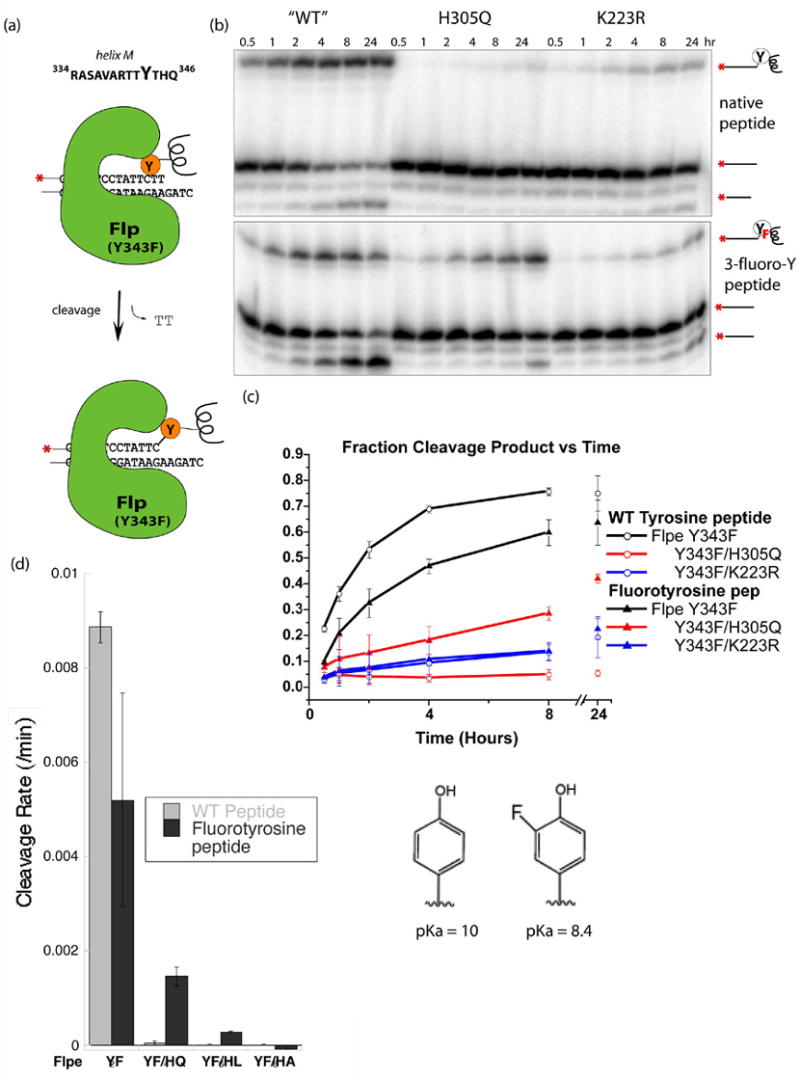
(a) Schematic describing the cleavage reaction with a 13 amino acide peptide corresponding to residues 334–346 of Flp, including helix M and the flanking loops. Peptides bearing tyrosine or 3-fluorotyrosine were reacted with Flp variants and a DNA suicide substrate containing a single Flp binding site which releases a TT dinucleotide upon cleavage. (b) The products of the cleavage reactions separated on sequencing gels. Hydrolysis of the phosphotyrosine linkage happens over time, especially with the fluorinated tyrosine analog. (c) Plot of Product vs. time. Product, shown as a percentage of total DNA, is the sum of the DNA-peptide complex and the subsequent hydrolysis product. The activity of both the Y343F and K223R decreased when the fluorotyrosine-containing peptide was used. The activity of H305Q, however, showed a significant increase. (d) Cleavage rates with several different Flpe H305 mutants.
