

Abstract
Understanding the early genesis of new enzymatic functions is one of the challenges in protein design, mechanistic enzymology, and molecular evolution. We have experimentally mimicked starting points in this process by introducing primitive iron and oxygen binding sites at various locations in thioredoxin, a small protein lacking metal centers, by using computational design. These rudimentary active sites show emerging enzymatic activities that select to varying degrees between different oxygen chemistries. Even within these nascent enzymes, mechanisms by which different reactions are controlled can be discerned. These involve both stabilizing and destabilizing interactions imposed on the metal center by the surrounding protein matrix.
Rational design is emerging as a technique to study general principles of protein structure and function (1–7). The design of enzyme activity has been a long-standing goal in structure-based protein design (8–12) and has met with varying degrees of success (13–17). Exploitation of the diverse reactivities of metal center cofactors presents a fruitful strategy to introduce catalytic activity into proteins, as demonstrated by the observation that about one-third of all enzymes use a metal (7). Metal centers often exhibit several different potential reactivities toward a single substrate. Control of activity is therefore an important aspect of biological metal chemistry and is the consequence of the interplay between the intrinsic properties of the metal center and the surrounding protein matrix. We have chosen to study the emergence of catalytic function, by using nonheme iron chemistry as an exemplar, because of its importance in many biological processes (18–21) and its role in early evolution (22). Here we present the construction of simple, mononuclear iron centers in a protein that is devoid of such centers. Their reactivity toward oxygen was determined to investigate whether these simple sites are catalytically active. Oxygen was selected for our experiments, because (i) the steric simplicity of this substrate does not require the construction of elaborate binding sites, (ii) it has general importance in biological processes, and (iii) there are several experimentally defined competing reactions (23, 24), including superoxide (O2⨪ dismutation, reductive production of O2⨪, and reductive cleavage of hydrogen peroxide (H2O2), allowing the emergence of mechanisms that control specificity to be investigated.
The reactions selected for study represent some of the important biological oxygen toxicity and protection mechanisms (25). Superoxide dismutase (SOD) catalyzes the conversion of the toxic O2⨪ radical into dioxygen (O2) and H2O2 (24, 26). This is an important physiological detoxification reaction and is catalyzed by single transition metal centers such as iron (27), copper (26), or manganese (28) in a two-step redox cycle: O2⨪ + Mn → Mn−1 + O2 and O2⨪ + 2 H+ + Mn−1 → Mn + H2O2. Metal aqua complexes readily catalyze this reaction (25, 29). However, free metals also catalyze the production of toxic hydroxyl radicals by reductive cleavage of H2O2 (Fenton chemistry, refs. 25 and 29: H2O2 + e− → OH− + ·OH) and, to a lesser extent, O2 (Udenfriend chemistry, refs. 21 and 23: O2 + e− → O2⨪ → → → ·OH). Structural destabilizing mutations in Cu,Zn SOD have been shown to be toxic because of a gain of Fenton activity (30), which has been implicated in the pathogenesis of familial amyotrophic lateral sclerosis (31). Mechanisms that determine the reaction specificity of the metal center are therefore an important aspect of SOD function.
The designs presented here were constructed by introducing minimalist, mononuclear iron binding sites and oxygen binding pockets into Escherichia coli thioredoxin, a stably folded protein lacking such sites (32, 33), by using an automated design procedure, dezymer (34). This program identifies regions where ligand binding sites of predefined geometry can be introduced by mutagenesis into proteins of known structure and has been used to successfully construct a variety of metal centers in thioredoxin and other proteins (35–39). The designed metal coordination sphere consists of three histidines, leaving at least one unsaturated position in the metal primary coordination sphere for ligand access, a motif commonly found in metalloenzymes and inorganic catalysts (20). Rudimentary oxygen binding sites were constructed by introducing pockets that are approximately sterically complementary to molecular oxygen, placed adjacent to the metal center. The reactivity of these sites toward oxygen is controlled by the microenvironment of the metal center, demonstrating that even within a protein matrix that lacks prior adaptations for the control of metal function, mechanisms for controlling biological function can readily emerge.
Materials and Methods
Molecular Modeling.
Metal binding sites were designed by using the dezymer algorithm (34). Metal-nitrogen (ND1 or NE2) bond lengths of 2.1 Å and nitrogen-metal-oxygen bond angles of 109.5° were used with a metal-O2 as the ligand [d(metal-O) = 1.8 Å, d(O-O) = 1.2 Å, ∠D(metal-O-O) = 120°].
Mutagenesis.
Designs chosen for construction are shown in Table 1 and were constructed by standard oligonucleotide-directed mutagenesis of the E. coli thioredoxin trxA gene. Mutations for complete designs and precursors lacking one or more of the histidines were constructed in a C32S,C35S,D2A,D26L E. coli thioredoxin maltose binding protein fusion as described (35). Proteins were expressed as maltose binding protein fusions, purified by amylose affinity chromatography, incubated with o-phenanthroline and EDTA, and purified by gel filtration as described (35).
Table 1.
