

Abstract
Protein misfolding and aggregation cause several diseases, by mechanisms that are poorly understood. The formation of amyloid aggregates is the hallmark of most of these diseases. Here, the properties and formation of amyloidogenic intermediates of transthyretin (TTR) were investigated by the use of hydrostatic pressure and spectroscopic techniques. Native TTR tetramers (T4) were denatured by high pressure into a conformation that exposes tryptophan residues to the aqueous environment. This conformation was able to bind the hydrophobic probe bis-(8-anilinonaphthalene-1-sulfonate), indicating persistence of elements of secondary and tertiary structure. Lowering the temperature facilitated the pressure-induced denaturation of TTR, which suggests an important role of entropy in stabilizing the native protein. Gel filtration chromatography showed that after a cycle of compression-decompression at 1°C, the main species present was a tetramer, with a small population of monomers. This tetramer, designated T4*, had a non-native conformation: it bound more bis-(8-anilinonaphthalene-1-sulfonate) than native T4, was less stable under pressure, and on decompression formed aggregates under mild acidic conditions (pH 5–5.6). Our data show that hydrostatic pressure converts native tetramers of TTR into an altered state that shares properties with a previously described amyloidogenic intermediate, and it may be an intermediate that lies on the aggregation pathway. This “preaggregated” state, which we call T4*, provides insight into the question of how a correctly folded protein may degenerate into the aggregation pathway in amyloidogenic diseases.
Comprehension of the mechanisms involved in protein aggregation is becoming crucial in structural and cell biology as well as in medicine. Several amyloidogenic diseases are caused by aggregation of soluble cellular proteins that undergo conformational changes leading to the formation of insoluble material (1–4). These diseases include transmissible spongiform encephalopathies (prion diseases), senile systemic amyloidosis, and familial amyloidotic polyneuropathy (5–7). The last two diseases are caused by the aggregation of either wild-type or mutant forms of the tetrameric human plasma protein transthyretin (TTR) (8–11).
Protein aggregation also has been a challenge for biotechnology, requiring the use of several time-consuming and expensive procedures to dissociate inclusion bodies extracted from heterologous cells (1, 12, 13). Recently, we showed that high pressure can dissociate large aggregates of bacteriophage P22 tailspike protein, increasing the yield of native trimers (14). High pressure has been used successfully to denature and dissociate proteins, protein–DNA complexes, and virus particles (15–18). A unique property of high-pressure denaturation is the formation of partially folded or molten-globule states at equilibrium (19–24).
TTR is a tetrameric protein composed of identical 127-residue subunits having a predominant β-sheet structure (25). TTR binds and transports thyroxine in the blood and cerebral spinal fluid and binds retinol binding protein (26–28). Several studies have revealed important features of the aggregation mechanism of TTR (3, 29–33). At very low pH, tetramers of TTR dissociate into partially folded monomers (A-state) (3, 30, 34). An interesting feature of TTR aggregation is its dependence on the history of the protein sample. Reconstituted protein previously denatured to the A-state at extremely acidic pH (pH 2, for instance) displays a higher yield of fibril formation at pH 3.5–4.5 than protein that is incubated continuously at pH 3.9–5.0 (29, 30). More recently, this hysteretic behavior also has been observed in the unfolding of TTR induced by guanidinium hydrochloride (31).
In this study we show that native TTR tetramer (T4) denatures under high pressure into a partially folded conformation. The pressure-denatured state binds bis-(8-anilinonaphthalene-1-sulfonate) (bis-ANS), suggesting persistence of some secondary and tertiary contacts. After return to atmospheric pressure, most of the protein is recovered as a tetramer that binds bis-ANS but displays lower stability. This altered species formed after a cycle of compression-decompression (T4*) undergoes aggregation under mild conditions where untreated native protein is stable and soluble (37°C, pH 5–5.6). We demonstrate that hydrostatic pressure can be a powerful tool to convert native protein into the amyloidogenic state, with the advantage over other methods that aggregation can be avoided, as long as pressure is maintained, and then obtained in a controlled way by returning to atmospheric pressure.
Experimental Procedures
Chemicals.
