

Abstract
The seven members of the human 14-3-3 protein family regulate a diverse range of cell signaling pathways by formation of protein–protein complexes with signaling proteins that contain phosphorylated Ser/Thr residues within specific sequence motifs. Previously, crystal structures of three 14-3-3 isoforms (zeta, sigma, and tau) have been reported, with structural data for two isoforms deposited in the Protein Data Bank (zeta and sigma). In this study, we provide structural detail for five 14-3-3 isoforms bound to ligands, providing structural coverage for all isoforms of a human protein family. A comparative structural analysis of the seven 14-3-3 proteins revealed specificity determinants for binding of phosphopeptides in a specific orientation, target domain interaction surfaces and flexible adaptation of 14-3-3 proteins through domain movements. Specifically, the structures of the beta isoform in its apo and peptide bound forms showed that its binding site can exhibit structural flexibility to facilitate binding of its protein and peptide partners. In addition, the complex of 14-3-3 beta with the exoenzyme S peptide displayed a secondary structural element in the 14-3-3 peptide binding groove. These results show that the 14-3-3 proteins are adaptable structures in which internal flexibility is likely to facilitate recognition and binding of their interaction partners.
Keywords: phosphorylation, signaling
The 14-3-3 protein family contains seven members that are highly conserved and ubiquitously expressed (1). Later, sequencing of the human genome revealed seven isoforms: beta (β), epsilon (ε), eta (η), gamma (γ), tau (τ), sigma (σ), and zeta (ζ), each encoded by a distinct gene. The 14-3-3 proteins form homo- and heterodimers (2, 3) that have been shown to interact with a large number of proteins, e.g., γ was shown in a yeast two-hybrid study to have as many as 130 potential binding partners (4). The 14-3-3 proteins are involved in the regulation of metabolism, signal transduction, cell-cycle control, apoptosis, protein trafficking, transcription, stress responses, and malignant transformation (5, 6) mainly through binding to phosphopeptides, thus modulating signaling events. Examples of well validated 14-3-3 interaction partners are the proto-oncogene RAF-1 (7–9) and the cell-cycle regulatory phosphatases Cdc25C/B (10, 11).
All isoforms recognize two high-affinity phosphorylation-dependent 14-3-3 binding motifs: RSXpSXP (mode 1) and RXXXpSXP (mode 2), where pS represents a phosphoserine (12–14). The 14-3-3 proteins can also recognize unmodified proteins such as the Pseudomonas aeruginosa virulence factor exoenzyme S (ExoS), p190RhoGEF, and the phage display-derived R18 peptide inhibitor (15–19). Recently, a small number of proteins that interact with 14-3-3 proteins via a C-terminal phosphorylation motif have also been identified (20, 21). Phosphorylation-dependent and -independent binding have been shown to be targeted to the same site of the 14-3-3 proteins (18).
The 14-3-3 proteins have been found to be up- or down-regulated in human disease, but their direct role in disease progression has not been clearly established. Examples of such disease conditions are as follows: (i) the σ isoform has been implicated in breast cancer (22); (ii) ζ has been implicated in neurological disorders (23); and (iii) γ in the cerebrospinal fluid (CSF) can be used as a marker for sporadic Creutzfeldt-Jakob disease (CJD) (24, 25), although studies on a γ-deficient mutant mouse showed that it is unlikely to play a causal role. In addition, β, ε, and η are also found in the CSF of CJD patients (26); (iv) β, γ, and τ expression levels are increased in lung cancer as compared with the equivalent normal tissues (27); (v) τ has been shown to be a specific marker of lung cancer (28); and (vi) σ is necessary for proper G2 checkpoint function (29).
Further evidence of the importance of 14-3-3 proteins has been provided in mouse studies. (i) Mice deficient in ε have defects in brain development and neuronal migration. Moreover, it has been shown in humans to be absent in certain human neuronal migration disorders such as the Miller–Dieker syndrome (30). (ii) Dominant-negative mutant forms of ζ were shown to inhibit ERK MAPK activation but increased the activation of JNK1 and p38 MAPK leading to increased apoptosis (31), and (iii) a dominant-negative η increased the basal activation of JNK1 and p38 MAPK and affected the ability of mice to compensate for pressure overload resulting in increased mortality, cardiomyopathy, and massive cardiomyocyte apoptosis (31). In contrast, the survival rate, anatomy, and cage behavior of γ-deficient mice were normal (32). A recent example displaying the physiological importance of 14-3-3 proteins demonstrated that they are required for lifespan extensions in C. elegans when promoted by extra copies of the sir-2.1 gene (33).
The mechanisms of 14-3-3 action have been suggested to include the following: (i) inducing conformational changes of the target protein, (ii) physically occluding sequence-specific or structural features, (iii) scaffolding, and (iv) changing cellular localization (34). Previously, a number of three-dimensional structures of 14-3-3 isoforms have been determined and deposited in the Protein Data Bank (PDB; www.pdb.org): ζ, in its apo form (35), as peptide complexes (14, 17, 18) and in complex with serotonin N-acetyltransferase (AANAT) (36); and σ in apo- and peptide-bound states (37, 38). In addition, the structure of τ was published (39) but not deposited in the PDB. These structural data showed that the 14-3-3 proteins adopted a similar conformation in the unliganded and peptide/protein-bound structures, to which the interaction partner adapts, and led to the hypothesis that the 14-3-3 proteins may act as “molecular anvils” (40).