The family of Fe⋅His3⋅O2 designs constructed in E. coli thioredoxin
| Site | Mutations*
|
Rb†, Å | Kd (Fe), μM | Fe: protein | ɛ350‡, M−1⋅cm−1 | pKa | E°, mV | Kd (N3−), mM | |
|---|---|---|---|---|---|---|---|---|---|
| Metal primary coordination sphere | Oxygen binding site | ||||||||
| G1 | V25H I72H T77H | L79G Y70T | 5.4 | <10 | 1:1 | 2,000 | 7.9 | +120 | 270 |
| G2 | V25H I72H T77H | L79G | 5.4 | <10 | 1:1 | 2,000 | 7.7 | +115 | 20 |
| G3 | V25H I72H F27H | L79G | 5.4 | <10 | 1:1 | 4,000 | 7.4 | +50 | 40 |
| S1 | L53H L80H L107H | 5.9 | 200 | 500 | 8.1 | +430 | 15 | ||
| S2 | L53H L80H L107H | A22G | 5.9 | <10 | 1:1 | 2,000 | 7.6 | +120 | 30 |
| D1 | L24H P76H L78H | L24A I38V L94T | 9.5 | <10 | 1:1 | 6,000 | 7.4 | +10 | 15 |
Precursor mutations lacking one or two of the histidines are not given as table entries, pre-GA: I72H, pre-GB: V25H; pre-SA: L53H; pre-SB: L53H, L80H; pre-D: P76H, L78H.
Distance of the metal center to the closest point on the solvent-accessible surface.
Shoulder.
Metal Binding.
Quantitative stoichiometries and semiquantitative metal affinities of the designed proteins were determined chromatographically. Metal-protein mixtures [50 μM metal, 25 μM protein in 50 mM Tris⋅Cl (pH 7.5), 100 mM NaCl] were incubated aerobically for 15 min and separated on a desalting 10-ml gel filtration column (Bio-Rad P-6). Colorimetic assays were used to determine protein (Bradford assay, ref. 41) and metal [Fe: o-phenanthroline (42); Cu: bathocuprine (43); Zn, Ni, Co, Mn: 4-(2-pyridylazo)resorcinal (44)] content in 1-ml fractions. Metal affinities and stoichiometries were divided into three classes: 1:1 complex of strong affinity, protein and metal peaks coincide (0.9–1.1:1 metal/protein stoichiometry; none of the metal complexes showed evidence for incorporation of more than one metal per protein; all apo-protein controls had <0.05 metal/protein content); moderate affinity, metal peak lagged relative to the protein (0.3–0.7:1 metal/protein content); none, no significant metal content in the first eight fractions past the protein peak (<0.05:1 metal/protein content, similar to apo-protein control).
FeIII affinities were quantified by spectrophotometric titrations: 0.1–2.0 equivalents of Fe(NH4)(SO4) were added to 50 μM protein, oxidized to completion by air (8 h incubation resulted in complexes that showed no further change in intensity upon prolonged air exposure), and centrifuged for 5 min to remove any unincorporated iron-oxide precipitate. The intensities of the resulting FeIIIHis3 charge transfer bands (ɛ350 500–6,000 M−1⋅cm−1; Table 1) were significantly stronger than FeIII Tris buffer (ɛ350 200 M−1⋅cm−1). In each case, the intensity at 350 nm decreased by 50–70% upon stoichiometric reduction with dithionite (relative to added Fe). Metal affinities were calculated based on changes in the absorbance at 350 nm, using a binding isotherm that takes into account protein concentration, as described (35). Proteins classified as strong Fe binders had dissociation constants of less than 10 μM [these conditions, KD(Fe) << [protein], preclude accurate determination of the KD(Fe), but allow estimation of an upper limit], whereas moderate Fe binders had dissociation constants between 10 μM and 1 mM. Similar results were obtained in CoII spectrophotometric titrations (not shown).
All subsequent characterizations of the metalloproteins described below were carried out under conditions where a 1:1 Fe/protein is formed, i.e., [protein] >> KD(Fe), with the exception of S1, where [protein] ≈ Kd(Fe).
Hydroxyl Binding.
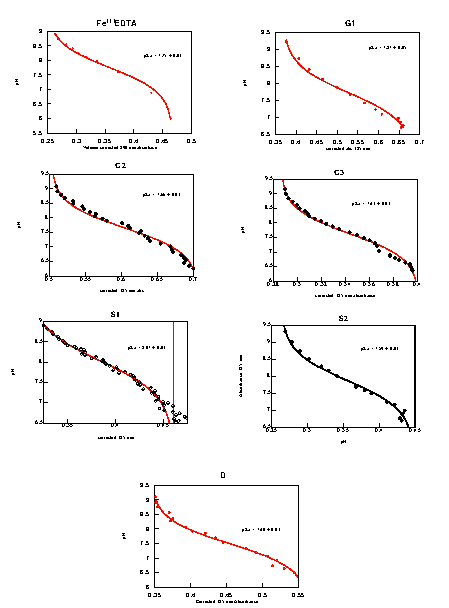
pKa values of FeIII(H2O) were determined spectrophotometrically: 1 mM NaOH was titrated (5 min equilibration) into a stirred 3 ml spectrophotometer cell (25°C) containing 100 μM 1:1 Fe/protein complex buffered with 50 mM each of Bis-Tris, Tris, and borate. The spectrum from 300 to 400 nm and pH (Microelectrodes) of the cell were recorded simultaneously. Changes in the intensities of the charge transfer bands between FeIII and H2O or OH− were fit to a one-proton Henderson–Hasselbach equation (see Figs. 9–15, which are published as supplementary material on the PNAS web site, www.pnas.org, for further detail) (45).