The results shown in Figure 2 clearly support the hypothesis that H305 is involved in general base catalysis in the DNA cleavage reaction. The rate for Y343F Flpe-catalyzed cleavage decreased with the change from tyrosine to the fluorinated analog. This is consistent with the WT enzyme utilizing a general base to aid deprotonation of the nucleophile, since the anionic form of the fluorinated analog is less nucleophilic than that of the native tyrosine. The rate for Y343F/H305Q-catalyzed cleavage increased significantly (at least 1–2 orders of magnitude; the exact ratio is hard to estimate due to the near-zero rate with the unmofidied tyrosine). Within error, the slow cleavage rate of the Y343F/K223R enzymes was unchanged. These results suggest that the lower pKa of the tyrosyl hydroxyl group specifically compensated for H305Q’s lack of a general base. We also tested the effects of two other changes at H305: to L and A (figure 2d). Mutation of H305 to A, which probably introduces a packing defect in the active site, destroyed all measurable cleavage activity with either peptide. Y343F/H305L also failed to catalyze measurable DNA cleavage with the tyrosine-containing peptide. However, measurable activity was rescued by the fluorotyrosine peptide. That the cleavage defect of H305L as well as H305Q can be at least partially overcome by lowering the pKa of the nucleophile argues against the possibility that the role of general base is played by a water molecule that is hydrogen bonded to the side chain at position 305. Similar enhancement of H305Q’s cleavage activity by the fluorotyrosine peptide was also seen between pH 7.5x and 9.5 (Figure S3). In this assay, all combinations of Flpe and peptide variants were essentially inactive above pH 9.5
Our analysis assumes that most of the DNA substrate is bound at any given time, and that the reaction is first order with respect to the concentration of protein-DNA complexes. We measured a Kd of 31.7 ± 3.96 nm for Flpe Y343F and the suicide DNA substrate (figure S1). This is similar to previously reported values of Kd for WT Flp binding to the full length DNA substrate [27]. This implies that under our reaction conditions ([DNA]=10nM and [Flp]=360nM), 91.5% of the DNA was bound. We also found that for DNA concentrations varying from 1 to 20nM, rate of product formation varied linearly with the DNA concentration, which can be taken as a surrogate for the protein-DNA complex concentration since the DNA is essentially fully occupied (Figure S2). This implies that a single Flp-DNA complex is sufficient to carry out catalysis with the trans helix peptide: even though dimerization is normally required for cleavage by Flp (i.e. without added peptide), it is not necessary here.
DNA religation
If H305 accepts a proton from the tyrosine nucleophile during DNA cleavage, it presumably also donates a proton to the leaving tyrosine group during ligation, which is chemically the reverse reaction. To isolate the ligation reaction, we used activated DNA substrates in which an analog of the tyrosine leaving group was esterified to the 3’ phosphate: one, cresol (or 4-methyl-phenol), has a pKa of 10, similar to that of tyrosine, and the other, pNP (or para-nitro-phenol), has a pKa of 7.1. The latter was used as a substrate for Cre- and vaccinia topoiosomerase-mediated DNA ligation [28], and should be oblivious to the presence of a general acid under our reaction conditions (pH 8). As shown in figure 3, ligation was initiated by adding an excess of an oligonucleotide complementary to the overhanging noncleaved strand.
Fig. 3. Ligation of modified DNA substrates.
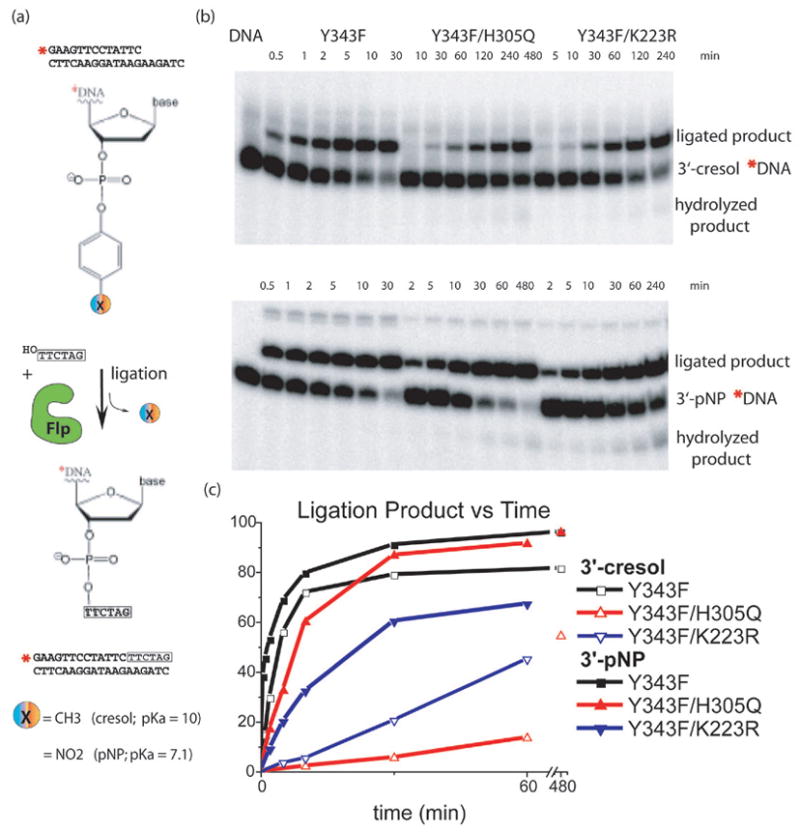
Schematic representation of the DNA ligation assay using a substrate carrying a single Flp binding site with a tyrosine mimic (cresol or pNP) esterified to the 3’ phosphate. A short oligo complementary to the overhang of the bottom strand, which mimics the incoming strand, is added in excess, displaces the tyrosine mimic from the 3’ end of the DNA, and produces a longer ligated product. (b) PAGE separation of ligation products. Note that different timepoints were taken for each Flpe variant. (c) Quantitation of gels showing ligated product as a percentage of total DNA. Reactions catalyzed by all three proteins displayed rate increases with the 3’-pNP compared to the 3’-cresol, when the pKa of the leaving group changed from 10 to 7.1. However, H305Q showed the most dramatic increase.