We set out to determine the structures of all remaining 14-3-3 isoforms (including the τ isoform, PDB ID 2BTP) to be able to conduct a thorough comparative structural analysis and in addition performed a study on dimerization preferences. Here we report the structures of apo-β, β in complex with an ExoS peptide, ε as a mode 2 peptide complex, η as both mode 1 and 2 peptide complexes, γ as a mode 2 complex, and τ as a mode 1 peptide complex, and we analyze them together with the available σ and ζ structures.
Results and Discussion
Structure Overview.
The overall structural features of the 14-3-3 protein family members were recently reviewed (41), and thus only a brief description is provided here. The determined 14-3-3 crystal structures were all homodimers. Each monomer consists of a bundle of nine α-helices (αA to αI) organized into groups of two, two, two, and three helices (39, 42). The first four are essential for formation of the dimer, which has a sizeable aperture at the subunit interface (Fig. 1). Helices αC, αE, αG, and αI form a conserved peptide-binding groove, which has a positively charged patch on one side and a hydrophobic patch on the other (39, 42). The positively charged patch is formed by a conserved triad of two arginines and a tyrosine residue (Arg-57, Arg-130, and Tyr-131; residue numbers will all refer to the ε isoform unless otherwise specified) that bind the phosphate group of the interacting phosphopeptide/protein. Tables 1 and 2, which are published as supporting information on the PNAS web site, contain the crystallization and data collection details for the seven structures generated in this study.
Fig. 1.

Overview of the dimeric 14-3-3 structure. Helices and loops involved in target domain interactions are labeled. Each monomer is colored blue to red from the N to C terminus. An aperture exists at the central dimeric interface, which is marked with a circle.
Dimerization Preferences.
Previous studies have shown that, apart from σ, several of the isoforms are able to form heterodimers (2, 3). The presence of unique residues at the σ dimer interface provided a clear structural explanation for its homodimerization preference (38). Using an in vivo-based approach, Chaudhri et al. (2) found that the ε isoform preferentially formed heterodimers with no observable homodimer formation. In addition phosphorylation influences the dimerization process (43, 44).
In this study, we investigated the dimerization equilibria of the β, ε, γ, η, and τ isoforms using an in vitro-based approach allowing a quantitative description of both the homo- and heterodimerization events.
Dimerization Equilibria of 14-3-3 Proteins.
Dimerization equilibria of β, ε, γ, η, and τ were investigated by nanoelectrospray ionization mass spectrometry under conditions where noncovalent complexes are preserved, i.e., gentle ionization and desolvation conditions and the appropriate pressure settings (see ref. 45). All five isoforms were detected as dimers (Fig. 6A, which is published as supporting information on the PNAS web site). The ε and γ isoforms were almost entirely dimeric, τ was mostly dimeric, and β and η were found to be in equilibrium with the monomeric form (70:30 ratio, dimer/monomer) under the same experimental conditions. This ratio reflects the relative stability of the dimeric interaction in solution, which appears to be higher for ε and γ as compared with β and η.
We also studied the formation of heterodimers between four different 14-3-3 isoforms. Different isoforms were mixed and left overnight before measurements were taken. Shorter incubation periods, for instance, of 1 h, did not allow the subunit exchange to proceed to completion. The results of this interaction study, presented as percentage of observed heterodimers, are as follows: β/ε, >95%; β/γ, 50%; β/τ, 50%; ε/γ, >95%; ε/τ, >95%; and γ/τ, 50%. The ε isoform showed a preference for binding of β, γ, and τ to an extent that virtually no homodimers were detected (Fig. 6B). Thus, the ε subunit has a higher affinity for the other subunits than for itself. In all of the other cases that do not involve the ε isoform, a statistical distribution of XX/XY/YY with a 1:2:1 ratio was formed with 50% heterodimer in the mixture (Fig. 6). The statistical formation of heterodimers between the β, γ, and τ isoforms suggests that their binding affinity to each other is similar, whereas the ε isoform shows a strong preference for heterodimerization.
Although 14-3-3 proteins commonly form dimers, the monomers are sometimes functional, depending on the target protein. For example, monomeric ζ is able to modulate the activity of a potassium channel (46) but not Raf kinase (47). Although these results were based on the use of a mutant ζ form that is unable to dimerize, our analysis shows that the native protein exists in both monomeric and dimeric states and suggests that these 14-3-3 proteins may use the dimerization process to control their cellular activities.
The molecular mechanism for dimerization and the basis for the preference for homo- and heterodimer formation can in part be deduced from the panel of crystal structures available. Recently, Gardino et al. (41) reviewed the structural determinants for dimer formation, and thus only a brief description of the main features is presented here. The dimerization interface around the aperture of all human 14-3-3 isoforms involves a conserved salt bridge between Arg-19 and Glu-92. Besides this interaction, two residues (ζ: Asp-21 and Lys-85) are conserved in all human isoforms except ε (equivalent ε residues being Glu-22 and Met-88). In all of the homodimer structures, the side chains at these positions do not interact. Only with ε as one subunit of a heterodimer is an additional hydrogen-bonding opportunity created, as illustrated schematically in Fig. 2, which is likely the contributing factor for its preference to form heterodimers.
Fig. 2.

Schematic representation of the heterodimerization process involving the ε (green) and ζ (yellow) isoforms. The lines between identified residues indicate specific interactions.
Binding of 14-3-3 Proteins to Interaction Partners.