Redox Potentials.
Reduction potentials were determined by spectroelectrochemical titrations with sodium dithionite in an anaerobic cell containing 100 μM 1:1 Fe/protein complex. Redox mediators (quinhydrone, dichloroindophenol, 1,2-naphthoquinone, thionine, and methylene blue) (46) were used to establish electrochemical equilibrium between the electrode and the metalloprotein (47–49). After the reduction potential (measured by using a Pt and Ag/AgCl combination microelectrode) had equilibrated (≈10–60 min), the changes in the electronic spectrum between 300 and 450 nm caused by loss of FeIII charge transfer bands were recorded. Reduction potentials were calculated by fitting the absorbance changes to the Nernst equation (see Figs. 9–15).
Azide Affinities.
Azide affinities were determined by spectrophotometric titration of 100 μM 1:1 Fe/protein complexes with sodium azide, recording changes in the absorbance at 350–450 nm caused by formation of the FeIII(N3−) charge transfer band. The observed charge-transfer bands have intensities between 250 and 400 M−1⋅cm−1, consistent with similar complexes (45, 50, 51). Affinities were calculated as reported (35).
Enzymatic Activity.
Enzymatic assays were carried out by using 10 μM 1:1 Fe/protein complexes. SOD was determined in an indirect assay (26, 52–54), using {3′-[1-(phenylamino)carbonyl]-3,4-tetrazolium}-bis(4-methoxy-6-nitro)benzenesulfonic acid (XTT) as the colorimetric O2⨪ indicator. Fenton and Udenfriend activities were determined by using a colorimetric assay based on the conversion of p-nitrophenol to p-nitrocatechol by ·OH-mediated oxidation (55). [Alternatively, p-nitrophenol could be oxidized by direct interaction with an FeIV-O species. However, p-nitrophenol titrations revealed no spectroscopic evidence of binding to the metal center.] Ascorbate was the exogenous reductant in both reactions. The Fenton reaction showed saturation kinetics with respect to H2O2, which was modeled by standard initial rate calculations (56). Udenfriend activity was determined in O2-saturated buffer (≈1 mM). No significant ·OH-mediated proteolytic degradation was observed during the 5–10 min time course of either enzymatic reaction, as monitored by denaturing gel electrophoresis.
Results and Discussion
Locations where Fe⋅His3⋅O2 sites could be introduced were identified by the dezymer program (34). From ≈200 predicted candidates, positioned throughout the protein interior and exterior, a subset of sites that were interpreted to have a local environment suitable for placement of an adjacent rudimentary oxygen binding pockets was selected by inspection. Six potential Fe⋅His3⋅O2 sites were identified in the protein (Table 1), positioned at three different locations in distinct classes of surface environment (Fig. 1): a groove (G1-G3), a shallow pocket (S1, S2), and a deep pocket (D1). The three sites at the groove location are variants of each other. G1 has an additional Y70T mutation relative to G2, thereby creating a different pocket. G3 differs in one of the three histidines in the primary coordination sphere. Consequently, in this site the Fe-O vector points toward the interior of the protein, whereas it points outward in the G1 and G2 sites. The two shallow sites differ in the nature of the oxygen binding pocket. The C2 atom of Ala-22 in the S1 site makes unfavorable steric contacts with the coordinated oxygen. This defect is removed in the S2 site by introduction of the A22G mutation. The differences between the family members generated sufficient variation in the behavior of the metal center to allow correlation between intrinsic physicochemical properties and activities, a strategy also used successfully in other design experiments (36). The full designs and precursors lacking one or two histidines (pre-GA, pre-GB, pre-SA, pre-SB, and pre-D; see Table 1 legend) were constructed by oligonucleotide-directed mutagenesis. All complete designs are folded as determined by circular dichroism, bind metal, and have catalytically active iron complexes.
Figure 1.

Models of Fe⋅His3⋅O2 sites designed in E. coli thioredoxin. (A) A tetrahedral coordination sphere (red sphere, Fe; cyan spheres, O2) was the predefined geometry used by the dezymer program (34) to generate the designs with a high-resolution x-ray structure of thioredoxin (32). (B) Six different sites were identified distributed between three different locations in the thioredoxin fold (red spheres mark the metal location in each design). (C) The variation in microenvironments around the metal centers, illustrated by the molecular surface: a groove (G1-G3), a shallow pocket (S1,2), and a deep pocket (D1). Molecular graphics were generated by the grasp program (40).
