

Abstract
Three-dimensional structures of functionally uncharacterized proteins may furnish insight into their functions. The potential benefits of three-dimensional structural information regarding such proteins are particularly obvious when the corresponding genes are conserved during evolution, implying an important function, and no functional classification can be inferred from their sequences. The Bacillus subtilis Maf protein is representative of a family of proteins that has homologs in many of the completely sequenced genomes from archaea, prokaryotes, and eukaryotes, but whose function is unknown. As an aid in exploring function, we determined the crystal structure of this protein at a resolution of 1.85 Å. The structure, in combination with multiple sequence alignment, reveals a putative active site. Phosphate ions present at this site and structural similarities between a portion of Maf and the anticodon-binding domains of several tRNA synthetases suggest that Maf may be a nucleic acid-binding protein. The crystal structure of a Maf-nucleoside triphosphate complex provides support for this hypothesis and hints at di- or oligonucleotides with either 5′- or 3′-terminal phosphate groups as ligands or substrates of Maf. A further clue comes from the observation that the structure of the Maf monomer bears similarity to that of the recently reported Methanococcus jannaschii Mj0226 protein. Just as for Maf, the structure of this predicted NTPase was determined as part of a structural genomics pilot project. The structural relation between Maf and Mj0226 was not apparent from sequence analysis approaches. These results emphasize the potential of structural genomics to reveal new unexpected connections between protein families previously considered unrelated.
Keywords: protein folding, structural genomics, sequence alignment, x-ray crystallography
Numerous completed and ongoing large-scale sequencing projects have provided a wealth of genetic information [Gaasterland, T. (1999) Genome Sequencing Projects, http://www.mcs.anl.gov/home/gaasterl/genomes.html; Kerlavage, A. R. (1999) The TIGR Microbial Genome Database, http://www.tigr.org/tdb/mdb/mdb.html; ref. 1]. In many cases, the function of the encoded gene products can be deduced from comparative sequence analysis (2, 3). Frequently, however, these methods do not permit a functional classification because of the absence of reliable similarity to proteins with known functions (4). Novel methods involving phylogenetic profiles, domain fusions, and gene localization can also provide information about functional relationships without relying on sequence matching (5). Because proteins may display similarities at the three-dimensional (3D) structural level even if their sequences appear nonhomologous, determination of the 3D structure of a functionally uncharacterized protein may provide functional insights (6–8). A number of demonstrations of the validity of this approach have been published recently (9–11). In addition, the availability of the structure of a protein may furnish valuable information even if the structure does not reveal its possible function(s). For example, structures of the products of novel genes may reveal new folding motifs or define a new superfamily within a known folding class (7).
Several strategies can be pursued to determine a so-called “basis set” of protein folds (6). One such strategy is based on sequence comparisons to identify protein families that are conserved across a wide range of genomes; these families have been termed clusters of orthologous groups (COGs) of proteins [Koonin, E. V. (2000) Phylogenetic Classification of Proteins Encoded in Complete Genomes; http://www.ncbi.nlm.nih.gov/COG; refs. 12, 13]. Along with many COGs whose members were functionally and structurally well characterized, this analysis revealed protein families with no apparent sequence similarity to those of known 3D structure. The maf gene (the name refers to the gene from Bacillus subtilis) was among a short list of around 200 COGs targeted for structural studies (14). The corresponding proteins appeared to be tractable for structural studies by using either x-ray crystallography or solution NMR and were considered to have a high probability of providing novel structural and evolutionary information. This approach has been exemplified recently by the determination of the structure of the Escherichia coli YciH protein, the bacterial ortholog of the eukaryotic translation factor eIF-1 (14).
The B. subtilis maf gene is the homolog of a putative E. coli morphogene orfE (of the mre operon) (15). Orthologous genes are present in most of the completely sequenced bacterial genomes as well as in eukaryotes and in some of the archaea. Despite this notable evolutionary conservation, no specific function has been assigned to the corresponding proteins. Multicopy plasmid overexpression of maf in B. subtilis cells led to elongated cell shape and formation of filamentous cells and appeared to arrest septum formation (15). However, insertional inactivation of the maf gene suggested that it was not essential for cell division. More recently, a functional characterization of 2,026 of the yeast Saccharomyces cerevisiae ORFs (more than one-third of the ORFs in the genome) by high-throughput gene deletion and parallel analysis found the maf homolog (yor111) to be nonessential for viability in either rich or minimal medium (16). To learn more about the relationships between Maf and other proteins of known structure and function, we determined the crystal structure of Maf at 1.85 Å resolution and examined the structure for potential clues as to the function and evolution of the protein.