Generally, proteins that interact with a 14-3-3 dimer are composed of a globular domain with an unstructured region that contains a phosphorylated Ser/Thr residue within a mode 1 or 2 binding motif. In the binding event, two processes likely occur: binding of the phosphorylated peptide to the conserved groove (αC, αE, αG, and αI), which we define as the primary interaction and interaction of the globular domain of the protein with the remaining sections of the 14-3-3, which we define as secondary interaction. The availability of three-dimensional structures for all human isoforms now allows us to rationalize this concept at a detailed structural level.
Primary Interaction.
The 14-3-3 family members have the potential to interact with a large but distinct number of phosphorylated proteins. In the phosphopeptide-interacting motif, the orientation of the phosphate group is fixed once bound to the Arg–Arg–Tyr triad; however, the phosphoserine Cα Cβ bond is free to rotate. Thus, for an extended peptide that binds along the groove, two orientations are possible, differing by a twofold rotation around the CαCβ bond. When analyzing the ligand complexes (ε, γ, η, τ, σ, and ζ), it is apparent that there is specificity in the relative orientation of the bound peptide. All of the phosphorylated peptides bind with the N to C termini being orientated similarly and interacting with three conserved features. (i) The Asn-176 side chain has an orientation that is fixed as a result of a hydrogen bond with the conserved Asp-127 as well as hydrogen bonds with an amide on the peptide backbone of the ligand. This interaction occurs only in the observed orientation that also allows the conserved Asn-227 to make hydrogen bonds with the ligand backbone (Fig. 3A). (ii) The extended phosphopeptide has most of its side chains pointing away from the 14-3-3 molecule, thus explaining the absolute requirement for peptide direction as if it were reversed (by 180°) steric clashes of the side chains would occur (Fig. 3B). (iii) A conserved hydrophobic patch (Leu-175, Leu-219, and Ile-220) within the 14-3-3 binding groove complements the hydrophobic character of the peptide on the C-terminal side of the phosphoserine (Fig. 3C).
Cβ bond is free to rotate. Thus, for an extended peptide that binds along the groove, two orientations are possible, differing by a twofold rotation around the CαCβ bond. When analyzing the ligand complexes (ε, γ, η, τ, σ, and ζ), it is apparent that there is specificity in the relative orientation of the bound peptide. All of the phosphorylated peptides bind with the N to C termini being orientated similarly and interacting with three conserved features. (i) The Asn-176 side chain has an orientation that is fixed as a result of a hydrogen bond with the conserved Asp-127 as well as hydrogen bonds with an amide on the peptide backbone of the ligand. This interaction occurs only in the observed orientation that also allows the conserved Asn-227 to make hydrogen bonds with the ligand backbone (Fig. 3A). (ii) The extended phosphopeptide has most of its side chains pointing away from the 14-3-3 molecule, thus explaining the absolute requirement for peptide direction as if it were reversed (by 180°) steric clashes of the side chains would occur (Fig. 3B). (iii) A conserved hydrophobic patch (Leu-175, Leu-219, and Ile-220) within the 14-3-3 binding groove complements the hydrophobic character of the peptide on the C-terminal side of the phosphoserine (Fig. 3C).
Fig. 3.

The selective nature of the primary interaction site. (A) Close-up view with side chain interactions highlighted for Pep1 in the binding groove of ε. The N- to C-terminal orientation is the same in all other 14-3-3 structures with phosphopeptides. (B) Same view as in A except with the ε peptide binding groove surface colored yellow. Reverse orientation of the same peptide (blue wire frame) about the phosphoserine Cα–Cβ bond results in major clashes. (C) Same view as in A, now with the character of the residues that make up the peptide binding groove color coded onto the surface as yellow (hydrophobic), red (negatively charged), blue (positively charged), and gray (neutral).
In conclusion, general features of 14-3-3–phosphopeptide interactions are that it relies on fixed positions of the binding pocket side chains and orientation of the peptide. Interactions are formed only with the main chain of the bound peptide and its phosphate group. For example, the hydrogen bond from Asn-176 to the backbone amide nitrogen in the +2 main chain atom position of the peptide would not be available with a peptide in the reversed orientation. In contrast, interactions with unphosphorylated peptides such as R18 and ExoS that have a reversed orientation are dependent on sequence-specific interactions with the peptide side chains.
Secondary Interaction.
According to this hypothesis, once the phosphopeptide has bound, the target protein would bind to the remaining sections of the 14-3-3 protein. Determination of the crystal structure of ζ in complex with AANAT (36) permitted the identification of the loop between helices αH and αI (HI loop) as being critical for binding. This interaction, as well as others potentially critical for binding, was recently further analyzed (41).
To complement the observations from the crystallographic data, we conducted a further analysis by applying the optimal docking area (ODA) methodology, which is based on atomic desolvation parameters adjusted for protein–protein docking (48). This method identifies continuous surface patches likely to be involved in protein–protein interactions and was initially validated by using a set of 66 unbound protein structures (48) and, later in the prediction “competition” CAPRI (critical assessment of predicted interactions), where it accurately predicted eight of nine protein–protein complexes (49)
The results from the ODA analysis are shown in Fig. 4, where red patches represent likely protein–protein interaction interfaces. No low-desolvation patches were observed for any of the isoforms in the conserved peptide binding groove or on the opposite face of each monomer. Two major ODA sites occur consistently for all 14-3-3 family members: site S1, located at the observed dimerization interface, and site S2 in the solvent accessible side of the helices αH and αI, located close to the previously identified specificity region (37).