A semiquantitative survey of metal binding properties of the biologically relevant first-row transition metals showed that all of the designed centers bound metals (Fig. 2A), with FeII/III, CuII, and ZnII forming the strongest complexes overall. The metal/protein stoichiometries of the high-affinity complexes were determined to be 1:1 in all cases. Neither wild-type thioredoxin, nor any of the design precursors, lacking one or two of the three histidines, bound metals with appreciable affinities (Kd > 500 μM). FeIII affinities and stoichiometries were further investigated by spectrophotometric titrations (Fig. 3). For the cases where Kd(Fe) < [protein], the 1:1 stoichiometry of Fe binding was confirmed, and upper limits for the Kd(Fe) values were established. FeIII affinities were below 5–10 μM except in the case of S1 (Table 1), where the binding is significantly weaker, presumably because of the steric defect associated with Ala-22, which is alleviated in S2. The extinction coefficients of the 1:1 FeIII/protein complexes (Table 1) are consistent with other, similarly coordinated ferric complexes (50). The designed His3 mutations therefore result in the gain of a metal binding site with the intended 1:1 metal/protein stoichiometry.
Figure 2.

Metal-binding properties of the designed proteins. (A) Representative gel filtration elution profiles upon which the classification of binding constants is based [solid line, strong binding (Kd <10 μM); dotted line, moderate binding (10 μM < Kd < 1,000 μM)]. The position of the protein peak is indicated. Free metal is found beyond fraction 10 (not shown). (B) Block diagram of binding. Dark gray box, strong binding; light gray box, moderate binding; white box, no binding. Significant deviations from the Irving–Williams series [MnII < FeII < CoII < NiII < CuII > ZnII] are observed (57).
Figure 3.

Representative examples of spectrophotometric titrations to determine iron affinities and extinction coefficients of the FeIII/protein complexes under conditions where Kd(Fe) < < [protein] (G1; Kd = 5 μM, an upper limit estimate), and Kd(Fe) > [protein] (S1; Kd = 200 μM). The gray line indicates the protein concentration at which the experiment was done. Lines were fit to a binding isotherm that takes the protein concentration into account (35).
In unconstrained metal centers the binding constants of first-row transition metals are ordered according to the Irving–Williams series (57). None of the designs obey this trend (Fig. 2B), indicating that the primary coordination sphere is relatively rigid as a consequence of interactions with the surrounding protein matrix.
The influence of the surrounding protein matrix on the intrinsic properties of the metal center also is revealed in the variation of binding interactions between the metal and ligands such as hydroxyl (Fig. 4A) and azide (Fig. 5A). In the aerobic resting state, the iron proteins form [His3FeIIIOH]2+ or [His3FeIII(H2O)]3+ complexes. There is significant variation in the pKa values of the bound hydroxyls (Table 1). The decrease in the dielectric constant of the protein matrix toward the protein center is expected to favor the formation of the hydroxide complex rather than water complex, thus lowering the pKa. Unfortunately, there is insufficient variation in the distance of the sites from the solvent-accessible surface to establish a quantitative relationship between depth of the site and pKa value (Table 1). Nevertheless, it is clear that the deepest site (D1) has the lowest pKa. Trends in other sites are also qualitatively consistent with this effect. The steric distortion caused by the presence of Ala-22 in S1 is expected to increase solvent accessibility. This defect is corrected in the S2 site, which has a correspondingly lower pKa than S1. Variations in the groove sites also can be explained in terms of variation in depth. The metal centers in the G1, G2, and G3 sites are located at approximately the same depth. G3 has a significantly lower pKa compared with either G1 or G2, however, presumably because of the change in orientation of Fe-O vector in the G3 site, which effectively results in a more deeply buried site.
Figure 4.

Representative examples of the determination of the pKa of the bound hydroxyl in the the FeIII complex (A) and the FeIII/FeII redox potential (B). In both cases the S2 design is shown. Absorbance changes were determined at 350 nm. The pKa (7.59 ± 0.03) was determined from the indicated fit to the Henderson–Hasselbach equation. The reduction potential, E° = 117 ± 2 mV, was determined from the indicated fit to the one-electron form of the Nernst equation. NHE, normal hydrogen electrode.
Figure 5.

Azide binding. (A) Determination of azide affinity by spectrophotometric titration (S2 design). (B) The relationship between azide binding and the pKa of the bound hydroxyl distinguishes between sites where binding is dominated by direct interactions between azide and the metal center (solid line) and sites affected by interactions with the surrounding protein matrix (arrows).
Azide binding (Fig. 5A) involves an exchange reaction with the metal-bound water or hydroxide ligand. If the azide binding were determined solely by direct interactions with the metal center, then a linear correlation between the pKa of the bound hydroxyl and the azide affinity should be observed. Several sites follow this behavior (Fig. 5B). However, there are two sites that show either favorable (D1) or unfavorable (G1) interactions between the bound azide and the surrounding protein matrix. The difference in azide affinities between G1 and G2 suggests that the hydroxyl introduced in the vicinity of the metal center by the Y70T mutation either sterically interferes with azide binding or makes a favorable interaction with the bound hydroxyl. The former interpretation is more likely, however, because the pKa value of the bound hydroxyl at G1 is somewhat higher than that of G2.
The FeII/III redox potentials also exhibit location-dependent variations (Fig. 4B). The values of most of the potentials fall into the +120 mV region, similar to that of an FeEDTA complex, except for three sites. The more buried D1 and G3 sites have significantly lower potentials, whereas the more exposed S1 site has a significantly higher potential. This again suggests the influence of a general effect that is determined by the degree of burial of a site within the protein matrix.