Methods
Overexpression and Purification of Wild-Type (wt) and Selenomethionine (Se-Met) Maf.
Isopropyl-β-d-thiogalactoside-induced overexpression of B. subtilis Maf by using the pQE30 vector transformed into the E. coli host strain M15 was performed as described (15). Cells were lysed in 100 mM NaCl/50 mM Tris⋅HCl, pH 8.0/1 mM EDTA/50 mM lysozyme/10 units/ml DNase I over the course of 30 min (room temperature). After ammonium sulfate precipitation, the protein was purified by histidine trap chelating chromatography. The initial buffer was 20 mM sodium phosphate, pH 7.2, and 500 mM NaCl, and for eluting the protein, the buffer was supplemented with 50 mM EDTA. The yield was about 10 mg pure protein per gram of cells. Se-Met-substituted Maf was prepared in a similar fashion by using a standard protocol to saturate the biosynthetic pathway for methionine production (17). In the case of Se-Met Maf, microcrystals were initially observed during dialysis of the protein against phosphate buffer after the ammonium sulfate precipitation step, and no further purification was required.
Crystallization and Data Collection.
Optimal crystallization conditions were screened by the sparse matrix technique (18) by using the Hampton Research (Riverside, CA) Crystal Screen I. A hanging droplet consisting of 2 μl of a protein solution (10 mg/ml) mixed with 2 μl of a reservoir solution containing 0.1 M Tris⋅HCl, pH 8.5, and 8% polyethylene glycol 8000 (solution 36) was equilibrated against 1 ml of the reservoir. Crystals suitable for data collection were obtained within 1 day with both wt and Se-Met Maf. The space group of the crystals is orthorhombic P212121 with cell constants a = 62.66 Å, b = 86.01 Å, c = 93.94 Å. The crystals were shock frozen in the above mother liquor plus 25% sucrose. Diffraction data for wt Maf were collected at 100 K on the 5-ID (insertion device) beamline of the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) at the Advanced Photon Source, Argonne, IL, to a maximum resolution of 1.85 Å (Table 1). Multiwavelength anomalous diffraction data were collected at four wavelengths on a single Se-Met Maf crystal to a resolution of 2.5 Å (Table 1). All data were integrated and scaled in the denzo/scalepack suite (19).
Table 1.
Data collection parameters
| Data collection | Wave length, Å | No. of reflections measured (unique) | % complete all (last shell) | Rmerge, %, all (last shell) | Phasing power λ1/λi (Friedel) |
|---|---|---|---|---|---|
| MAD* | λ1 = 1.1000 | 116,513 (16,209) | 90.4 (88.1) | 6.4 (26.7) | — (0.52) |
| λ2 = 0.9796 | 112,523 (15,649) | 87.7 (83.0) | 6.7 (30.2) | 2.33 (2.40) | |
| λ3 = 0.9794 | 116,198 (15,811) | 88.2 (82.3) | 8.3 (38.4) | 1.93 (2.16) | |
| λ4 = 0.9500 | 113,313 (15,595) | 86.7 (80.9) | 7.1 (36.2) | 1.08 (1.30) | |
| WT† | λ = 1.1004 | 186,600 (43,834) | 99.4 (96.3) | 6.8 (44.9) | — |
Maximum resolution 2.5 Å (last shell 2.59 − 2.50 Å).
†Maximum resolution 1.85 Å (last shell 1.92 − 1.85 Å); figure of merit, 0.64.
Structure Determination and Refinement.