Ligation reactions were carried out with the same Flp mutants as the cleavage reactions, and the results of the two sets of experiments were in good agreement (Figure 3). When comparing product formation with the cresol and pNP substrates, reactions catalyzed by Y343F and Y343F/K223R mutants displayed minor rate changes with pNP, which is a better leaving group; however, the effect of the lower pKa of pNP was far more dramatic for the Y343F/H305Q reaction. The Y343F/H305Q ligation reaction has a ~27x rate enhancement with pNP compared to cresol, while the rate enhancements for the Y343F and Y343F/K223R ligation reactions were both less than 4x, as shown in Figure 3. This implies that the phosphotyrosyl oxygen leaves more readily when H305 can participate by releasing a well-positioned proton.
The results of both sets of experiments are consistent with a general acid/base model for Flp catalysis where H305 is involved in the abstraction of a proton from and the donation of a proton to the catalytic tyrosyl oxygen in the DNA cleavage and ligation reactions, respectively. The role of H305 has been more difficult to place than that of other catalytic residues, and this is not surprising. Though H305 is clearly important for catalysis, it is less conserved and its mutants show smaller decreases in activity – especially cleavage activity - compared to the conserved lysine and arginines [20]. This suggests that, in the cleavage step, the catalytic power derived from providing a proton acceptor may be less than that derived from providing a proton donor to the leaving hydroxyl group. This may be due in part to the lower pKa of tyrosine (10) relative to that of a 5’-hydroxyl group (~15). In addition, model reactions of nucleophilic substitution at phosphodiesters show less dependence on the basicity of the nucleophile (βnuc ~0.2−0.3) than the leaving group (βlg ~−0.8) [8] [6]. Finally, it is worth noting that even in the paradigmatic serine proteases, mutation or modification of the active site histidine (which assists a serine; closer in pKa to a 5’ hydroxyl) does not utterly destroy catalytic activity: kcat for the His/Asp double alanine mutant of subtilisn was ~104 fold lower than that for the WT, but still ~104 fold above that for the uncatalyzed reaction [29, 30].
From comparison of Flp to the type IB topoisomerases, it is clear that there is more than one way for these related enzymes to assist the tyrosine in catalysis: H305 is important in Flp, whereas it is apparently replaced by a well-ordered water molecule in the topoisomerases [13, 15]. Recent studies on Flp’s W330 have found that the relative catalytic contribution of a specific residue may vary among related enzymes even while the general catalytic strategy is preserved. However, despite Flp’s unique trans active site composition, the orientation of its active site components is quite similar to those of Cre and γ integrase which cleave DNA in cis [18, 31] The equivalent histidines, H289 and H308 respectively, lie in similar positions relative to the phosphotyrosine linkage and DNA backbone. Although more complicated assays may give different secondary effects of their mutation, such as altering the preferred direction of Holliday junction resolution in Cre, their close structural similarity implies that they are likely to perform a similar underlying chemical function in general acid/base catalysis [32].