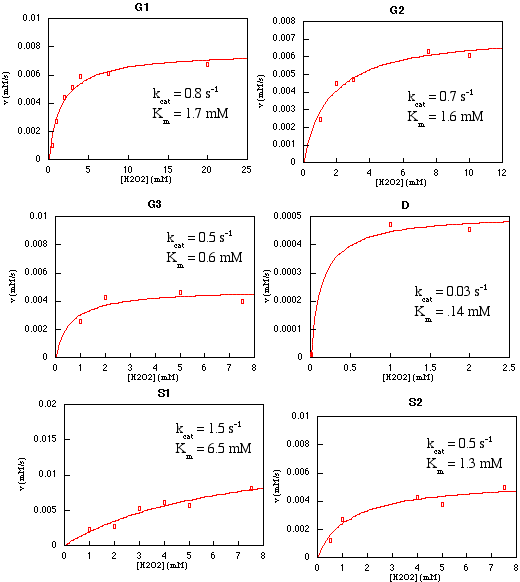
Steady-state kinetic analyses (Table 2) of the SOD (Fig. 6), Udenfriend (Fig. 7), and Fenton (Fig. 6) reactions all reveal the influence of the protein matrix on metal center reactivity. Two of the designs (S2 and D1) exhibit SOD activities appreciably above background, with S2 approaching 1% of wild-type E. coli iron SOD activity. We observe an apparent correlation between SOD activity and the number of positive charges in the vicinity of the site (Fig. 6A). SOD activity is determined in an indirect assay using enzymatically generated, low concentrations of O2⨪ (≈10 nM; ref. 58). Under these conditions, the rate of Fe⋅O2⨪ complex formation is rate limiting, because the O2⨪ concentration is much less than the ligand affinities of the metal centers (compare to azide affinities that are presumed to be strongly correlated with O2⨪ affinities, as observed in natural SODs, ref. 50). Electrostatic stabilization of complex formation would mimic the mechanism of natural SODs that have an arrangement of charges that create a funnel-shaped electrostatic field, attracting the negatively charged O2⨪ ion (59).
Table 2.
Observed catalytic activities for designed proteins
| Site | Fenton*
|
SOD
|
Udenfriend†
|
|
|---|---|---|---|---|
| kcat, s−1 | Km, mM | k, 106 M−1⋅s−1 | k, s−1 | |
| G1 | 0.80 | 1.70 | 0.75 | 9.70 |
| G2 | 0.70 | 1.60 | 0.10 | 0.01 |
| G3 | 0.50 | 0.60 | 0.10 | 16.00 |
| S1‡ | 1.50 | 6.50 | 2.30 | 0.01 |
| S2 | 0.50 | 1.30 | 6.40 | 0.01 |
| D1 | 0.03 | 0.14 | 3.30 | 30.00 |
Two types of background rates were determined: the free metal salt in buffer and precursor protein metal complexes. Background activity upper limits: SOD, k ∼ 1⋅106 M−1⋅s−1; Udenfriend, k ∼ 0.050 s−1; Fenton, kcat ∼ 0.005 s−1, Km(H2O2) ∼ 20 mM.
Absence of ascorbate reduces rates 100- to 1,000-fold, indicating that intrinsic H2O2 disproportionation is not a major contributing factor.
Pseudo first-order rate constant: M catechol, M−1; 1:1 Fe/protein, s−1.
Although results for S1 are reported, this design is omitted from the analysis of trends (see text), because its weak metal affinity precludes formation of a 1:1 complex in the absence of substantial concentrations of free metal.
Figure 6.

SOD activity. (A) Variation of the activity with respect to the weighted total, Q, of the charges, qi, contributed by all surface ionizable groups, i, within a radius, ri, from the designed site (Q = Σ qi/rI; qi = ±1). (B) Wild-type SOD reaction mechanism. (C) Rendering of charged residues in the vicinity of the designed sites by the grasp program (40), where blue and red lobes designate residues of positive or negative charge, respectively.
Figure 7.

Udenfriend activity. (A) Variation of activity with respect to redox potential. (B) Reaction mechanism. O2⨪ generated by this reaction goes on to produce ·OH in the presence of ascorbate through Haber–Weiss chemistry (23) or via the reduction of the [FeO2]2+ intermediate by ascorbate to form an FeIIIOOH intermediate, similar to activated-bleomycin (21), which decomposes to form ·OH and a transient FeIVOH species. NHE, normal hydrogen electrode.
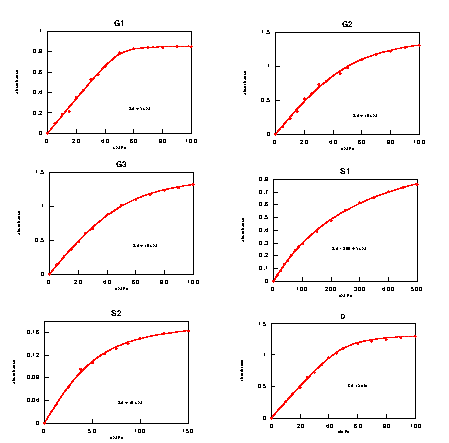
Three designs catalyze the reductive activation of O2 with rates significantly above background (Udenfriend reaction). The activities are correlated with the redox potential of the metal (Fig. 7A). This presumably reflects stabilization of the transition state of the reaction, the FeIII⋅O2⨪ resonance form (Fig. 7B).