All reagents were of analytical grade. Bis-ANS was purchased from Molecular Probes. Distilled water was filtered and deionized through a Milli Q water purification system (Millipore). TTR was purchased from Sigma and used without further purification. The purity of each protein batch used was checked by SDS/PAGE and gel filtration chromatography in a HPLC system. Protein concentration was determined by using an extinction coefficient of 7.76 × 104 M−1⋅cm−1 at 280 nm (26). The high-pressure experiments were performed in the following buffers: 50 mM Bis-Tris⋅HCl/100 mM KCl, pH 5.0 or 5.6 and 50 mM Tris⋅HCl/100 mM KCl, pH 7.5. We emphasize that Tris and Bis-Tris buffers were chosen for pressure experiments because the pH does not change significantly under high pressure (35).
Spectroscopic Measurements Under Pressure.
The high-pressure cell equipped with optical windows has been described (19) and was purchased from ISS (Champaign, IL). Fluorescence spectra were recorded on an ISS K2 spectrofluorometer. The pressure was increased in steps of 200 bar. At each step the sample was allowed to equilibrate for 15 min before making measurements. There were no time-dependent changes in fluorescence spectra between 10 and 60 min. Tryptophan emission spectra were obtained by setting the excitation at 280 nm and collecting the emission in the 300- to 400-nm range. Bis-ANS spectra were recorded by exciting the sample at 360 nm and collecting emission from 400–600 nm. Fluorescence spectra at pressure p were quantified by the center of spectral mass 〈νp〉.
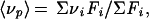 |
1 |
where Fi is the fluorescence emitted at wavenumber νi (19). The degree of dissociation (α) is related to 〈νp〉 by the expression:
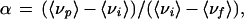 |
2 |
where 〈νi〉 and 〈νf〉 are the initial and final values of center of spectral mass, respectively, whereas 〈νp〉 is the center of spectral mass at pressure p. Thyroxine binding was evaluated by its ability to quench tryptophan fluorescence upon binding (8). All experiments were performed at least twice by using different batches of protein and a representative result is shown.
Aggregation Measurements and Fibril Formation.
Light scattering was measured by exciting the samples at 320 nm and collecting the light at 90° through the monochromator. Aggregation also was evaluated by absorption at 330 nm, measured after sample removal from the high-pressure cuvette. The presence of typical fibrils was based on the binding of Congo red (30). Congo red (10 μM) was prepared in 50 mM phosphate buffer, 100 mM NaCl, pH 7.5. A small volume (50 μl) of a fibril suspension was added, and absorbance was measured at 477 and 540 nm. The extent of Congo red binding was calculated from (A540nm/25295) − (A477nm/46306) (30). To measure fibril formation induced by pH shift, a solution of TTR (3.5 μM) was first diluted in pH 2 (50 mM maleic acid, 100 mM KCl), and after 30 min the pH was raised to 3.7 by the addition of a predetermined volume of 2 M KOH. Aggregation or fibril formation was followed by light scattering, absorption at 330 nm, and Congo red binding. This experiment was performed at 37°C without stirring, and the extent of aggregation was used as a point of reference for the pressure-induced aggregation. In Fig. 2 each set of experiments was performed with the same batch of protein, allowing internal comparison in each panel.
Figure 2.

Pressure-induced aggregation of TTR as measured by light scattering. (A) At pH 5, 3.5 μM (●) or 1 μM (○) TTR was compressed at 3.5 kbar for 60 min at 37°C. Pressure was released at time 0 on the graph, and the light scattering was measured. The extent of aggregation is plotted as the ratio between light scattering at any given time (LS) and the light scattering value obtained immediately after decompression (LS0). Also shown for comparison is the increase in LS when 3.5 μM TTR was incubated at atmospheric pressure at pH 2.0 and then transferred to pH 3.7 (ΔpH, □). (B) At pH 5.0, 3.5 μM TTR was compressed at 3.5 kbar for 60 min at 37°C (●) or 1°C (○). After the return to atmospheric pressure, light scattering was measured with time. At the arrow, the sample that was pressurized at 1°C was heated up to 37°C, triggering the aggregation reaction. (C) At pH 5.6, 3.5 μM TTR was compressed at 37°C for 60 min (●) or 72 h (▵) and then pressure was released and aggregation was followed by the LS increase. Light scattering was measured by exciting the samples at 320 nm and collecting the scattered light from 315 to 325 nm. Because experiments were performed inside the pressure cell, no stirring was allowed. (D) At the end, experiments like those in A–C were removed from the pressure cell and the absorbance was measured at 330 nm (hollow bars) or Congo red binding (filled bars) was evaluated. The pressurized samples are marked P; the controls, C. Condition 1, ΔpH; condition 2, pH 5 and 1 μM TTR, 37°C; condition 3, pH 5 and 3.5 μM TTR, 37°C; condition 4, pH 5 and 3.5 μM TTR, 1°C → 37°C; condition 5, pH 5.6 and 3.5 μM TTR, 37°C.