Se sites were determined with the program solve (20) based on 2.8 Å anomalous data (Table 1), and 8 selenium atoms per asymmetric unit could be located. The asymmetric unit consists of two Maf molecules, each containing six Se atoms. The two missing Se atoms per molecule are located at the structurally poorly defined N terminus and in a relatively flexible loop region located near the surface. The Se sites were refined with the program sharp (21), but no new Se sites were found. Electron density maps were calculated in the CCP4 suite (22), and about 85% of the polypeptide backbones of both molecules were readily built by using the program o (23). Although there is noncrystallographic symmetry between subunits, no averaging was used for improving the electron density maps, and the two molecules were built separately. Cycles of manual rebuilding were followed by positional simulated annealing and temperature factor refinements with the program cns (24), which gradually improved the model. The free R factor was monitored by setting aside 10% of the reflections as a test data set (25). The initial experimental phases based on multiwavelength anomalous diffraction data to 2.8 Å resolution were progressively replaced by model phases combined with wt data to a maximum resolution of 1.85 Å. Along with 2 Maf molecules, 304 water molecules, 3 phosphate ions, and 1 sucrose molecule were built into the electron density maps. The Ramachandran plot calculated with the program procheck (22) shows that 100% of the nonglycine and nonproline residues in the final model lie in the most favored and additional allowed regions. The final R factor is 19.5% (Rfree 22.3%) for 40,372 reflections in the 25.0- to 1.85-Å resolution range (bulk solvent correction). The average B factors for Maf atoms and water molecules are 32.8 (18.0) and 37.2 (9.9) Å2 (standard deviations in parentheses), respectively. The r.m.s. deviations from standard geometries for bond lengths and angles are 0.009 Å and 1.4°, respectively.
Analysis of the Maf–dUTP Complex.
Diffraction data for the Maf–dUTP complex were collected at 100 K on the 5-BM (bending magnet) beamline of the DND-CAT at the Advanced Photon Source, Argonne, IL, to a maximum resolution of 2.7 Å. The final R factor and Rfree are 19.7% and 26.0%, respectively, for 12,260 reflections (85.5% completeness) in the resolution range of 17 Å to 2.7 Å. The r.m.s. deviations for bonds and angles from standard values are 0.007 Å and 1.40°, respectively.
Coordinates.
The coordinates and structure factors of the Maf and Maf–dUTP complex structures have been deposited in the Protein Data Bank [PDB ID codes 1EX2 (Maf) and 1EXC (complex)].
Results and Discussion
Overall Structure and Topology.
The Maf protein from B. subtilis analyzed here comprises 189 amino acids and a 6-residue N-terminal histidine tag. The crystal structure of Maf was determined by the multiwavelength anomalous diffraction technique based on data with 2.8 Å resolution and refined at 1.85 Å to a crystallographic R factor of 19.5% (Fig. 1A). The asymmetric unit of the orthorhombic crystal form consists of two independent Maf molecules related by a noncrystallographic 2-fold axis. The present model comprises 185 amino acids per molecule, with the four C-terminal residues and the N-terminal His tag undefined in the electron density maps. A primary feature of the Maf structure is an extended three-stranded antiparallel β sheet (β3, β5, and β6) that forms the core of the protein and, along with β7, connects two mostly α helical lobes, termed 1 (α1, α2, α6, α7, β1, and β2) and 2 (α3, α4, α5, and β4) (Fig. 1B Right and Left, respectively; a stereo diagram of the α-carbon trace is shown in Fig. 5A). This gives the protein an elongated appearance with approximate overall dimensions 28 × 36 × 50 Å. In lobe 1, the central β sheet is extended by two short parallel β strands (β1 and β2) on the β3 side. These are packed against the helices α1 and α7 (Fig. 1B). In addition, a 20-residue-long loop (colored orange in Fig. 1B Right), interrupted only by a short β segment (β2, three residues), is inserted between helices α1 and α2 in lobe 1. The extended polypeptide loop arches over lobe 1 and leads to the longest helical portion in the structure (α2, 19 residues), which is located on the other side of the central β sheet. In the smaller lobe 2, helices α3 and α4, both 10 residues long, adopt a roughly perpendicular relative orientation. A 10-residue relatively flexible loop then joins helix α4 and the short helix α5 (Fig. 1B Left). The relative orientation of the two lobes creates an approximately 10-Å-wide cleft between them. Although Maf is most likely an intracellular protein, a disulfide bridge between residues Cys-74 (β3) and Cys-79 (β4) on the surface of lobe 2 appears to restrict its position relative to lobe 1 and the β-stranded core of the molecule (Fig. 1B).