Fig. 4.

Calculated desolvation energies mapped onto the monomeric surface representations for each of the seven human isoforms. All views look down onto the dimerization interface (Left), central binding groove, and the αG–αI helices (Right). The desolvation energies are color coded from high (blue) to low (red). The sequence conservation is mapped onto a tube representation of the τ isoform, color coded from green (100%) to white. The variable CD and HI loops are labeled V1 and V2, respectively, and the two conserved low-energy desolvation sites are labeled S1 (S1a and S1b) and S2, respectively.
Based on the analysis of a few 14-3-3 interactions, the sequence variability within the HI loop was postulated to be of critical importance for isoform/target specificity. The S1 patch constitutes a recognition motif involved in the dimerization interface. However, analysis of target structures revealed that the S1 patch could be distinguished into two contributing “subsites.” We suggest that the first (S1a), is directly involved in the dimerization process, whereas the second site (S1b) contributes to target protein interactions. The S1b region has several similarities to S2: (i) the residues responsible for the low desolvation energy are conserved; (ii) the immediate vicinity of the desolvation patch consists of flexible loops; (iii) the residues in the loops are not well conserved; and (iv) there is a preference for charged residues in the loops. Validating this approach, the S1b and S2 patches are extensively involved in forming protein–protein contacts within the crystals (Table 3, which is published as supporting information on the PNAS web site).
We suggest that the target protein is attracted first by general protein–protein interaction motifs (the desolvation patches) followed by chain/loop rearrangements that contribute to the formation of specific contacts. In keeping with this hypothesis, the CD and HI loops appear to be flexible: in all crystal structures of the human 14-3-3 proteins, they appear either as regions with high thermal motion or as unstructured regions.
Binding Mechanism.
The crystal structures of the first three 14-3-3 proteins (τ, σ, and ζ) adopted a similar conformation in both apo- and ligand-bound forms (here denoted as the “closed state”). These initial data gave rise to the hypothesis that the 14-3-3 proteins may have a fairly rigid conformation and that the interacting protein had to adapt to enable the interaction. However, the structure determination of the apo-β isoform in this study clearly shows that the peptide-binding site on the 14-3-3 protein allows flexible adaptation. In the apo-β structure, one of the monomers was captured in the closed state conformation, whereas the phosphopeptide binding site in the opposite subunit had an open conformation following a ≈20° rotation of the αG to αI helices, producing a shallow and exposed groove (Fig. 5A). Because β does not contain any unusual sequence motifs, it is possible that such flexibility is a feature of all isoforms and the variation in size of the peptide binding groove allows binding to peptides of diverse sequence and differing secondary structures. Based on the structural data, another suggested mechanism by which the 14-3-3 proteins can facilitate binding of diverse target proteins is through conformational heterogeneity to alter the angle between the two subunits. Superimposition of one subunit for all of the closed state 14-3-3 dimers shows that there is a significant difference in relative position of the partner subunit (Fig. 5B). This is particularly evident for the β (PDB ID 2C23; blue) and τ (PDB ID 2BTP; green) isoforms.
Fig. 5.

Dynamic nature of the 14-3-3 dimers. (A) Crystal structure of the apo-β isoform looking down the peptide binding grooves, which are labeled open and closed for the individual monomers. (B) Superimposition of all seven closed state 14-3-3 isoforms using only one monomer as the reference, with β shown in blue and τ in green. The other 14-3-3 monomers, which have intermediate positions, are colored transparent gray.
To test whether the β isoform would undergo a conformational change upon binding of a nonphosphorylated target, we determined the structure of β in complex with a peptide based on the sequence of the P. aeruginosa ExoS. In this structure, the ExoS peptide is bound to the conserved binding groove with both 14-3-3 subunits adopting a closed conformation. We obtained this structure by soaking crystals of apo-β with the ExoS peptide. The conformational change took place in the crystalline form and resulted in a change of crystal symmetry to one in which both 14-3-3 subunits are identical. The structure of this complex also allowed us to further rationalize target specificity dependent on the phosphorylation status of the interaction partner. Both ExoS (this study) and R18 (17, 18) bind with their N to C termini in the opposite direction compared with phosphorylated peptides. In addition, when ExoS is bound to the 14-3-3 partner, it forms a helical structure (Fig. 7, which is published as supporting information on the PNAS web site), showing a secondary structural element in the 14-3-3 peptide binding groove, as predicted for the general mode of 14-3-3 binding by Liu et al. (35), before the phosphorylated 14-3-3 binding sequence motifs were discovered.
The orientation and structure of the ExoS helix are stabilized by the interaction of β Asn-175 with backbone carbonyl (Asp-427) and amide (Ala-429) groups at the C terminus of the ExoS fragment peptide. These interactions define the end of the helix while the C terminus is in an extended conformation. The interaction is stabilized by a hydrogen bond between Tyr in the conserved 14-3-3 Arg–Arg–Tyr triad and the negatively charged Asp-427 of ExoS, a hydrogen bond between the conserved Lys-51 and Asp-424 of ExoS, and interactions between hydrophobic side chains in the ExoS peptide and the conserved hydrophobic patch on the surface of the 14-3-3 peptide binding groove. A superimposition of the closed state of apo-β and the closed state of ExoS-β showed that the ExoS-β interface was rotated ≈4°, displaying further flexibility most likely incurred by the specific binding partner, but we cannot exclude that this is due to crystal packing effects. However, the helical conformation of the ExoS peptide appears to push the 14-3-3 αH and αI helices backwards, which results in the 4° rotation difference.