HPLC Gel Filtration Measurements.
The average size of TTR after a cycle of compression-decompression was evaluated by size exclusion chromatography on a Superdex 75 HR column (Amersham Pharmacia Biotech) using a HPLC system (Shimadzu SPD-10A). The elution was monitored by fluorescence emission at 330 nm with excitation set at 280 nm.
Results and Discussion
Effects of High Pressure on T4: pH and Temperature Dependence.
Dissociation and denaturation of TTR by high pressure was investigated at pH 7.5 and 5.6 at 37°C (Fig. 1A, ▴ and ●, respectively). These two pH values were chosen to approximate conditions found in blood and during cellular processing of proteins in lysosomes. TTR has two tryptophan residues per monomer, located far apart in the tertiary structure. Tryptophan 79 is highly quenched in the tetramer at pH 7 whereas tryptophan 41 makes the major contribution to the fluorescence emission spectrum (30). High pressure promoted a red shift of the tryptophan emission, resulting in a decrease of the center of spectral mass (Fig. 1 A and B). After decompression, the initial center of mass was completely restored, indicating reversibility of the conformational changes induced by pressure (Fig. 1A, □).
Figure 1.

Pressure-induced dissociation-unfolding of TTR. (A) The center of mass of tryptophan emission was used to follow dissociation-denaturation of TTR as a function of pressure at pH 5.6 and 37°C (●), at pH 5.6 and 1°C (■), and at pH 7.5 and 37°C (▴). Protein concentration was 1 μM. The center of mass obtained after decompression was identical on all three cases (□). (Inset) The extent of dissociation-unfolding (α) was calculated as described in Experimental Procedures (Eq. 2) by using the center of mass values presented in A. The symbols used in the curves were the same. (B) Normalized fluorescence emission spectra of tryptophan at pH 5.6, 1°C at (solid line) atmospheric pressure or (dotted line) at 3.5 kbar. (C) Size exclusion chromatography of TTR after a cycle of compression. TTR (1 μM) was pressurized at pH 5.6 for 60 min at 1°C. After decompression at 1°C, the sample was injected immediately into the HPLC (chromatogram 2) or kept on ice for 30 min after decompression (chromatogram 3). The control (1 h at pH 5.6 not subjected to pressure) is shown in chromatogram 1. (Inset) An expansion of the monomer peak in these three conditions. The elution was followed by setting the excitation at 280 nm and collecting the emission at 330 nm.
TTR exists mainly as a tetramer in the pH range 5 to 7 (8, 30). As seen in Fig. 1A Inset, the stability of T4 at pH 5.6 (●) and pH 7.5 (▴) was comparable, suggesting that the stability of T4 is not strongly influenced by the acidification of the solution in the pH range 7.5 to 5.6. Therefore, in vivo, a much lower pH would be necessary to induce massive dissociation of the tetramers into monomers at atmospheric pressure.
Lowering the temperature facilitates the pressure-induced dissociation and denaturation of several proteins and macromolecular assemblages (17, 21, 36, 37). The combined effects of high pressure and low temperature on TTR were investigated at pH 5.6 (Fig. 1A). TTR denatured at much lower pressures at 1°C (▪) than at 37°C (○), demonstrating the entropic character of folding and association of TTR. The p1/2 values (pressure that causes 50% denaturation) were 1.9 and 1.3 kbar at 37°C and 1°C, respectively (Fig. 1A Inset).
Information about the state of association after return to atmospheric pressure was obtained by gel filtration chromatography on a sample of TTR subjected to 3.5 kbar at 1°C for 60 min. After decompression the sample was maintained on ice and injected into a HPLC-gel filtration column (Fig. 1C). The control sample at pH 5.6 (not subjected to pressure) eluted as a single peak at approximately 11 min, compatible with a tetramer the size of TTR (chromatogram 1). After pressure treatment at 1°C, the peak corresponding to the tetramers was again the prominent species but a new peak eluting around 17.4 min was observed (chromatogram 2). This peak, approximately 20% of the total protein present after decompression, is compatible with TTR monomers. After 30 min at 1 bar on ice, when this same sample was reinjected into the HPLC the monomer population decreased to less than 10% of the total protein present, whereas the tetramer peak exhibited a small increase, suggesting reassociation of the monomers (chromatogram 3).