Figure 1.

(A) Final (2 Fo − Fc) electron density at 1.85 Å resolution [1σ level, drawn with turbo frodo (37)], depicting the phosphate ion bound at the Maf putative active site. Atoms of selected side chains are colored yellow, blue, and red for carbon, nitrogen, and oxygen, respectively, and oxygen atoms of water molecules are shown as small red spheres. (B) Overall structure of the Maf protein drawn with the program ribbons (38). The α helices and β strands are colored cyan and green, respectively, and are numbered. Loop regions are colored orange, N and C termini are labeled, and a yellow dot indicates the location of the disulfide bridge.
Figure 5.

(A) Stereo diagram depicting superimposed Maf–dUTP (thick line) and Mj0226–dATP complexes. Selected residues of Maf are numbered. (B) Stereo diagram depicting orientation and interactions of dUTP at the Maf putative active site. The protein α-carbon trace is drawn with thin lines, side chains of conserved residues are drawn with thick lines and are labeled, nucleotide bonds are gray, and hydrogen bonds are dashed lines.
The two independent Maf molecules adopt rather similar conformations, and notable deviations are found only in the long loop of lobe 1 and portions of lobe 2 (for an illustration of the differences between the conformations of the two molecules, see Fig. 6, published as supplementary material on the PNAS web site, www.pnas.org). The r.m.s. deviation between all 185 Cα atoms of the two subunits is 0.74 Å. The regions with the largest conformational deviations between Maf molecules are also those that display the highest temperature factors in the individual molecules. By using dynamic light-scattering experiments, we found that Maf forms a dimer in solution (data not shown). The orientation of the noncrystallographic 2-fold axis combined with the crystal packing result in potential dimeric interfaces in the orthorhombic lattice (for an illustration of these interfaces, see Fig. 7, published as supplementary material on the PNAS web site, www.pnas.org). The surfaces buried by the three interfaces are 2,452 Å2, 1,228 Å2, and 940 Å2, respectively [Fig. 7 A–C; calculated with the program cns (24)]. Neither displays conserved residues, and dimerization is most likely not a shared feature of the Maf family of proteins from different organisms.
Active Site Location.
We have pursued several strategies for locating a putative active site of Maf based on its structure alone and in combination with sequence comparisons. The crevice formed between the two lobes is of obvious interest in this respect (Fig. 1B), because the largest cavity on enzyme surfaces often corresponds to the catalytic center (26). To assess this further in the case of Maf, phylogenetically conserved residues were identified by aligning the sequences of the Maf protein from B. subtilis and those of 18 homologous proteins from archaea, prokaryotes, and eukaryotes (Fig. 2). When mapped onto the surface defined by the 3D structure of Maf, exposed amino acids that are conserved in all or in 80% of related sequences lie inside the cleft (Fig. 3A). These include Ser-9, Arg-14, Lys-53, and Asp-70 from lobe 1 and Lys-82 from lobe 2 (all residue numbers refer to the Maf protein from B. subtilis) (Fig. 2). The side chain of Glu-34, a further strictly conserved residue that resides in the loop connecting β2 and α2, points into the cleft and is positioned in close vicinity of the above residues (Figs. 1B, 3A, and 5B).
Figure 2.

Sequence alignment of the Maf protein from B. subtilis (Upper) and homologous proteins from 18 selected archaea, prokaryotes, and eukaryotes. The sequence of Mj0226 from M. jannaschii is shown (Lower); gaps in its sequence indicate regions with larger deviations between the Maf and Mj0226 structures, preventing meaningful structure-based alignment. Conserved residues are highlighted (blue, basic; red, acidic; yellow, hydrophobic; magenta, all others), secondary structure elements observed in the Maf crystal structure are indicated (Upper), and selected conserved residues in Maf are numbered.
Figure 3.

(A) Arrangement of conserved residues on the 3D surface of Maf. Green, yellow, and cyan patches indicate the locations of amino acids that are conserved in 18 or more, in 17 or 16 of 19, and in 15 of 19 analyzed proteins, respectively (see Fig. 2). All other residues are white. (B) Electrostatic surface potential of Maf calculated with the program grasp (39). Blue and red patches indicate regions of positive and negative charge, respectively. The views in A and B are similar to that in Fig. 1B.