Significance
This work uses the tools of chemical biology to probe how Flp, a tyrosine recombinase, catalyzes phosphoryl transfer. The catalytic mechanism of these enzymes is somewhat unusual for DNA phosphotransferases in that it does not require divalent cations. For the first time, we have experimentally addressed the question of which residue, if any, acts as a general acid/base to assist the tyrosine in catalysis. We were able to examine both the forward and reverse steps of the phosphoryl transfer because Flp’s unique trans active site geometry allows it to accept an exogenous tyrosine. The critical tyrosine could thus be replaced with synthetic analogs that have altered pKas, either incorporated into a peptide, as the nucleophile in the forward reaction, or esterified to the DNA, as the leaving group in the reverse reaction. In both reactions, the tyrosine analog with the lowered pKa, which should be less dependent on a general acid/base catalyst, largely restored the activity of a Flp variant lacking the conserved histidine. Our experiments, together with previous structural and sequence conservation data, provide strong support for the hypothesis that this residue functions as the general acid/base that assists the tyrosine during catalysis.
Methods
Protein mutagenesis and purification
Y343F Flpe with a C-terminal His6 tag was used as ‘wild type’ in our studies. Point mutagenesis was performed using QuickChange Site-Directed Kit from Stratagene and the results were confirmed by DNA sequencing. The proteins were purified with a previously-published protocol [10], using a Talon column for the His tag and a DNA affinity column containing Flp-binding sequences. The purification procedure for each protein was completed within one day starting from cell pellet. The protein was flash-frozen in liquid nitrogen and stored at −80°C. It was later dialyzed against 50mM HEPES (pH7.1), 1mM EDTA, 1mM DTT, and 15% glycerol and 1M NaCl overnight and aliquoted before usage.
Peptide Synthesis
The 13 residue peptides (RASAVARTTYTHQ and RASAVARTTYFTHQ, YF=3-fluorotyrosine) were made manually by “in situ neutralization” Boc chemistry with stepwise solid phase peptide synthesis on -OCH2-Pam-resins, as described previously [33]. Peptide compositions were confirmed by reversed-phase HPLC and LCMS on an Agilent 1100 Series chromatography instrument equipped with an MSD ion trap, using Vydac C4 columns (5 ím, 0.46 25 cm). Chromatographic separations were performed using a linear gradient (10–60%) of buffer B in buffer A over 30 min at a flow rate of 1 mL/min. Buffer A = 0.1% TFA in water; buffer B = 0.08% TFA in acetonitrile. Preparative HPLC was performed on a Waters Prep LC 4000 system using Vydac C4 column (12 ím, 2.2 25 cm) at a flow rate of 10 mL/min, with a gradient of 20–50% buffer B in buffer A over 60 min. Fractions were pooled based on LC-MS analysis.
Synthesis of 3’-cresol/pNP substrate DNA
Oligonucleotides (6 X 1 □mol) were synthesized using 3’-PO4 CPG (Glen Research, Sterling, VA) and standard DNA phosphoramidites on an ABI 392 automated DNA synthesizer. Oligonucleotide synthesis was completed with automated removal of the 5’-dimethoxytrityl protecting group. The resin was incubated in concentrated ammonium hydroxide at 65°C for 12 h. DNA was precipitated with the addition of 0.1 vol of 3 M sodium acetate and 3 vol of absolute ethanol, and the DNA pellet was resuspended in 1 ml of 2 mM MgCl2, 100 mM MES (pH 5.5). Aliquots of 1 ml 99% para-cresol (Aldrich, Milwaukee, WI) and 0.048 g of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC) (Aldrich) were added sequentially. The resulting two immiscible layers were mixed with a magnetic stir bar at 65°C to form an emulsion. After 20–24 h, the aqueous layer was extracted three times with 2 ml each of ethyl acetate and the DNA was ethanol precipitated. This EDC-derivatization protocol was repeated twice more. Typically, ~5% of the 3’- PO4 (starting material) was converted to 3’-cresol DNA.
The 3’-cresol derivatized DNA was purified by anion exchange chromatography on a Hitachi analytical HPLC using a DNAPac PA-100 4 mm X 250 mm column (Dionex, Sunnyvale, CA) at 1 ml/min, 5-60% 2 M sodium chloride gradient (buffered with 20 mM sodium phosphate, pH 7.0) over 15 min. Peak fractions were pooled, precipitated, resuspended and stored at 4°C. The synthesis of 3’-pNP substrate was similar to that of 3’-cresol.