The reductive splitting of H2O2 involves a two-step redox cycle of the iron in which the metal is regenerated by an exogenous reductant such as ascorbate (Fenton reaction; Fig. 8B). All designs show saturation binding kinetics with respect to H2O2. The Km values increase with kcat (Fig. 8A), indicating the presence of an intermediate beyond the initial ES complex (56). Such a mechanism dictates that KS > Km⋅ N3− is an analogue of HOO−, and in many systems KS(HOO−) ≈ Kd(N3−) to within an order of magnitude (45, 50, 51). In all designs, the Km(HOO−) exceeds the Kd(N3−) 10- to 100-fold, which, even given the uncertainties in the relationship between KS(HOO−) and Kd(N3−), is consistent with the buildup of an intermediate. A minimalist mechanism (23, 24) consistent with these observations is a scheme in which the rate-limiting step is the regeneration of the metal center by ascorbate-mediated reduction (Fig. 8B). This involves electron transfer between a nonspecifically bound ascorbate complex and the metal center. In this model, k3 is expected to depend on distance and driving force according to the Marcus model of electron transfer (60). kcat and Km both are correlated with the redox potential (Fig. 8A), with higher potentials being more favorable, indicative of a driving force dependence of the rate-limiting step. Distance-dependent effects are again difficult to establish quantitatively, because of the lack in variation in the degree of burial of the sites and unknown location(s) of ascorbate complexes. Nevertheless, it is apparent that the more buried G3 and D1 sites are slower, with the shallow sites again being the exception.
Figure 8.

Fenton activity. (A) Variation of kcat (black) and Km (gray) values for Fenton activity with respect to measured redox potential. (B) Minimalist model for the reaction mechanism. NHE, normal hydrogen electrode.
Suppression of toxic hydroxyl radical production by reductive cleavage of H2O2 is a critical aspect to the proper functioning of SODs. The SOD assay used in this work precludes full characterization of the reaction steady-state parameters. To compare the SOD and Fenton activities, we therefore extrapolate the rates of these two reactions to the approximate physiological concentrations of the two substrates: 6 μM for of O2⨪ (61) and 1 mM for H2O2 (62). Under these conditions, the ratio of the calculated velocities correlates with the degree of burial of the site within the protein matrix, with more buried sites preferentially selecting SOD over Fenton activity. Because these buried sites do not have the highest SOD activities, the effect is caused by suppression of Fenton activity, rather than enhancement of SOD. As shown above, the rate-limiting step in the Fenton chemistry is the exogenous reduction of the metal center, which is controlled both by driving force (redox potential) and distance of electron transfer through the protein matrix. The deeper sites have more favorable driving forces than the surface sites. This suggests that the Fenton reaction is predominantly suppressed by a distance-dependent insulating effect in which the protein matrix acts as an insulator preventing nonspecific electron transfer. Indeed, all of the metal centers in natural SODs as well as many other metalloenzymes are deeply buried within their protein matrix (25), presumably reflecting a general mechanism for the suppression of nonspecific redox reactions.
Conclusions
The nascent metalloenzymes presented here exhibit the emergence of mechanisms for controlled catalytic activity. This is the consequence of a hierarchy of structure-function relationships inherent even within a protein that has no prior adaptations for the control of metal center activity: location of a metal center determines its microenvironment, and hence catalytic activities, ultimately leading to a degree of specificity required for biological function. The emergence of both stabilizing and destabilizing interactions are observed, including stabilization of complex formation by surface electrostatics (SOD), stabilization of transition states by metal reduction potential (Udenfriend and Fenton), and destabilization of nonspecific electron transfer-dependent reactions (Fenton).
Of particular interest is the importance of competing state destabilization, or “negative design” (37, 63–65), which can be considered to be a cryptic protein function, because it cannot be identified until the competing reactions have been uncovered. Such mechanisms are particularly difficult to establish in an evolved protein, especially because the destabilization mechanism may involve multiple, long-range interactions, as appears to be the case in preventing damaging free radical production in SODs via the Fenton reaction. Rational design experiments are increasingly demonstrating that competing state destabilization can be a dominant factor in the control of specificity (37, 63–65).
The nascent metalloenzymes also exhibit divergence of activity and could serve as potential starting points for evolution pathways. Several other, nonenzymatic redox-active or structural metal centers also have been successfully introduced into the thioredoxin fold (35–39), further illustrating the potential of a given protein to evolve in different directions. This experimental demonstration of multiple, independent, multifunctional starting points for potentially parallel paths of evolution within a single protein fold has implications both for protein design and understanding the early genesis of new functions in molecular evolution.
Supplementary Material
Acknowledgments
We thank Dr. G. G. Hammes for discussions. This work was supported by National Institutes of Health Grant GM49871 (H.W.H.) and National Institutes of Health Fellowship GM19459–01A1 (D.E.B.).