Computation of the electrostatic surface potential (27) for Maf reveals that the region with the most conserved amino acids exhibits a positive charge (Fig. 3B). We take this as evidence that the overall charge or at least that of a moiety of the ligands recognized by Maf is negative. An independent confirmation of the insights gained from protein shape, sequence–structure correlation, and distribution of electrostatic surface potential (Figs. 2 and 3) is provided by the fact that phosphate ions coordinate inside the cleft in both subunits. In the first subunit, one ordered phosphate ion coordinates to several of the conserved residues (Fig. 1A). In the second, the phosphate is disordered and occupies two alternative positions (for details of the interactions between this phosphate ion and Maf residues, see Fig. 8, published as supplementary material on the PNAS web site, www.pnas.org). A third site of phosphate ion binding is located on a noncrystallographic 2-fold axis. The ion is coordinated to Lys-51 and Lys-55 from both subunits and is thus located above the dimeric interface involving β6 strands (Fig. 7B). Both the identification of negatively charged ions at a key site and the fact that this putative active site is positively charged provide evidence that Maf could be a nucleotide- or nucleic acid-binding protein.
Structural Comparison with Other Proteins.
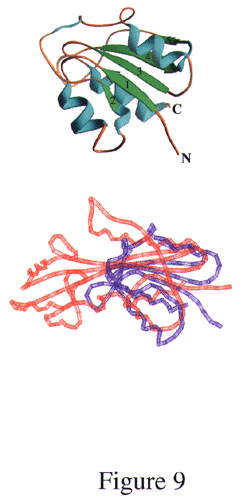
Potential structural homologs of Maf in the PDB were searched with the program dali (28). The database search revealed a good similarity between the structure of lobe 1 (including the antiparallel β sheet) and the C-terminal anticodon-binding domain of glycyl-tRNA synthetase (29) (PDB ID code 1ati; z score, 4.9) (Fig. 4A). Slightly lower z scores of 4.1 and 3.3 were found for threonyl-tRNA synthetase (30) (PDB ID code 1qf6) and histidyl-tRNA synthetase (31) (PDB ID code 1adj), respectively. In addition, α-d-glucose-1,6-bisphosphate phosphoglucomutase (32) (PDB ID code 3pmg) exhibited a z score of 3.5 (for this structural comparison, see Fig. 9, published as supplementary material on the PNAS web site, www.pnas.org). Among the 15 proteins with the highest z scores (cutoff 2.6), no fewer than seven are nucleotide- or RNA-binding proteins, including four aminoacyl-tRNA synthetases. Although not among the list of proteins with structural similarity recovered by dali, it was found on the basis of visual inspections that the C-terminal portion of E. coli RNase H (33) (PDB ID code 1lav) and the N-terminal portion of Maf share certain structural features (N. V. Grishin and L. Aravind, personal communication). However, the order of secondary structure elements is not the same along the sequence in the two proteins, as they are circularly permuted. As shown in Fig. 4B, RNase H also exhibits the extensive antiparallel β core sandwiched between α helical regions. All these observations lend support to the idea that Maf may indeed be involved in the recognition and processing of nucleotides or oligonucleotides.
Figure 4.

Structural comparisons between Maf and selected proteins. (A) Secondary structure of the C-terminal putative anticodon-binding domain of glycyl-tRNA synthetase (Upper) and structural comparison between Maf and glycyl-tRNA synthetase [red and blue, respectively (Lower)]. (B) Secondary structure of E. coli RNase H (Upper) and structural comparison between the N-terminal portion of Maf and the C-terminal portion of RNase H [red and blue, respectively (Lower)]. The β strands 3, 4, and 5 in glycyl-tRNA synthetase and phosphoglucomutase correspond to strands 3, 5, and 6 in Maf (Fig. 1B). Similarly, β strands 1, 2, and 3 in RNase H correspond to strands 3, 5, and 6 in Maf.