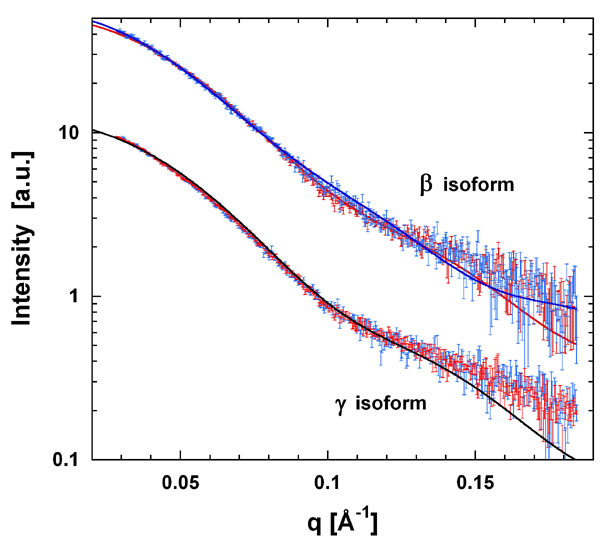
To analyze any change in conformation on peptide binding we measured the radius of gyration (Rg) of β and γ with and without the phosphoserine peptide Pep2 by small angle x-ray scattering. The observed changes in Rg upon peptide binding were from 30.7 ± 0.3 Å to 30.2 ± 0.3 Å and 31.3 ± 0.2 Å to 31.1 ± 0.2 Å for β and γ, respectively. A change from a fully open to a closed conformation as indicated by the apo-β and peptide-bound β structures would correspond to a reduction in Rg of 7%. The 1–2% reduction observed is smaller, although still significant, and consistent with a dynamic 14-3-3 dimer in solution. The apo measurements represent a conformational average, with Rg in the presence of Pep2 only slightly smaller than this average. Comparing the experimental scattering data with the crystal structures shows that the peptide-bound closed conformations also provide acceptable fits to the scattering curves of the apo-forms (Fig. 8, which is published as supporting information on the PNAS web site).
Based on the results from this structural comparison of all 14-3-3 members, we suggest that conformational flexibility in both the peptide binding site and in the dimer interface facilitates binding of 14-3-3 proteins to diverse peptides and proteins of varying size and sequence. Specifically, we propose that the initial interaction involves conformational changes of the three C-terminal helices (αG to αI). Through these changes, the 14-3-3 protein could adapt to the type of bound peptide whether it is phosphorylated and extended or nonphosphorylated and helical. The flexibility at the dimerization interface provides a mechanism for the 14-3-3 proteins to bind proteins of varying size. This versatility is most likely enhanced by the presence of desolvation sites (S1b and S2) providing nonspecific protein–protein interaction motifs, with specificity features primarily from the variable V1 and V2 regions (see Fig. 4). Further specificity in 14-3-3 regulation of intracellular signaling is most likely provided by its tissue (50, 51), temporal distribution (52, 53), and subcellular localization (41, 54), allowing this small protein family to be a central and specific regulator through its ability to adapt its conformation to interact with a variety of binding partners.
Materials and Methods
Protein Expression and Purification.
DNA for β, ε, η, γ, and τ was PCR amplified from clones in the Mammalian Gene Collection (I.M.A.G.E. Consortium Clone IDs 3051079, 2900956, 3915246, 3543571, and 6164592, respectively) and inserted into vectors containing either an N- or C-terminal hexahistidine tag and TEV protease tag cleavage site (Table 4, which is published as supporting information on the PNAS web site). The resulting plasmids were transformed into E. coli BL21 (DE3) cells for expression. Cultures were grown in Terrific Broth media at 37°C until an OD600 of 0.6 was reached. The temperature was then reduced to 25°C for 1 h before induction by addition of 1 mM isopropyl-β-d-thiogalactopyranoside. Protein expression was allowed to continue for 4 h before cell harvesting by centrifugation. The cells were resuspended in 20–50 mM Tris·HCl (pH 8.0), 200–500 mM NaCl, 5% glycerol, 10 mM imidazole, 0.5 mM TCEP, and Complete EDTA-free protease inhibitor mixture (Roche Applied Science, Burgess Hill, U.K.). The cells were lysed by high-pressure homogenization, and the insoluble debris was removed by centrifugation.
The cell extracts were passed over Ni2+ resin, and the resin was then washed with 50 mM Tris·HCl (pH 8.0), 150–500 mM NaCl, 5% glycerol, 25 mM imidazole (pH 8.0), and 0.5 mM TCEP. Proteins were eluted with 50 mM Hepes (pH 8.0), 150–500 mM NaCl, 5% glycerol, 250 mM imidazole (pH 8.0), and 0.5 mM TCEP. The eluted fractions were purified by gel filtration in 50 mM Hepes (pH 8.0), 100–500 mM NaCl, and 0.5 mM TCEP. Removal of the hexahistidine tag was accomplished by using TEV protease at 4°C overnight followed by passing the solution over Ni2+ resin. Purified proteins were concentrated and stored at −80°C before crystallization.
Peptides Used in Crystallization.