It is noteworthy that no monomers were detected when TTR was pressurized at 37°C and then injected into the HPLC, presumably because of rapid reassociation aided by the high temperature (not shown). A monomeric fraction could be detected only when the reassociation was slowed by keeping the temperature at 1°C.
Fibril Formation After Pressure Treatment.
Aggregation of TTR into fibrils as a result of a pH shift has been shown in previous reports (29, 30). It occurred to us that high pressure might have a similar effect, populating an intermediate that lies at the juncture between the folding and aggregation pathways. To examine this possibility, we first incubated samples under high pressure and then used the light scattering to monitor the kinetics of aggregation after returning to atmospheric pressure (Fig. 2). The experiments were performed at pH 5, 5.6, 6.0, and 7.5. An experiment showing the increase in aggregation triggered only by a pH shift (pH 2.0→pH 3.7) was included for comparison (Fig. 2A, □).
After pressure release (Fig. 2 A and C, circles), TTR at pH 5 or 5.6 (but not at pH 6 or 7.5) underwent aggregation that was highly sensitive to protein concentration, indicating a reaction of high molecular order. Fig. 2B shows that low temperature has a striking effect on aggregation under these conditions. TTR (3.5 μM, pH 5) was pressurized at 1°C or 37°C for 60 min. After decompression, aggregation occurred in the 37°C sample but not at 1°C (Fig. 2B, ○). However, when the temperature was raised rapidly to 37°C in this sample (arrow) aggregation was triggered, suggesting that an amyloidogenic state is induced by pressure at both temperatures but is trapped in this state unless the temperature is raised. This finding is important, because it opens the possibility of carrying out structural studies (such as high-resolution NMR) after pressure treatment at 1°C to characterize this amyloidogenic intermediate. Recently, Smeller et al. (38) has shown that myoglobin, a nonamyloidogenic protein, also undergoes aggregation after pressure denaturation at 12 kbar.
To check whether prolonged pressure treatment would interfere with the aggregation process, TTR was pressurized at pH 5.6 for 72 h at 37°C, and then atmospheric pressure was restored and the changes in the light scattering were followed (Fig. 2C, ▵). Compared with a sample that was under pressure for only 60 min (Fig. 2C, ●), the extent of aggregation was very similar, although it occurred more slowly and with a pronounced lag phase (Fig. 2C Inset). It may be that prolonged pressure treatment produces an amyloidogenic state that has to undergo additional structural adjustments before aggregation.
Fig. 2D shows the absorbance at 330 nm (open bars) as well as the binding of Congo red (filled bars). As expected, the samples that were subjected to high pressure (P) displayed higher absorbance at 330 nm when compared with the control (C) samples indicating the presence of fibrils. The binding of Congo red, a specific dye for amyloid fibrils with cross β-sheet (39), was very pronounced in the pressure-treated samples with values comparable to those obtained for the fibrils induced by a pH shift without pressure. Pressure-treated samples also bound thioflavin (not shown), a property of fibrils.
Characterizing the Species Trapped Under High Pressure.
Binding of ANS and bis-ANS to accessible hydrophobic pockets in native proteins or molten-globule structures is followed by a large increase in fluorescence emission of these probes (19, 24, 40). The native state of TTR at pH 7 binds ANS in its hydrophobic channel, which is formed by juxtaposition of the four subunits of where the binding sites for thyroxine and ANS overlap (41). ANS binding was greatly enhanced when TTR was incubated at pH 2.0, where the A-state (molten-globule state) is formed (30). Here we used bis-ANS instead of ANS because bis-ANS seems to be more specific for partially folded states (19, 24, 40), and lower concentrations can be used.
Fig. 3 shows the binding of 10 μM bis-ANS during the pressure titration of 1 μM TTR at pH 7.5, 5.6, or 5.0 at 37°C. The conformational states induced by pressure at pH 5.0 (triangles) or pH 5.6 (circles) were able to bind this probe, suggesting the existence of long-range tertiary interactions in these states. It is interesting to note that pressure-induced binding of bis-ANS was much smaller at pH 7.5 (squares) than at pH 5.6 or 5.0. This suggests that different conformational states are populated under pressure in these different conditions despite the fact that the tryptophan residues of TTR report similar environments under pressure at those pHs (Fig. 1A). Fig. 3 Inset shows the displacement to short wavelengths for the emission of the bis-ANS bound to TTR as a function of pressure at pH 5.6, indicating that the pocket that binds bis-ANS in this partially folded state is more hydrophobic.