Peptide mediated cleavage assays
Synthentic oligonucleotides were purchased from the Keck biotechnology facility at Yale University and purified by urea polyacrylamide gel electrophoresis. S3 (TAA TCC AGT GGA AGT TCC TAT TCT T) was 5’ end-labeled with 32P and purified on a BioRad Spin 6 Chromatography Column. It was then annealed with S4P (5’-phos-CTA GAA GAA TAG GAA CTT CCA CTG GAT TA) to form a suicide substrate containing one Flp-binding site. The final substrate for the DNA cleavage assays consisted of both radioactively-labeled and unlabeled suicide substrates at a ratio of 1:9. The 5ul reaction contains 10nM DNA substrate, 360nM Flpe protein, 2mM peptide, 25mM Taps (pH8.0), 200mM NaCl, 5% glycerol, 2mM EDTA, 1 μg/mL salmon sperm DNA and 100 μg/ml BSA. Cleavage assays with varying concentrations of DNA shown in Figure S2 contained 1-20nM DNA substrate. The assay was carried out at 30ºC and terminated with formamide. Products were separated on sequencing gels and quantified using a Molecular Dynamics phosphorimager. Experiments were conducted in triplicate, and the percent of total complex was calculated by adding together the covalent peptide-DNA complex and the hydrolysis products and dividing by the total DNA. The pH dependence assays were carried out with the following buffers: pH 6-7: 25mM MES, 25mM Acetic Acid + 50mM Tris pH 8-9: 50mM TAPS pH 10: 50mM CAPS. Rates were calculated by fitting a line to the slope of the cleavage product vs time plot, and only early timepoints taken every 5–10 minutes from the first 30–60 minutes were included. Rates for the H305Q, H305A and H305L were arrived at in the same way, these and the pH dependence assays were repeated at least twice.
DNA ligation assay
The top strand of the activated DNA substrate was radioactively labeled and purified as described above. It was then annealed with S43P (5’-phos-CTA GAA GAA TAG GAA CTT C). The final substrate for the ligation assays consisted of both radioactively-labeled and unlabeled suicide substrates at a ratio of 1:9. The 5’-overhang was phosphorylated so it could not attack the phospho-phenyl bond. A 6nt oligo complementing the single-strand spacer region was used as the incoming strand and supplied the 5’-hydroxyl group that cleaved the 3’-phospho-phenyl linkage, resulting in a longer ligation DNA product. The 10ul reaction contains 10nM DNA substrate, 360nM Flpe protein, 6uM incoming strand, 25mM Taps (pH8.0), 200mM NaCl, 12% glycerol, 2mM EDTA, and 1mg/ml BSA. The protein was pre-incubated with the activated DNA substrate before the addition of the incoming strand, which was in large excess to the activated substrate. The assay was carried out at 30ºC and terminated with 0.4% SDS. Products were separated on 15% urea gels and quantified using Molecular Dynamics phosphorimager. Results were averaged from two sets of experiments.
Electrophoretic Mobility Shift Analysis
Flpe YF was diluted into dialysis buffer (15% glycerol, 1M NaCl, 50mM Hepes, 1mM EDTA, 1mM DTT, pH 7.1) in two independent serial dilutions leading to final concentrations of protein from 0 to 1μM. A 20 μL sample was prepared under the same conditions as the DNA cleavage assays described above, including 1nM radiolabelled DNA suicide substrate and 2μL of the protein dilution. After a two hour incubation at 30ºC, 10μl sample was loaded onto a native 16x18cm gel containing 10% 29:1 acrylamide:bis-acrylamide (w/w), 5% glycerol and 0.5x TBE (45mM tris-Borate, 1mM EDTA) at 4ºC and run for 1.5 hours at 140V. Gels were dried and visualized following exposure to phosphorimager screens (Kodak) using a Molecular Dynamics phosphorimager. Free DNA and DNA-protein complex intensities in each lane were quantified with the volume analysis function in rectangle mode of ImageQuant V5.0 (Molecular Dynamics). Free protein concentration was assumed to be equal to total protein concentration, because it was in excess over the DNA concentration. The equilibrium dissociation constant was calculated with Kaleidagraph 3.6.4 by fitting data to the equation: θ = (Bmax*[P])/([P] + Kd) where θ = fraction DNA bound, Bmax = maximum DNA bound, and P = Flpe YF. Experiments were repeated a minimum of three times.