While the structure-based functional analysis of Maf was under way, the structure-based identification of an NTPase from Methanococcus jannaschii was reported (10). Interestingly, Maf and Mj0226 adopt similar folds, although there are some differences between the structures of lobe 2 (our nomenclature; Fig. 5A). However, by using a structure-based alignment, the maf and mj0226 genes exhibit only 14% sequence identity (Fig. 2). Despite the fact that both protein structures feature the large cleft, the locations of conserved residues differ, and there are no obvious similarities between the amino acids that make up the floor and the walls of the two cavities. Moreover, although both proteins form homodimers, the dimeric interfaces are clearly different in the two cases (data not shown).
Crystal Structure of the Complex Between Maf and dUTP.
To examine whether Maf can bind nucleotides, a variety of nucleoside mono-, di-, and triphosphates were either soaked into Maf crystals or cocrystallized with the protein. The structure of a crystal soaked with dUTP was determined at 2.7 Å resolution. Two binding sites per subunit were identified and, for both, the locations of the γ-phosphates nearly coincide with those of the previously observed phosphate ions. The interactions of dUTP inside the cleft are depicted in Fig. 5. The side chains of residues Ser-9, Arg-14, and Lys-53 interact with the γ-phosphate. The β-phosphate is contacted by Lys-53 and Lys-82, whereas no contacts between the protein and the α-phosphate are observed (Fig. 5B). An additional hydrogen bond exists between uracil N3 and Glu-34.
Although the three phosphate groups of dUTP can obviously be accommodated in the cleft, the pocket-like shape of the Maf-binding site and the manner in which the γ-phosphate is being stabilized suggest that the Maf substrate(s) most likely feature a terminal phosphate or a phosphate monoester. Assuming that a terminal phosphate group is part of a nucleic acid molecule, it could be either at the 5′- or the 3′-oxygen of the ribose (or 2′-deoxyribose) moiety. In the case of the dUTP molecule bound inside the cleft, the carboxylates of Glu-34 and Asp-70 are positioned 9.8 Å and 7.4 Å, respectively, from the γ-phosphorus (Fig. 5B). These distances are similar to the spacing between adjacent intrastrand phosphate groups in oligodeoxynucleotide duplexes (up to ca. 7.5 Å). Thus, it is conceivable that Glu-34 and Asp-70 participate in the hydrolysis of the 5′-phosphate (assuming that the 3′-terminal phosphate is bound at the site currently occupied by the γ-phosphate of dUTP) or the 3′-phosphate (assuming that the 5′-terminal phosphate is bound at the site currently occupied by the γ-phosphate of dUTP) of a nucleotide that is part of a di- or oligomeric nucleic acid fragment. The topology of the cleft is compatible with binding of either dimers or longer single-stranded oligonucleotides. However, based on just the two structures, it is impossible to establish whether Maf substrates are composed of ribonucleotides or 2′-deoxyribonucleotides.
Regarding the question of the type of reaction that may be catalyzed by Maf, it is intriguing that Maf (Asp-70), RNase H from E. coli (Asp-10), and Mj0226 from M. jannaschii (Asp-73) all feature a conserved aspartate that protrudes from the floor of the β core that is a hallmark of these enzymes (Fig. 4). In RNase H, this aspartate has been implicated in serving a key role in the metal-ion-dependent hydrolysis of oligoribonucleotides (34). Although it was not mentioned as a key residue in the original paper, we propose that Asp-73 in the case of Mj0226 is involved in the conversion of xanthine or inosine triphosphates to the corresponding monophosphates, the type of reaction established by the structural and biochemical experiments (10). Despite the fact that details of the reaction mechanisms in these two cases have not been worked out, it is reasonable to also predict an important role of Asp-70 in the reaction catalyzed by Maf. Moreover, the side chain of Lys-82 that is hydrogen-bonded to the β phosphate in the dUTP complex could easily move toward Glu-34 and Asp-70 and participate in a cleavage reaction. We would like to point out that the lack of an interaction with the α-phosphate of the nucleotide in the case of Maf suggests that the enzyme does not catalyze a type of reaction similar to Mj0226. Thus, we did not observe cleavage of dUTP or dATP in the presence of Mg2+. Moreover, the orientations of the dATP and dUTP ligands inside the clefts of Mj0226 and Maf, respectively, differ considerably (Fig. 5B).
Conclusions.