Figure 3.

Binding of bis-ANS to the pressure-dissociated/denatured TTR at different pHs. Bis-ANS (10 μM) was added to each sample containing 1 μM TTR. The spectral area (A) of bis-ANS emission measured after each increase in pressure was divided by the initial spectral area (A0) at atmospheric pressure. The samples were pressurized at pH 7.5 (squares), pH 5.6 (circles), or pH 5 (triangles). The decompression pathway is represented by empty symbols. All experiments were performed at 37°C and excitation was set at 360 nm and emission collected from 400–600 nm. (Inset) Shift in the center of mass of the bis-ANS fluorescence emission induced by pressure at pH 5.6, 37°C.
Upon decompression, the binding of bis-ANS was not reversible and the refolded tetramers retained an increased capacity to bind this probe (Fig. 3, empty symbols). This indicates the persistence of the contacts produced by high pressure after refolding of the tetramers. These data clearly suggest the formation of a stable, altered protein that is tetrameric as seen by the HPLC experiments (Fig. 1C) and by its ability to bind thyroxine (data not shown), one where the tryptophan residues retains their native environment (Fig. 1A, isolated symbols), but with a persistent capacity to bind bis-ANS. This species will be called T4* and may be similar to the species obtained by Lai et al. (31) after refolding TTR that had been denatured by guanidine.
Stability of T4*.
The spectroscopic changes promoted by pressurizing T4 presented two well-defined transitions (Fig. 1A and Inset). At pH 5.6 and 1°C, the first transition extends from approximately 750 to 1,500 bar. A second transition ranges from 1,500 to 3,500 bar. Only the first transition was sensitive to protein concentration (not shown), a feature that denotes dissociation of the tetramers. However, displacement of the pressure curves on increasing the protein concentration 10-fold was smaller than that expected on theoretical grounds (17). The reduced concentration dependence can be explained either by heterogeneity of the interactions among the subunits, as observed in large oligomers (15), or by incomplete dissociation of the tetramers, for example by concomitant formation of denatured tetramer. Denaturation without dissociation recently has been reported for other proteins (42, 43).
To check the stability of T4* against pressure after pressure treatment, two cycles of compression-decompression were performed. Fig. 4 shows that in a second cycle of compression (▪) the changes in the center of mass occurred at lower pressures, demonstrating hysteresis. Thus the recovered tetramer (T4*) was less stable than the native, nonpressurized tetramer. Fig. 4 Inset shows the extent of the reaction (α) for the two compression cycles. The p1/2 values for the first and second cycles were 2,275 and 1,850 bar, respectively.
Figure 4.

Pressure stability of the pressurized, refolded tetramers. TTR (1 μM) was subjected to a cycle of compression (●) and decompression (○) at pH 5.6, 37°C and then, after the return to atmospheric pressure, another cycle of compression (■) and decompression (□) was performed. The shift in the center of spectral mass of tryptophan emission was followed and used to calculate the extent of unfolding (α) (Inset). Other conditions were as in Fig. 1.
Hysteresis also was found for the guanidine-induced denaturation of TTR (31) and may be a characteristic of TTR regardless of the perturbing agent used. Hysteresis previously has been described for the association-refolding processes of several proteins (for review see refs. 15 and 17) and could be an indication that complete equilibrium has not been reached. In the case of chemical-induced denaturation of TTR (31), luciferase (44), and tailspike protein (45, 46), as well as with pressure-induced dissociation of many proteins (ref. 15 and references therein), the interconversion between species is slow leading to hysteris. In the 1980s, Gregorio Weber proposed an elegant hypothesis to explain the hysteresis, termed conformational drift. His proposal accounts for the rapid dissociation of an oligomeric protein coupled to a slow isomerization process (a first order-reaction) that takes place after dissociation (47, 48). The partial loss of affinity between subunits results from this progressive conformational change upon dissociation. Reciprocally, conformational adjustments restoring the original properties of the oligomer occur upon reassociation (47, 48).
Pressure-Induced Formation of Amyloidogenic States and Implications for the Formation of Amyloids.