All peptides were synthesized by Thermo Electron Corporation (Ulm, Germany), dissolved to a concentration of 40 mM in water, and stored at −20°C. The sequences of phosphoserine peptide I (Pep I) and II (Pep II) are RRQRpSAP and RSIpSLP, respectively, where pS represents a phosphorylated serine. The sequence of the ExoS fragment is GLLDALDLASK. The peptides and 14-3-3 proteins were mixed to a molar ratio of 3:1 peptide/protein for crystallization.
Crystallization and Data Collection.
Crystals were grown at 20°C by using the sitting drop method (Table 1). All crystals were mounted in loops and cryo-cooled before data collection. X-ray data were collected at 100 K. Diffraction data were collected to 2.5 and 2.8 Å resolution on a Rigaku/MSC FR-E rotating anode generator equipped with an R-AXIS HTC image plate for 14-3-3β and τ, respectively. The data sets for ε and γ were collected to 1.6- and 2.5-Å resolution, respectively, at the Swiss Light Source (beamline SLS-X10). The data were processed with MOSFLM (55) and the CCP4 suite (56). Structures were solved by molecular replacement using PHASER (57), and crystallographic models were rebuilt by using O (58) or COOT (59). Refinements were performed by using Refmac5 (60) (Table 2).
Mass Spectrometry of Intact Complexes.
Mass spectra were recorded by using either an oTOF (Waters, Manchester, U.K., LCT) or a tandem-mass spectrometer (Waters Q-Tof 2) modified for high mass operation (61). Both instruments were fitted with a standard offline nanoelectrospray source. Sample solutions were sprayed from borosilicate glass capillaries with a tapered tip that was cut under a microscope to an inner diameter of ≈2–5 μm. Approximately 2 μl of protein solution (10–30 μM) was loaded per capillary. Protein samples were buffer exchanged into 20 mM aqueous ammonium acetate (pH 7.0) using gel-filtration spin columns (microbiospin; Bio-Rad, Hercules, CA).
A low backing pressure of nitrogen gas was used to initiate and maintain flow through the capillary, and a nitrogen cone gas flow was used to aid desolvation of the spray. The conditions within the mass spectrometer were adjusted to preserve noncovalent interactions (61). All data were acquired in positive ion mode and processed by using MassLynx software (Waters). Experimental conditions for the monomer/dimer study were: Waters Q-Tof 2; capillary voltage 1.4 kV, cone 100–120 V, extractor 0–2 V, source temperature 25°C, drying gas rate 150 liters/h and Pirani pressure 5 × 10−3 mbar. Experimental conditions for the heterodimerization study were: Waters LCT oTOF mass spectrometer, capillary voltage 1.4 kV, cone 120 V, extractor 0 V, source temperature 25°C, drying gas rate 150 liters/h, and Pirani pressure 6 × 10−1 mbar.
Small Angle X-Ray Scattering.
Data were collected at station 2.1 of the Synchrotron Radiation Source (Daresbury, U.K.) of β and γ isoforms (at concentrations between 2 and 3 mg/ml) and buffer in the momentum transfer interval 0.02 Å−1 ≤ q ≥ 0.19 Å−1 where q = 4πsinθ/λ, (2θ is the scattering angle and λ is the x-ray wavelength 1.5 Å) according to published procedures (62). Measurements with peptides were performed at a 9-fold excess of peptide. The calculation of the Rg as well as scattering pattern simulations based on crystal structure information were carried out with the program CRYSOL (63).
ODA Calculations.
The default ODA algorithm as described by Fernandez-Recio et al. (48) and implemented in the program ICM version 3.4-3 (www.molsoft.com) was used to calculate the optimal docking areas for all members of the human 14-3-3 family.
Supplementary Material
Acknowledgments
We thank members of the Structural Genomics Consortium (SGC) for assistance with plasmid preparation and diffraction data collection. The SGC is a registered charity (no. 1097737) funded by the Wellcome Trust, GlaxoSmithKline, Genome Canada, the Canadian Institutes of Health Research, the Ontario Innovation Trust, the Ontario Research and Development Challenge Fund, the Canadian Foundation for Innovation, VINNOVA, The Knut and Alice Wallenberg Foundation, The Swedish Foundation for Strategic Research, and Karolinska Institutet.
Abbreviations
- ExoS
exoenzyme S
- ODA
optimal docking area.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
Data deposition: The crystal structures reported in this paper have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2BR9, 2BTP, 2B05, 2C74, 2C63, 2C23, and 2BQ0).