Abbreviations
- O2⨪
superoxide
- SOD
superoxide dismutase
References
- 1.Hellinga H W. In: Protein Engineering: Principles and Practice. Cleland J L, Craik C S, editors. New York: Wiley–Liss; 1996. pp. 369–398. [Google Scholar]
- 2.Bryson J W, Betz S F, Lu H S, Suich D J, Zhou H X, O'Neil K T, DeGrado W F. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 3.Regan L. Trends Biochem Sci. 1995;20:280–285. doi: 10.1016/s0968-0004(00)89044-1. [DOI] [PubMed] [Google Scholar]
- 4.Malakauskas S M, Mayo S L. Nat Struct Biol. 1998;5:470–475. doi: 10.1038/nsb0698-470. [DOI] [PubMed] [Google Scholar]
- 5.Hellinga H W. Nat Struct Biol. 1998;5:525–527. doi: 10.1038/776. [DOI] [PubMed] [Google Scholar]
- 6.DeGrado W F, Summa C M, Pavone V, Nastri F, Lombardi A. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 7.Lu Y, Valentine J S. Curr Opin Struct Biol. 1997;7:495–500. doi: 10.1016/s0959-440x(97)80112-1. [DOI] [PubMed] [Google Scholar]
- 8.Pabo C. Nature (London) 1983;301:200. doi: 10.1038/301200a0. [DOI] [PubMed] [Google Scholar]
- 9.Knowles J R. Science. 1987;236:1252–1258. doi: 10.1126/science.3296192. [DOI] [PubMed] [Google Scholar]
- 10.DeGrado W F. Nature (London) 1993;365:488–489. doi: 10.1038/365488a0. [DOI] [PubMed] [Google Scholar]
- 11.Distefano M D, Kuang H, Qi D F, Mazhary A. Curr Opin Struct Biol. 1998;8:459–465. doi: 10.1016/s0959-440x(98)80123-1. [DOI] [PubMed] [Google Scholar]
- 12.Benson D E, Wisz M S, Hellinga H W. Curr Opin Biotechnol. 1998;9:370–376. doi: 10.1016/s0958-1669(98)80010-4. [DOI] [PubMed] [Google Scholar]
- 13.Hahn K W, Klis W A, Stewart J M. Science. 1990;248:1544–1547. doi: 10.1126/science.2360048. [DOI] [PubMed] [Google Scholar]
- 14.Corey M J, Corey E. Proc Natl Acad Sci USA. 1996;93:11428–11434. doi: 10.1073/pnas.93.21.11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies R R, Distefano M D. J Am Chem Soc. 1997;119:11643–11652. [Google Scholar]
- 16.Kuang H, Davies R R, Distefano M D. Bioorg Med Chem Lett. 1997;7:2055–2060. [Google Scholar]
- 17.Quemeneur E, Moutiez M, Charbonnier J B, Menez A. Nature (London) 1998;391:301–304. doi: 10.1038/34687. [DOI] [PubMed] [Google Scholar]
- 18.Kaim W, Schewderski B. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life. New York: Wiley; 1994. [Google Scholar]
- 19.Que L J, Ho R Y N. Chem Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 20.Kitajima N, Tolman W B. Prog Inorg Chem. 1995;43:419–531. [Google Scholar]
- 21.Feig A L, Lippard S J. Chem Rev. 1994;94:759–805. [Google Scholar]
- 22.Bilinski T. Biosystems. 1991;24:305–312. doi: 10.1016/0303-2647(91)90049-q. [DOI] [PubMed] [Google Scholar]
- 23.Sawyer D T. Oxygen Chemistry. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 24.Valentine J S. In: Bioinorganic Chemistry. Bertini I, Gray H B, Lippard S J, Valentine J S, editors. Mill Valley, CA: University Science; 1994. pp. 253–313. [Google Scholar]
- 25.Valentine J, Wertz D, Lyons T, Liou L, Goto J, Gralla E. Curr Opin Chem Biol. 1998;2:253–262. doi: 10.1016/s1367-5931(98)80067-7. [DOI] [PubMed] [Google Scholar]
- 26.McCord J M, Fridovich I. J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 27.Yost F J, Fridovich I. J Biol Chem. 1973;248:4905–4908. [PubMed] [Google Scholar]
- 28.Gregroy E M, Yost F J, Jr, Fridovich I. J Bacteriol. 1973;115:987–991. doi: 10.1128/jb.115.3.987-991.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walling C. Acc Chem Res. 1975;8:125–132. [Google Scholar]
- 30.Goto J J, Gralla E B, Valentine J S, Cabelli D E. J Biol Chem. 1998;273:30104–30109. doi: 10.1074/jbc.273.46.30104. [DOI] [PubMed] [Google Scholar]
- 31.Waggoner D J, Bartnikas T B, Gitlin J D. Neurobiol Dis. 1999;6:221–230. doi: 10.1006/nbdi.1999.0250. [DOI] [PubMed] [Google Scholar]
- 32.Katti S K, LeMaster D M, Eklund H. J Mol Biol. 1990;212:167–184. doi: 10.1016/0022-2836(90)90313-B. [DOI] [PubMed] [Google Scholar]
- 33.Hellinga H W, Wynn R, Richards F M. Biochemistry. 1992;31:11203–11209. doi: 10.1021/bi00160a034. [DOI] [PubMed] [Google Scholar]