Supplementary Material
Supplementary Figure 1. Binding of Flpe Y343F and the suicide substrate shown in Figure 2. A typical gel mobility shift assay is shown, and the averaged results of several such assays are plotted above. The Kd=31.7±3.96 with a maximum binding of 0.87, while the Kd=40.8±4.2 when the maximum binding is fixed at 1.
Supplementary Figure 2. Cleavage assay with varying DNA suicide substrate concentrations. The cleavage product (normalized as a fraction of the total DNA in each reaction) is plotted for reactions containing 1-20nM DNA and 360nM Flp Y343F. Since the DNA should be essentially saturated at this protein concentration (see figure S1), this serves as a surrogate for the Flp-DNA complex concentration. The linear relationship between [DNA] and rate, evident because of the superimposable slopes in the time vs normalized [product] plot seen here, shows that Flp oligomerization is not necessary during the peptide-mediated cleavage reaction.
Supplementary Figure 3. Peptide cleavage assays from pH 6 to 10. Cleavage rates (/min) at a range of pH with Flpe Y343F, Flpe Y343F/H305Q with both the WT and Fluorotyrosine peptide (each individual rate was determined 2 or 3 times). Rescue of the histidine mutant (YF/HQ) by the fluorotyrosine is consistent across the range of pH where the reaction was fast enough for product to be measured. Although the enzyme might be expected to become independent of the histidine above the pK of tyrosine (∼10); all Flp variants were unfortunately inactive at high pH.
Acknowledgments
We would like to thank Ying Zhang Pigli for technical assistance, and Kerren Swinger, Adam Conway and Kent Mouw for helpful discussions. Alex Burgin and Steve Kent both generously allowed access to their laboratories for the synthesis of the DNA and peptide substrates, Brad Pentelute from the Kent lab was essential for the synthesis of a second and difficult batch of fluorotyrosine peptide. Yuen-Ling Chan contributed electrophoresis troubleshooting and advice. Joe Piccirilli and his lab (especially Jimmy Hougland and Subha Das) made insightful suggestions. Daniel Whiteson provided statistical guidance in the estimation of the rates. This work was supported by NIH grant GM058827.
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Katrine L. Whiteson, Biochemistry and Molecular Biology Department, The University of Chicago, 929 E. 57th St. CIS W125, Chicago, IL 60637, Katrine@uchicago.edu.
Yu Chen, Current: Dept. of Pharmaceutical Chemistry, UCSF, 1700 4th St, QB3 Building, San Francisco, CA 94158, chen@blur.compbio.ucsf.edu.
Neeraj Chopra, Biochemistry and Molecular Biology Department, University of Chicago, 929 E. 57th St. CIS W125, Current: Department of Pathology, The University of Chicago, N344, 5841 South Maryland Avenue, Chicago, Illinois 60637, nchopra@bsd.uchicago.edu.
Amy C. Raymond, deCODE biostructures, 7869 NE Day Road West, Bainbridge Island, WA 98110, Current: Sloan-Kettering Institute 1275 York Ave., Box 73, New York, NY 10021, raymonda@mskcc.org
Phoebe A. Rice, Biochemistry and Molecular Biology Department, The University of Chicago, 929 E. 57th St. CIS W125, Chicago, IL 60637, price@uchicago.edu, phone: 773 834 1723, fax: 773 702 0439
References
- 1.Grindley ND, Whiteson KL, Rice PA. Mechanisms of Site-Specific Recombination. Annu Rev Biochem. 2006 doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- 2.Chen JW, Lee J, Jayaram M. DNA cleavage in trans by the active site tyrosine during Flp recombination: switching protein partners before exchanging strands. Cell. 1992;69:647–658. doi: 10.1016/0092-8674(92)90228-5. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Narendra U, Iype LE, Cox MM, Rice PA. Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol Cell. 2000;6:885–897. [PubMed] [Google Scholar]
- 4.Futcher AB. The 2 micron circle plasmid of Saccharomyces cerevisiae. Yeast. 1988;4:27–40. doi: 10.1002/yea.320040104. [DOI] [PubMed] [Google Scholar]