References
- 1.Moore BW, Perez VJ. Specific Acidic Proteins of the Nervous System. Englewood Cliffs, NJ: Prentice Hall; 1967. [Google Scholar]
- 2.Chaudhri M, Scarabel M, Aitken A. Biochem Biophys Res Commun. 2003;300:679–685. doi: 10.1016/s0006-291x(02)02902-9. [DOI] [PubMed] [Google Scholar]
- 3.Jones DH, Ley S, Aitken A. FEBS Lett. 1995;368:55–58. doi: 10.1016/0014-5793(95)00598-4. [DOI] [PubMed] [Google Scholar]
- 4.Jin J, Smith FD, Stark C, Wells CD, Fawcett JP, Kulkarni S, Metalnikov P, O'Donnell P, Taylor P, Taylor L, et al. Curr Biol. 2004;14:1436–1450. doi: 10.1016/j.cub.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 5.Darling DL, Yingling J, Wynshaw-Boris A. Curr Top Dev Biol. 2005;68:281–315. doi: 10.1016/S0070-2153(05)68010-6. [DOI] [PubMed] [Google Scholar]
- 6.van Heusden GP. IUBMB Life. 2005;57:623–629. doi: 10.1080/15216540500252666. [DOI] [PubMed] [Google Scholar]
- 7.Fantl WJ, Muslin AJ, Kikuchi A, Martin JA, MacNicol AM, Gross RW, Williams LT. Nature. 1994;371:612–614. doi: 10.1038/371612a0. [DOI] [PubMed] [Google Scholar]
- 8.Freed E, Symons M, Macdonald SG, McCormick F, Ruggieri R. Science. 1994;265:1713–1716. doi: 10.1126/science.8085158. [DOI] [PubMed] [Google Scholar]
- 9.Fu H, Xia K, Pallas DC, Cui C, Conroy K, Narsimhan RP. Science. 1994;266:126–129. doi: 10.1126/science.7939632. [DOI] [PubMed] [Google Scholar]
- 10.Forrest A, Gabrielli B. Oncogene. 2001;20:4393–4401. doi: 10.1038/sj.onc.1204574. [DOI] [PubMed] [Google Scholar]
- 11.Peng C-Y, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 12.Muslin AJ, Tanner JW, Allen PM, Shaw AS. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 13.Rittinger K, Budman J, Xu J, Volinia S, Cantley LC, Smerdon SJ, Gamblin SJ, Yaffe MB. Mol Cell. 1999;4:153–166. doi: 10.1016/s1097-2765(00)80363-9. [DOI] [PubMed] [Google Scholar]
- 14.Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 15.Henriksson ML, Francis MS, Peden A, Aili M, Stefansson K, Palmer R, Aitken A, Hallberg B. Eur J Biochem. 2002;269:4921–4929. doi: 10.1046/j.1432-1033.2002.03191.x. [DOI] [PubMed] [Google Scholar]
- 16.Masters SC, Pederson KJ, Zhang L, Barbieri JT, Fu H. Biochemistry. 1999;38:5216–5221. doi: 10.1021/bi982492m. [DOI] [PubMed] [Google Scholar]
- 17.Petosa C, Masters SC, Bankston LA, Pohl J, Wang B, Fu H, Liddington RC. J Biol Chem. 1998;273:16305–16310. doi: 10.1074/jbc.273.26.16305. [DOI] [PubMed] [Google Scholar]
- 18.Wang B, Yang H, Liu Y.-C., Jelinek T, Zhang L, Ruoslahti E, Fu H. Biochemistry. 1999;38:12499–12504. doi: 10.1021/bi991353h. [DOI] [PubMed] [Google Scholar]
- 19.Zhai J, Lin H, Shamim M, Schlaepfer WW, Cañete-Soler R. J Biol Chem. 2001;276:41318–41324. doi: 10.1074/jbc.M107709200. [DOI] [PubMed] [Google Scholar]
- 20.Coblitz B, Shikano S, Wu M, Gabelli SB, Cockrell LM, Spieker M, Yoshiro H, Fu H, Amzel LM, Li M. J Biol Chem. 2005;280:36263–36272. doi: 10.1074/jbc.M507559200. [DOI] [PubMed] [Google Scholar]
- 21.Ganguly S, Weller JL, Ho A, Chemineau P, Malpaux B, Klein DC. Proc Natl Acad Sci USA. 2005;102:1222–1227. doi: 10.1073/pnas.0406871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urano T, Saito T, Tsukui T, Fujita M, Hosoi T, Muramatsu M, Ouchi Y, Inoue S. Nature. 2002;417:871–875. doi: 10.1038/nature00826. [DOI] [PubMed] [Google Scholar]
- 23.Zanusso G, Fiorini M, Farinazzo A, Gelati M, Benedetti MD, Ferrari S, Dalla Libera A, Capaldi S, Monaco HL, Rizzuto N, Monaco S. Neurology. 2005;64:1618–1620. doi: 10.1212/01.WNL.0000160397.81314.84. [DOI] [PubMed] [Google Scholar]
- 24.Van Everbroeck BRJ, Boons J, Cras P. J Neurol Neurosurg Psychiatry. 2005;76:100–102. doi: 10.1136/jnnp.2003.032037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peoc'h K, Schroder HC, Laplanche J.-L., Ramljak S, Müller WEG. Neurosci Lett. 2001;301:167–170. doi: 10.1016/s0304-3940(01)01619-6. [DOI] [PubMed] [Google Scholar]
- 26.Wiltfang J, Otto M, Baxter HC, Bodemer M, Steinacker P, Bahn E, Zerr I, Kornhuber J, Kretzschmar HA, Poser S, et al. J Neurochem. 1999;73:2485–2490. doi: 10.1046/j.1471-4159.1999.0732485.x. [DOI] [PubMed] [Google Scholar]
- 27.Qi W, Liu X, Qiao D, Martinez JD. Int J Cancer. 2005;113:359–363. doi: 10.1002/ijc.20492. [DOI] [PubMed] [Google Scholar]
- 28.Qi W, Martinez JD. Radiat Res. 2003;160:217–223. doi: 10.1667/rr3038. [DOI] [PubMed] [Google Scholar]