The data presented here show that high pressure can replace a pH shift in perturbing the structure of TTR so that the protein aggregates. We demonstrate that massive aggregation of wild-type TTR can be triggered at relatively mild pH values provided that the subunit stability has been affected by populating an altered oligomer. This is achieved by using pressure (3.5 kbar) as a tool to poise the conformation to the amyloidogenic form. After the return to atmospheric pressure and physiological temperature, the protein exhibits a “spoiled” tetrameric structure, susceptible to aggregation (T4*). The stability of T4* is much lower (Fig. 4) than native TTR. At neutral pH, TTR is extremely stable to guanidine, and extrapolation of the dissociation kinetics suggests a half-life of about 293 years (31). From our data, we cannot discern whether aggregation proceeds from T4* or from the monomeric species generated from the microscopic dissociation of T4*, especially in light of our result showing a small fraction of monomers after return to atmospheric pressure (Fig. 1C). Fig. 5 depicts aggregation as a thermodynamic sink for T4*. Aggregation does not occur as long as pressure is maintained. Dissociation and prevention of aggregation of some protein aggregates by pressure recently have been described (14, 49).
Figure 5.

Free-energy diagram for dissociation, denaturation, and aggregation. The native tetramer and the monomer are represented by circles, the altered tetramer, monomer and aggregates by squares, and the denatured, unfolded monomer as a line. Distance along vertical axis indicates differences in Gibbs free energy among different TTR states.
In conclusion, T4* produced by a cycle of compression and decompression seems to have the properties of a “conformationally drifted” state. The free-energy diagram in Fig. 5 illustrates the dissociation and isomerization to an altered conformation and reassociation to form a loose tetramer (preaggregated state). A substantial activation barrier between the species T4 and T4* should be expected. In fact, a large barrier was proposed by Lai et al. (31) in the case of reassociated tetramer obtained by renaturing guanidine-denatured TTR. Pressure causes denaturation of proteins by inducing the penetration of molecules of water into the interior of the protein structure (50, 51), which facilitates the population of partially folded states. The lower stability of T4* may be caused by a more hydrated state. In fact, the preaggregated state has hydrophobic holes exposed to water that permit the binding of nonpolar dyes such as bis-ANS. We can speculate that under physiological conditions during the lifetime of TTR, any alteration in tetrameric structure that exposes hydrophobic patches (such as aging) and loosening of the subunit interactions could serve as the starting point for the aggregation process. The amyloidogenic state obtained under pressure or the altered tetramer could be the target for the further development of an antagonist capable of blocking formation of T* or T4*, a potential drug against the amyloid disease.
In addition to providing insights into the mechanism involved in TTR aggregation, this work opens avenues for investigation of pressure effects on single-amino acid mutant forms of TTR associated with familial amyloid polyneuropathy as well as other amyloidogenic proteins. The concept of a “preaggregated” state provides insights into the question of how correctly folded proteins sink into the aggregation pathway in amyloidogenic diseases.
Acknowledgments
We thank Martha Sorenson for critical reading of the manuscript and suggestions and Emerson Gonçalves for his competent technical assistance. This work was supported in part by an International Grant from the Howard Hughes Medical Institute (75197–553402) (to J.L.S.) and by grants from Programa de Núcleos de Excelência (PRONEX), Programa de Apoio ao Desenvolvimento Científico e Tecnológico (PADCT), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Apoio à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Financiadora de Estudos e Projetos (FINEP) of Brazil (to D.F. and J.L.S.), and by the Third World Academy of Sciences (to D.F.). J.L.S is a Howard Hughes Medical Institute International Researcher.