- 34.Hellinga H W, Richards F M. J Mol Biol. 1991;222:763–785. doi: 10.1016/0022-2836(91)90510-d. [DOI] [PubMed] [Google Scholar]
- 35.Benson D E, Wisz M S, Liu W, Hellinga H W. Biochemistry. 1998;37:7070–7076. doi: 10.1021/bi980583d. [DOI] [PubMed] [Google Scholar]
- 36.Wisz M S, Garrett C Z, Hellinga H W. Biochemistry. 1998;37:8269–8277. doi: 10.1021/bi980718f. [DOI] [PubMed] [Google Scholar]
- 37.Hellinga H W. J Am Chem Soc. 1998;120:10055–10066. [Google Scholar]
- 38.Coldren C D, Hellinga H W, Caradonna J P. Proc Natl Acad Sci USA. 1997;94:6635–6640. doi: 10.1073/pnas.94.13.6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinto A L, Hellinga H W, Caradonna J P. Proc Natl Acad Sci USA. 1997;94:5562–5567. doi: 10.1073/pnas.94.11.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicholls A, Honig B. J Comp Chem. 1991;12:435–445. [Google Scholar]
- 41.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 42.Lovenberg W, Buchanan B B, Rabinowitz J C. J Biol Chem. 1963;238:3899–3913. [PubMed] [Google Scholar]
- 43.Evans P J, Halliwell B. Methods Enzymol. 1994;233:82–92. doi: 10.1016/s0076-6879(94)33010-7. [DOI] [PubMed] [Google Scholar]
- 44.Hunt J B, Neece S H, Ginsburg A. Anal Biochem. 1985;146:150–157. doi: 10.1016/0003-2697(85)90409-9. [DOI] [PubMed] [Google Scholar]
- 45.Vance C K, Miller A-F. Biochemistry. 1998;37:5518–5527. doi: 10.1021/bi972580r. [DOI] [PubMed] [Google Scholar]
- 46.Fultz M L, Durst R A. Anal Chim Acta. 1982;140:1–8. [Google Scholar]
- 47.Stankovich M T. Anal Biochem. 1980;109:295–308. doi: 10.1016/0003-2697(80)90652-1. [DOI] [PubMed] [Google Scholar]
- 48.Dutton P L. Methods Enzymol. 1978;54:411–435. doi: 10.1016/s0076-6879(78)54026-3. [DOI] [PubMed] [Google Scholar]
- 49.Wilson G S. Methods Enzymol. 1978;54:396–410. doi: 10.1016/s0076-6879(78)54025-1. [DOI] [PubMed] [Google Scholar]
- 50.Bull C, McClune G J, Fee J A. J Am Chem Soc. 1983;105:5290–5300. [Google Scholar]
- 51.Bull C, Fee J A. J Am Chem Soc. 1985;107:3295–3304. [Google Scholar]
- 52.Sutherland M W, Learmonth B A. Free Radical Res. 1997;27:283–289. doi: 10.3109/10715769709065766. [DOI] [PubMed] [Google Scholar]
- 53.Ukeda H, Maeda S, Ishii T, Sawamura M. Anal Biochem. 1997;251:206–209. doi: 10.1006/abio.1997.2273. [DOI] [PubMed] [Google Scholar]
- 54.Goldstein S, Michel C, Bors W, Saran M, Czapski G. Free Radical Biol Med. 1988;4:295–303. doi: 10.1016/0891-5849(88)90050-0. [DOI] [PubMed] [Google Scholar]
- 55.Goscin S A, Fridovich I. Arch Biochem Biophys. 1972;153:778–783. doi: 10.1016/0003-9861(72)90398-0. [DOI] [PubMed] [Google Scholar]
- 56.Fersht A. Enzyme Structure and Mechanism. New York: Freeman; 1999. [Google Scholar]
- 57.Shriver D F, Atkins P W, Langford C H. Inorganic Chemistry. New York: Freeman; 1990. [Google Scholar]
- 58.Bull C, Fee J A, O'Neill P, Fielden E M. Arch Biochem Biophys. 1982;215:551–555. doi: 10.1016/0003-9861(82)90115-1. [DOI] [PubMed] [Google Scholar]
- 59.Getzoff E D, Cabelli D E, Fisher C L, Parge H E, Viezzoli M S, Banci L, Hallewell R A. Nature (London) 1992;358:347–350. doi: 10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- 60.Marcus R A, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 61.Imlay J A, Fridovich I. J Biol Chem. 1991;266:6957–6965. [PubMed] [Google Scholar]
- 62.Starke D W, Chen Y, Bapna C P, Lesnefsky E J, Mieyal J J. Free Radical Biol Med. 1997;23:373–384. doi: 10.1016/s0891-5849(97)00009-9. [DOI] [PubMed] [Google Scholar]
- 63.Hecht M H, Richardson J S, Richardson D C, Ogden R C. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 64.Quinn T P, Tweedy N B, Williams R W, Richardson J S, Richardson D C. Proc Natl Acad Sci USA. 1994;91:8747–8751. doi: 10.1073/pnas.91.19.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richardson J S, Richardson D C, Tweedy N B, Gernert K M, Quinn T P, Hecht M H, Erickson B W, Yan Y, McClain R D, Donlan M E, et al. Biophys J. 1992;63:1185–1209. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
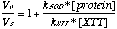{kind=link}