- 29.Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. Nature. 1999;401:616–620. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- 30.Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai LH, Dobyns W, Ledbetter D, et al. Nat Genet. 2003;34:274–285. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- 31.Xing H, Zhang S, Weinheimer C, Kovacs A, Muslin AJ. EMBO J. 2000;19:349–358. doi: 10.1093/emboj/19.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steinacker P, Schwarz P, Reim K, Brechlin P, Jahn O, Kratzin H, Aitken A, Wiltfang J, Aguzzi A, Bahn E, et al. Mol Cell Biol. 2005;25:1339–1346. doi: 10.1128/MCB.25.4.1339-1346.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L. Cell. 2006;125:1165–1177. doi: 10.1016/j.cell.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 34.Bridges D, Moorhead GB. Sci STKE. 2004:re10. doi: 10.1126/stke.2422004re10. [DOI] [PubMed] [Google Scholar]
- 35.Liu D, Bienkowska J, Petosa C, Collier RJ, Fu H, Liddington R. Nature. 1995;376:191–194. doi: 10.1038/376191a0. [DOI] [PubMed] [Google Scholar]
- 36.Obsil T, Ghirlando R, Klein DC, Ganguly S, Dyda F. Cell. 2001;105:257–267. doi: 10.1016/s0092-8674(01)00316-6. [DOI] [PubMed] [Google Scholar]
- 37.Benzinger A, Popowicz GM, Joy JK, Majumdar S, Holak TA, Hermeking H. Cell Res. 2005;15:219–227. doi: 10.1038/sj.cr.7290290. [DOI] [PubMed] [Google Scholar]
- 38.Wilker EW, Grant RA, Artim SC, Yaffe MB. J Biol Chem. 2005;280:18891–18898. doi: 10.1074/jbc.M500982200. [DOI] [PubMed] [Google Scholar]
- 39.Xiao B, Smerdon SJ, Jones DH, Dodson GG, Soneji Y, Aitken A, Gamblin SJ. Nature. 1995;376:188–191. doi: 10.1038/376188a0. [DOI] [PubMed] [Google Scholar]
- 40.Yaffe MB. FEBS Lett. 2002;513:53–57. doi: 10.1016/s0014-5793(01)03288-4. [DOI] [PubMed] [Google Scholar]
- 41.Gardino AK, Smerdon SJ, Yaffe MB. Semin Cancer Biol. 2006;16:173–182. doi: 10.1016/j.semcancer.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 42.Liu MY, Cai S, Espejo A, Bedford MT, Walker CL. Cancer Res. 2002;62:6475–6480. [PubMed] [Google Scholar]
- 43.Gu YM, Jin YH, Choi JK, Baek KH, Yeo CY, Lee KY. FEBS Lett. 2006;580:305–310. doi: 10.1016/j.febslet.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 44.Woodcock JM, Murphy J, Stomski FC, Berndt MC, Lopez AF. J Biol Chem. 2003;278:36323–36327. doi: 10.1074/jbc.M304689200. [DOI] [PubMed] [Google Scholar]
- 45.Sobott F, Robinson CV. Curr Opin Struct Biol. 2002;12:729–734. doi: 10.1016/s0959-440x(02)00400-1. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Y, Reddy S, Murrey H, Fei H, Levitan IB. J Biol Chem. 2003;278:10073–10080. doi: 10.1074/jbc.M211907200. [DOI] [PubMed] [Google Scholar]
- 47.Tzivion G, Luo Z, Avruch J. Nature. 1998;394:88–92. doi: 10.1038/27938. [DOI] [PubMed] [Google Scholar]
- 48.Fernandez-Recio J, Totrov M, Skorodumov C, Abagyan R. Proteins. 2005;58:134–143. doi: 10.1002/prot.20285. [DOI] [PubMed] [Google Scholar]
- 49.Fernández-Recio J, Abagyan R, Totrov M. Proteins. 2005;60:308–313. doi: 10.1002/prot.20575. [DOI] [PubMed] [Google Scholar]
- 50.Perego L, Berruti G. Mol Reprod Dev. 1997;47:370–379. doi: 10.1002/(SICI)1098-2795(199708)47:4<370::AID-MRD3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 51.Watanabe M, Isobe T, Ichimura T, Kuwano R, Takahashi Y, Kondo H, Inoue Y. Brain Res Mol Brain Res. 1994;25:113–121. doi: 10.1016/0169-328x(94)90285-2. [DOI] [PubMed] [Google Scholar]
- 52.Tien A-C, Hsei H-Y, Chien C-T. Mech Dev. 1999;81:209–212. doi: 10.1016/s0925-4773(98)00238-x. [DOI] [PubMed] [Google Scholar]
- 53.Watanabe M, Isobe T, Ichimura T, Kuwano R, Takahashi Y, Kondo H. Brain Res Mol Brain Res. 1993;17:135–146. doi: 10.1016/0169-328x(93)90082-z. [DOI] [PubMed] [Google Scholar]
- 54.Shikano S, Coblitz B, Wu M, Li M. Trends Cell Biol. 2006;16:370–375. doi: 10.1016/j.tcb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 55.Leslie AGW. Acta Crystallogr D. 1999;55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- 56.Collaborative Computational Project No 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 57.Storoni LC, McCoy AJ, Read RJ. Acta Crystallogr D. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 58.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 59.Emsley P, Cowtan K. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 60.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Acta Crystallogr D. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 61.Sobott F, Hernández H, McCammon MG, Tito MA, Robinson CV. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 62.Grossmann JG, Hall JF, Kanbi LD, Hasnain SS. Biochemistry. 2002;41:3613–3619. doi: 10.1021/bi015955o. [DOI] [PubMed] [Google Scholar]
- 63.Svergun D, Barberato C, Koch MHJ. J Appl Cryst. 1995;28:768–773. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}