The structure determination of Maf from B. subtilis, a protein whose function is unknown, has furnished several important insights: (i) A new overall fold together with the Mj0226 protein; (ii) location of a putative active site and identification of amino acids potentially involved in substrate binding and catalysis; (iii) evidence that Maf is a nucleotide- or nucleic acid-binding protein; (iv) ligands or substrates of Maf likely feature a terminal phosphate group; (v) there appears to be an evolutionary relationship between Maf and Mj0226 from M. jannaschii; and (vi) there may be a distant linkage between NTPases (by virtue of the Mj0226 homology) and enzymes involved in sugar metabolism (by virtue of the phosphoglucomutase homology). The current structure-based ideas regarding the function of Maf can be explored by using further crystallographic experiments as well as biochemical and computational methods. Before the determination of the structure, there were no clear directions to take in investigating the functions of this conserved family of proteins.
Considering everything we know at the moment about Maf from the structural data and previous assays that probed its function, one could envision a role for the protein in the broadly defined area of DNA repair. Notably, a role in repair can be predicted also for the Mj0226 protein, given the phenotype of the yeast mutants in the ham1 gene, which is the ortholog of mj0226 (35). Because DNA repair is of crucial importance to ensure accurate biological information transfer, failure to correct or prevent DNA damage via one particular pathway may be compensated for by an alternative set of proteins that can act as a backup (36). It appears likely that Maf is part of such a redundant pathway, because the ortholog of this gene is missing in some bacteria, such as Haemophilus influenzae and the spirochetes, as well as in many of the archaea, and also because of the lack of an obvious phenotype in the yeast knockouts.
If the Maf protein were part of such a redundant pathway, deleting its gene may not be of much consequence in a wt background. Therefore, assaying the growth of cells that lack the maf gene in a limited variety of media will likely fail to provide any insights regarding the function of the gene product. Taken together, the example of Maf demonstrates that a single 3D structure of a protein can provide a multitude of insights even in the absence of any other biochemical or functional data. However, it is also apparent that a structure alone will provide conclusive functional information only in very rare cases. Therefore, it is imperative that any large-scale effort to determine the 3D structures of proteins identified in the current sequencing projects be followed by an endeavor of similar magnitude, focused on the sequence- and structure-guided elucidation of function.
Supplementary Material
Acknowledgments
We are grateful to Drs. L. Shuvalova and V. Tereshko for help with protein overexpression and data collection, respectively, and to Drs. C. A. Muchmore and J. S. Brunzelle for advice with various programs. Thanks also to Drs. N. V. Grishin and L. Aravind for their help in the comparative analysis of the Maf structure, to Dr. P. A. Bash for interesting discussions, and to the reviewers for their comments. This work was supported by National Institutes of Health (NIH) Grant GM57049 (G.C.S.) and partially supported by NIH grant GM55237 (M.E.). The DuPont–Northwestern–Dow Collaborative Access Team Synchrotron Research Center at the Advanced Photon Source, Argonne, Illinois, is supported by E. I. DuPont de Nemours & Co., the Dow Chemical Company, the National Science Foundation, and the State of Illinois.
Abbreviations
- 3D
three-dimensional
- Se-Met
selenomethionine
- wt
wild type
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org [PDB ID codes 1EX2 (Maf) and 1EXC (complex)].