- 5.Kimball AS, Lee J, Jayaram M, Tullius TD. Sequence-specific cleavage of DNA via nucleophilic attack of hydrogen peroxide, assisted by Flp recombinase. Biochemistry. 1993;32:4698–4701. doi: 10.1021/bi00069a002. [DOI] [PubMed] [Google Scholar]
- 6.Cassano AG, Anderson VE, Harris ME. Understanding the transition states of phosphodiester bond cleavage: insights from heavy atom isotope effects. Biopolymers. 2004;73:110–129. doi: 10.1002/bip.10517. [DOI] [PubMed] [Google Scholar]
- 7.Maegley KA, Admiraal SJ, Herschlag D. Ras-catalyzed hydrolysis of GTP: a new perspective from model studies. Proc Natl Acad Sci U S A. 1996;93:8160–8166. doi: 10.1073/pnas.93.16.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagarajan R, Kwon K, Nawrot B, Stec WJ, Stivers JT. Catalytic Phosphoryl Interactions of Topoisomerase IB. Biochemistry. 2005;44:11476–11485. doi: 10.1021/bi050796k. [DOI] [PubMed] [Google Scholar]
- 9.Krogh BO, Shuman S. Proton relay mechanism of general acid catalysis by DNA topoisomerase IB. J Biol Chem. 2002;277:5711–5714. doi: 10.1074/jbc.C100681200. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Rice PA. The role of the conserved Trp330 in Flp-mediated recombination. Functional and structural analysis. J Biol Chem. 2003;278:24800–24807. doi: 10.1074/jbc.M300853200. [DOI] [PubMed] [Google Scholar]
- 11.Stivers JT, Jagadeesh GJ, Nawrot B, Stec WJ, Shuman S. Stereochemical outcome and kinetic effects of Rp- and Sp-phosphorothioate substitutions at the cleavage site of vaccinia type I DNA topoisomerase. Biochemistry. 2000;39:5561–5572. doi: 10.1021/bi992429c. [DOI] [PubMed] [Google Scholar]
- 12.Petersen BO, Shuman S. Histidine 265 is important for covalent catalysis by vaccinia topoisomerase and is conserved in all eukaryotic type I enzymes. J Biol Chem. 1997;272:3891–3896. doi: 10.1074/jbc.272.7.3891. [DOI] [PubMed] [Google Scholar]
- 13.Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 14.Tian L, Claeboe CD, Hecht SM, Shuman S. Guarding the genome: electrostatic repulsion of water by DNA suppresses a potent nuclease activity of topoisomerase IB. Mol Cell. 2003;12:199–208. doi: 10.1016/s1097-2765(03)00263-6. [DOI] [PubMed] [Google Scholar]
- 15.Davies DR, Mushtaq A, Interthal H, Champoux JJ, Hol WG. The structure of the transition state of the heterodimeric topoisomerase I of Leishmania donovani as a vanadate complex with nicked DNA. J Mol Biol. 2006;357:1202–1210. doi: 10.1016/j.jmb.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 16.Conway AB, Chen Y, Rice PA. Structural plasticity of the Flp-Holliday junction complex. J Mol Biol. 2003;326:425–434. doi: 10.1016/s0022-2836(02)01370-0. [DOI] [PubMed] [Google Scholar]
- 17.Van Duyne G. A Structural View of Tyrosine Recombinase Site-Specific Recombination. In: Craig N, Craigie R, Gellert M, Lambowitz A, editors. Mobile DNA II. Washington, D.C.: ASM Press; 2002. pp. 93–117. [Google Scholar]
- 18.Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T. A structural basis for allosteric control of DNA recombination by lambda integrase. Nature. 2005;435:1059–1066. doi: 10.1038/nature03657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacDonald D, Demarre G, Bouvier M, Mazel D, Gopaul DN. Structural basis for broad DNA-specificity in integron recombination. Nature. 2006;440:1157–1162. doi: 10.1038/nature04643. [DOI] [PubMed] [Google Scholar]
- 20.Parsons RL, Prasad PV, Harshey RM, Jayaram M. Step-arrest mutants of FLP recombinase: implications for the catalytic mechanism of DNA recombination. Mol Cell Biol. 1988;8:3303–3310. doi: 10.1128/mcb.8.8.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu XD, Sadowski PD. Cleavage-dependent ligation by the FLP recombinase. Characterization of a mutant FLP protein with an alteration in a catalytic amino acid. J Biol Chem. 1995;270:23044–23054. doi: 10.1074/jbc.270.39.23044. [DOI] [PubMed] [Google Scholar]
- 22.Buchholz F, Angrand PO, Stewart AF. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat Biotechnol. 1998;16:657–662. doi: 10.1038/nbt0798-657. [DOI] [PubMed] [Google Scholar]