Abbreviations
- TTR
transthyretin
- T4
TTR tetramer
- bis-ANS
bis-(8-anilinonaphthalene-1-sulfonate)
References
- 1.Wetzel R. Trends Biotechnol. 1994;12:193–198. doi: 10.1016/0167-7799(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 2.Harper J D, Lansbury P T., Jr Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 3.Kelly J W. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 4.Koo E H, Lansbury P T, Jr, Kelly J W. Proc Natl Acad Sci USA. 1999;96:9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sipe J D. Annu Rev Biochem. 1992;61:947–975. doi: 10.1146/annurev.bi.61.070192.004503. [DOI] [PubMed] [Google Scholar]
- 6.Horwich A L, Weissman J S. Cell. 1997;89:499–510. doi: 10.1016/s0092-8674(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 7.Cohen F E, Prusiner S B. Annu Rev Biochem. 1998;67:793–819. doi: 10.1146/annurev.biochem.67.1.793. [DOI] [PubMed] [Google Scholar]
- 8.McCutchen S L, Kelly J W, Colon W. Biochemistry. 1993;32:12119–12127. doi: 10.1021/bi00096a024. [DOI] [PubMed] [Google Scholar]
- 9.McCutchen S L, Lai Z, Miroy G, Kelly J W, Colon W. Biochemistry. 1995;34:13527–13536. doi: 10.1021/bi00041a032. [DOI] [PubMed] [Google Scholar]
- 10.Saraiva M J. Hum Mutat. 1995;5:191–196. doi: 10.1002/humu.1380050302. [DOI] [PubMed] [Google Scholar]
- 11.Goldsteins G, Andersson K, Olofsson A, Dacklin I, Edvinsson A, Baranov V, Sandgren O, Thylen C, Hammarstrom S, Lundgren E. Biochemistry. 1997;36:5346–5352. doi: 10.1021/bi961649c. [DOI] [PubMed] [Google Scholar]
- 12.Mitraki A, Fane B, Haase-Pettingell C, Sturtevant J, King J. Science. 1991;253:54–58. doi: 10.1126/science.1648264. [DOI] [PubMed] [Google Scholar]
- 13.King J, Haase-Pettingel C, Robinson A S, Speed M, Mitraki A. FASEB J. 1996;10:57–66. doi: 10.1096/fasebj.10.1.8566549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foguel D, Robinson C R, deSousa P C, Jr, Silva J L, Robinson A S. Biotechnol Bioeng. 1999;63:552–558. doi: 10.1002/(sici)1097-0290(19990605)63:5<552::aid-bit5>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 15.Silva J L, Weber G. Annu Rev Phys Chem. 1993;44:89–113. doi: 10.1146/annurev.pc.44.100193.000513. [DOI] [PubMed] [Google Scholar]
- 16.Jonas J, Jonas A. Annu Rev Biophys Biomol Struct. 1994;23:287–318. doi: 10.1146/annurev.bb.23.060194.001443. [DOI] [PubMed] [Google Scholar]
- 17.Silva J L, Foguel D, Da Poian A T, Prevelige P E. Curr Opin Struct Biol. 1996;6:166–175. doi: 10.1016/s0959-440x(96)80071-6. [DOI] [PubMed] [Google Scholar]
- 18.Heremans K, Smeller L. Biochim Biophys Acta. 1998;1386:353–370. doi: 10.1016/s0167-4838(98)00102-2. [DOI] [PubMed] [Google Scholar]
- 19.Silva J L, Silveira C F, Correa A, Pontes L. J Mol Biol. 1992;223:545–555. doi: 10.1016/0022-2836(92)90669-b. [DOI] [PubMed] [Google Scholar]
- 20.Peng X, Jonas J, Silva J L. Proc Natl Acad Sci USA. 1993;90:1776–1780. doi: 10.1073/pnas.90.5.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nash D P, Jonas J. Biochemistry. 1997;36:14375–14383. doi: 10.1021/bi970881v. [DOI] [PubMed] [Google Scholar]
- 22.Ruan K, Lange R, Bec N, Balny C. Biochem Biophys Res Commun. 1997;239:150–154. doi: 10.1006/bbrc.1997.7313. [DOI] [PubMed] [Google Scholar]
- 23.Vidugiris G J, Royer C A. Biophys J. 1998;75:463–470. doi: 10.1016/S0006-3495(98)77534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foguel D, Silva J L, Prat-Gay G. J Biol Chem. 1998;273:9050–9057. doi: 10.1074/jbc.273.15.9050. [DOI] [PubMed] [Google Scholar]