References
- 1.Rost B. Structure (London) 1998;6:259–263. doi: 10.1016/s0969-2126(98)00029-x. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S F, Madden T L, Schäffer A A, Zhang J H, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bork P, Koonin E V. Nat Genet. 1998;18:313–318. doi: 10.1038/ng0498-313. [DOI] [PubMed] [Google Scholar]
- 4.Murzin A G, Patthy L. Curr Opin Struct Biol. 1999;9:359–361. [Google Scholar]
- 5.Marcotte E M, Pellegrini M, Thompson M J, Yeates T O, Eisenberg D. Nature (London) 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- 6.Kim S-H. Nat Struct Biol. 1998;5:643–645. doi: 10.1038/1334. [DOI] [PubMed] [Google Scholar]
- 7.Sali A. Nat Struct Biol. 1998;5:1029–1031. doi: 10.1038/4136. [DOI] [PubMed] [Google Scholar]
- 8.Teichmann S A, Chothia C, Gerstein M. Curr Opin Struct Biol. 1999;9:390–399. doi: 10.1016/S0959-440X(99)80053-0. [DOI] [PubMed] [Google Scholar]
- 9.Zarembinski T I, Hung L-W, Mueller-Dieckmann H-J, Kim K-K, Yokota H, Kim R, Kim S-H. Proc Natl Acad Sci USA. 1998;95:15189–15193. doi: 10.1073/pnas.95.26.15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang K Y, Chung J H, Kim S-H, Han Y S, Cho Y. Nat Struct Biol. 1999;6:691–696. doi: 10.1038/10745. [DOI] [PubMed] [Google Scholar]
- 11.Boggon T J, Shan W-S, Santagata S, Myers S C, Shapiro L. Science. 1999;286:2119–2125. doi: 10.1126/science.286.5447.2119. [DOI] [PubMed] [Google Scholar]
- 12.Tatusov R L, Koonin E V, Lipman D J. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 13.Tatusov R L, Galperin M Y, Natale D A, Koonin E V. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cort J R, Koonin E V, Bash P A, Kennedy M A. Nucleic Acids Res. 1999;27:4018–4027. doi: 10.1093/nar/27.20.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler Y X, Abhayawardane Y, Stewart G C. J Bacteriol. 1993;175:3139–3145. doi: 10.1128/jb.175.10.3139-3145.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winzeler E A, Shoemaker D D, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke J D, Bussey H, et al. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 17.Doublié S. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 18.Jancarik J, Kim S-H. J Appl Crystallogr. 1991;24:409–411. [Google Scholar]
- 19.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Terwilliger T C. Methods Enzymol. 1997;276:530–537. [PubMed] [Google Scholar]
- 21.de La Fortelle E, Bricogne G. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 22.Collaborative Computational Project Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 23.Jones T A, Kjeldgaard M. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- 24.Brünger A T. New Haven, CT: Yale Univ.; 1998. , Version 0.5. [Google Scholar]
- 25.Brünger A T. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 26.Laskowski R A, Luscombe N M, Swindells M B, Thornton J M. Protein Sci. 1996;5:2438–2452. doi: 10.1002/pro.5560051206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Honig B. Am Chem Soc. 1998;216:U635. [Google Scholar]
- 28.Sander C, Schneider R. Proteins Struct Funct Genet. 1991;9:56–68. doi: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- 29.Logan D T, Mazauric M-H, Kern D, Moras D. EMBO J. 1995;14:4156–4167. doi: 10.1002/j.1460-2075.1995.tb00089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sankaranarayanan R, Dock-Bregeon A-C, Romby P, Caillet J, Springer M, Rees B, Ehresmann C, Ehresmann B, Moras D. Cell. 1999;97:371–381. doi: 10.1016/s0092-8674(00)80746-1. [DOI] [PubMed] [Google Scholar]
- 31.Aberg A, Yaremchuk A, Tukalo M, Rasmussen B, Cusack S. Biochemistry. 1997;36:3084–3090. doi: 10.1021/bi9618373. [DOI] [PubMed] [Google Scholar]
- 32.Ray Jr W J, Post C B, Liu Y, Rhyu G I. Biochemistry. 1993;32:48–57. doi: 10.1021/bi00052a008. [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa K, Nakamura H, Morikawa K. Biochemistry. 1993;32:6171–6178. [PubMed] [Google Scholar]
- 34.Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Ikehara M, Matsuzaki T, Morikawa K. Nature (London) 1990;347:306–309. doi: 10.1038/347306a0. [DOI] [PubMed] [Google Scholar]
- 35.Noskov V N, Staak K, Shcherbakova P V, Kozmin S G, Negishi K, Ono B C, Hayatsu H, Pavlov Y I. Yeast. 1996;12:17–29. doi: 10.1002/(SICI)1097-0061(199601)12:1%3C17::AID-YEA875%3E3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 36.Friedberg E C, Walker G C, Siede W, editors. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. pp. 191–226. [Google Scholar]
- 37.Cambillau C, Roussel A. Marseille, France: Université Aix–Marseille II; 1997. , Version OpenGL 1. [Google Scholar]
- 38.Carson M. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]
- 39.Nicholls A, Bharadwaj R, Honig B. Biophys J. 1993;64:166–170. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}