- 23.Knudsen BR, Lee J, Lisby M, Westergaard O, Jayaram M. Alcoholysis and strand joining by the Flp site-specific recombinase. Mechanistically equivalent reactions mediated by distinct catalytic configurations. J Biol Chem. 1998;273:22028–22036. doi: 10.1074/jbc.273.34.22028. [DOI] [PubMed] [Google Scholar]
- 24.Cole PA, Sondhi D, Kim K. Chemical approaches to the study of protein tyrosine kinases and their implications for mechanism and inhibitor design. Pharmacol Ther. 1999;82:219–229. doi: 10.1016/s0163-7258(98)00046-1. [DOI] [PubMed] [Google Scholar]
- 25.Nunes-Duby SE, Matsumoto L, Landy A. Half-att site substrates reveal the homology independence and minimal protein requirements for productive synapsis in lambda excisive recombination. Cell. 1989;59:197–206. doi: 10.1016/0092-8674(89)90881-7. [DOI] [PubMed] [Google Scholar]
- 26.Qian XH, Inman RB, Cox MM. Protein-based asymmetry and protein-protein interactions in FLP recombinase-mediated site-specific recombination. J Biol Chem. 1990;265:21779–21788. [PubMed] [Google Scholar]
- 27.Ringrose L, Lounnas V, Ehrlich L, Buchholz F, Wade R, Stewart AF. Comparative kinetic analysis of FLP and cre recombinases: mathematical models for DNA binding and recombination. J Mol Biol. 1998;284:363–384. doi: 10.1006/jmbi.1998.2149. [DOI] [PubMed] [Google Scholar]
- 28.Woodfield G, Cheng C, Shuman S, Burgin AB. Vaccinia topoisomerase and Cre recombinase catalyze direct ligation of activated DNA substrates containing a 3'-para-nitrophenyl phosphate ester. Nucleic Acids Res. 2000;28:3323–3331. doi: 10.1093/nar/28.17.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henderson R. Catalytic activity of -chymotrypsin in which histidine-57 has been methylated. Biochem J. 1971;124:13–18. doi: 10.1042/bj1240013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carter P, Wells JA. Dissecting the catalytic triad of a serine protease. Nature. 1988;332:564–568. doi: 10.1038/332564a0. [DOI] [PubMed] [Google Scholar]
- 31.Van Duyne GD. A structural view of cre-loxp site-specific recombination. Annu Rev Biophys Biomol Struct. 2001;30:87–104. doi: 10.1146/annurev.biophys.30.1.87. [DOI] [PubMed] [Google Scholar]
- 32.Gelato KA, Martin SS, Baldwin EP. Reversed DNA strand cleavage specificity in initiation of Cre-LoxP recombination induced by the His289Ala active-site substitution. J Mol Biol. 2005;354:233–245. doi: 10.1016/j.jmb.2005.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bang D, Kent SB. A one-pot total synthesis of crambin. Angew Chem Int Ed Engl. 2004;43:2534–2538. doi: 10.1002/anie.200353540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Binding of Flpe Y343F and the suicide substrate shown in Figure 2. A typical gel mobility shift assay is shown, and the averaged results of several such assays are plotted above. The Kd=31.7±3.96 with a maximum binding of 0.87, while the Kd=40.8±4.2 when the maximum binding is fixed at 1.
Supplementary Figure 2. Cleavage assay with varying DNA suicide substrate concentrations. The cleavage product (normalized as a fraction of the total DNA in each reaction) is plotted for reactions containing 1-20nM DNA and 360nM Flp Y343F. Since the DNA should be essentially saturated at this protein concentration (see figure S1), this serves as a surrogate for the Flp-DNA complex concentration. The linear relationship between [DNA] and rate, evident because of the superimposable slopes in the time vs normalized [product] plot seen here, shows that Flp oligomerization is not necessary during the peptide-mediated cleavage reaction.
Supplementary Figure 3. Peptide cleavage assays from pH 6 to 10. Cleavage rates (/min) at a range of pH with Flpe Y343F, Flpe Y343F/H305Q with both the WT and Fluorotyrosine peptide (each individual rate was determined 2 or 3 times). Rescue of the histidine mutant (YF/HQ) by the fluorotyrosine is consistent across the range of pH where the reaction was fast enough for product to be measured. Although the enzyme might be expected to become independent of the histidine above the pK of tyrosine (∼10); all Flp variants were unfortunately inactive at high pH.