- 25.Blake C C, Geisow M J, Oatley S J, Rerat B, Rerat C. J Mol Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 26.Van Jaarsveld P, Branch W T, Robbins J, Morgan F J, Kanda Y, Canfield R E. J Biol Chem. 1973;248:7898–7903. [PubMed] [Google Scholar]
- 27.Rosen H N, Moses A C, Murell J, Liepnieks J J, Benson M D. J Clin Endocrinol Metab. 1993;77:370–374. doi: 10.1210/jcem.77.2.8345041. [DOI] [PubMed] [Google Scholar]
- 28.Almeida M R, Saraiva M J. Eur J Endocrinol. 1996;135:226–230. doi: 10.1530/eje.0.1350226. [DOI] [PubMed] [Google Scholar]
- 29.Colon W, Kelly J W. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 30.Lai Z, Colon W, Kelly J W. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 31.Lai Z, McCulloch J, Lashuel H A, Kelly J W. Biochemistry. 1997;36:10230–10239. doi: 10.1021/bi963195p. [DOI] [PubMed] [Google Scholar]
- 32.Quintas A, Saraiva M J, Brito R M. FEBS Lett. 1997;418:297–300. doi: 10.1016/s0014-5793(97)01398-7. [DOI] [PubMed] [Google Scholar]
- 33.Lashuel H A, Lai Z, Kelly J W. Biochemistry. 1998;37:17851–17864. doi: 10.1021/bi981876+. [DOI] [PubMed] [Google Scholar]
- 34.Kelly J W, Colon W, Lai Z, Lashuel H A, McCulloch J, McCutchen S L, Miroy G J, Peterson A S. Adv Protein Chem. 1997;50:161–181. doi: 10.1016/s0065-3233(08)60321-6. [DOI] [PubMed] [Google Scholar]
- 35.Neuman R C, Kauzmann W, Zipp A. J Phys Chem. 1973;77:2687–2691. [Google Scholar]
- 36.Zhang J, Peng X, Jonas A, Jonas J. Biochemistry. 1995;34:8631–8641. doi: 10.1021/bi00027a012. [DOI] [PubMed] [Google Scholar]
- 37.Foguel D, Weber G. J Biol Chem. 1995;270:28759–28766. doi: 10.1074/jbc.270.48.28759. [DOI] [PubMed] [Google Scholar]
- 38.Smeller L, Rubens P, Heremans K. Biochemistry. 1999;38:3816–3820. doi: 10.1021/bi981693n. [DOI] [PubMed] [Google Scholar]
- 39.Glenner G G, Eanes E D, Page D L. J Histochem Cytochem. 1972;20:821–826. doi: 10.1177/20.10.821. [DOI] [PubMed] [Google Scholar]
- 40.Shi L, Palleros D R, Fink A L. Biochemistry. 1994;33:7536–7546. doi: 10.1021/bi00190a006. [DOI] [PubMed] [Google Scholar]
- 41.Cheng S Y, Pages R A, Saroff H A, Edelhoch H, Robbins J. Biochemistry. 1977;16:3707–3713. doi: 10.1021/bi00635a031. [DOI] [PubMed] [Google Scholar]
- 42.Robinson C R, Rentzeperis D, Silva J L, Sauer R T. J Mol Biol. 1997;273:692–700. doi: 10.1006/jmbi.1997.1342. [DOI] [PubMed] [Google Scholar]
- 43.Mohana-Borges R, Silva J L, Ruiz-Sanz J, de Prat-Gay G. Proc Natl Acad Sci USA. 1999;96:7888–7893. doi: 10.1073/pnas.96.14.7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinclair J F, Ziegler M M, Baldwin T O. Nat Struct Biol. 1994;1:320–326. doi: 10.1038/nsb0594-320. [DOI] [PubMed] [Google Scholar]
- 45.Fuchs A, Seiderer C, Seckler R. Biochemistry. 1991;30:6598–6604. doi: 10.1021/bi00240a032. [DOI] [PubMed] [Google Scholar]
- 46.Schuler B, Rachel R, Seckler R. J Biol Chem. 1999;274:18589–18596. doi: 10.1074/jbc.274.26.18589. [DOI] [PubMed] [Google Scholar]
- 47.Weber G. Biochemistry. 1986;25:3626–3631. doi: 10.1021/bi00360a022. [DOI] [PubMed] [Google Scholar]
- 48.Weber G. Protein Interactions. New York: Chapman & Hall; 1992. [Google Scholar]
- 49.St John R J, Carpenter J F, Randolph T W. Proc Natl Acad Sci USA. 1999;96:13029–13033. doi: 10.1073/pnas.96.23.13029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hummer G, Garde S, Garcia A E, Paulaitis M E, Pratt L R. Proc Natl Acad Sci USA. 1998;95:1552–1555. doi: 10.1073/pnas.95.4.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oliveira A C, Gaspar L P, Da Poian A T, Silva J L. J Mol Biol. 1994;240:184–187. doi: 10.1006/jmbi.1994.1433. [DOI] [PubMed] [Google Scholar]