

Abstract
Peptide methionine sulfoxide reductase (MsrA; EC 1.8.4.6) reverses the inactivation of many proteins due to the oxidation of critical methionine residues by reducing methionine sulfoxide, Met(O), to methionine. MsrA activity is independent of bound metal and cofactors but does require reducing equivalents from either DTT or a thioredoxin-regenerating system. In an effort to understand these observations, the four cysteine residues of bovine MsrA were mutated to serine in a series of permutations. An analysis of the enzymatic activity of the variants and their free sulfhydryl states by mass spectrometry revealed that thiol–disulfide exchange occurs during catalysis. In particular, the strictly conserved Cys-72 was found to be essential for activity and could form disulfide bonds, only upon incubation with substrate, with either Cys-218 or Cys-227, located at the C terminus. The significantly decreased activity of the Cys-218 and Cys-227 variants in the presence of thioredoxin suggested that these residues shuttle reducing equivalents from thioredoxin to the active site. A reaction mechanism based on the known reactivities of thiols with sulfoxides and the available data for MsrA was formulated. In this scheme, Cys-72 acts as a nucleophile and attacks the sulfur atom of the sulfoxide moiety, leading to the formation of a covalent, tetracoordinate intermediate. Collapse of the intermediate is facilitated by proton transfer and the concomitant attack of Cys-218 on Cys-72, leading to the formation of a disulfide bond. The active site is returned to the reduced state for another round of catalysis by a series of thiol—disulfide exchange reactions via Cys-227, DTT, or thioredoxin.
Keywords: oxidation, cysteine, thioredoxin, Alzheimer's disease, aging
Aerobic metabolism produces many reactive oxygen species that can oxidize free methionine and methionine in proteins to methionine sulfoxide, Met(O) (1–4). This posttranslational modification of proteins has been shown to alter biological activity, cellular signaling processes, and the virulence determinants of mammalian and plant pathogens (5–9). Evidence also suggests that methionine residues may function as endogenous antioxidants, which could be important in the aging process and the onset of Alzheimer's disease (3, 10, 11). Therefore, it is of interest to elucidate the mechanism of the enzymatic reduction of Met(O) to methionine by peptide methionine sulfoxide reductases (MsrAs; EC 1.8.4.6).
MsrA is a ubiquitous enzyme found in a wide variety of organisms and animal tissues (9, 12, 13). The importance of MsrAs in protecting cells against oxidative damage is supported by the decreased viability of the null mutants of both Escherichia coli and yeast during oxidative stress (14, 15). Moreover, the overexpression of MsrA in E. coli, yeast, and stably transfected human T cells provided higher resistance to hydrogen peroxide treatment (14, 16).
MsrAs have broad substrate specificity and can reduce a variety of methyl or ethyl sulfoxide compounds including l-Met(O), d-Met(O), N-Ac-l-Met(O), dimethyl sulfoxide, and l-ethionine sulfoxide (12, 17). However, the most important physiological role of MsrAs is to reduce Met(O) residues in proteins (12, 17, 18). The enzymatic activity of MsrAs depends on the reducing equivalents from DTT or a thioredoxin-regenerating system (12, 17), suggesting the potential involvement of one or more cysteine residues. To test this involvement, the four cysteine residues of bovine MsrA (bMsrA, Fig. 1) were mutated to serine in a series of permutations.
Figure 1.

Sequence alignment of MsrAs and proteins containing putative MsrA domains. The longer N-terminal sequences preceding the region of strong homology are not shown. Filled squares mark those sequences that have been truncated at the C terminus. The locations of the cysteine residues of bMsrA are indicated by filled circles. The alignment was generated by using the clustal w algorithm in macvector 6.0 (Oxford Molecular Group). Dark and light background shading illustrate regions having greater than 51% identity or similarity, respectively. Spaces added for insertions are shown by dashed lines. The sequence of Neisseria gonorrhoeae (NEIGO) MsrA was redetermined (see text) and the region corresponding to Cys-72 in bMsrA was found to be G205GCFWGLEAYFQRIDGVVDAVSG227, which differs from that determined previously (A205AASGAWGAWKPISNASTAWLTRYR226) (22). The remainder of the sequence within the MsrA domain was unchanged. Abbreviations: ARATH, Arabidopsis thaliana (accession number, P54150); BACSU, Bacillus subtilis (P54154); BOVIN, Bos taurus (P54149); BRANA, Brassica napus (P54151); DROME, Drosophila melanogaster (P08761); ERWCH, Erwinia chrysanthemi (AJ012716); FRAAN, Fragaria ananassa (P54152); GRACI, Gracilaria gracilis (AAD43253); HAEIN, Haemophilus influenzae (P45213); ECOLI, Escherichia coli (P27110); HELPY, Helicobacter pylori (O25011); HUMAN, Homo sapiens (CAB59628); LYCES, Lycopersicon esculentum (P54153); MYCGE, Mycoplasma genitalium (P47648); MYCPN, Mycoplasma pneumoniae (P75188); MYTB, Mycobacterium tuberculosis (CAB07043); NEIGO, Neisseria gonorrhoeae (P14930); SCHIPO, Schizosaccharomyces pombe (Q09859); STRPN, Streptococcus pneumoniae (P35593); YEAST, Saccharomyces cerevisiae (P40029).
Materials and Methods
Expression and Purification of Recombinant bMsrA.
An N-terminal poly(His)-tagged form of bMsrA was obtained by overexpression in E. coli. The expression vector was constructed by amplifying the bMsrA gene (17) with PCR mixtures containing Deep Vent DNA Polymerase (New England Biolabs). The flanking restriction sites, NdeI and XhoI, were added with the following primers: 5′ end primer, 5′-G GAA TTC CAT ATG CTC TCG GTC ACC CGT CGT GCC CTC CAG C-3′; 3′ end primer, 5′-ATC TTA CTC GAG TTA CTT TTT AAT ACC C-3′. In addition, the tandem rare Arg codons (AGG) that code for residues 6 and 7 of the protein were replaced by CGU, preferred by E. coli (19). The PCR product was cloned into the Novagen pet28b expression vector, and the sequence was verified. Overexpression of bMsrA from pet28b resulted in the addition of a His-tag to the N terminus of the protein, i.e., GSSHHHHHHSSGLVPRGSH-. Cys to Ser mutants were made by using the QuickChange Site-Directed Mutagenesis Kit (Stratagene) with the following primers and their complement: Cys-72 → Ser, 5′-GCT GTA TTT GGA ATG GGC TCT TTC TGG GGA GGC-3′; Cys-107 → Ser, 5′-CCC AAT CCT ACT TAT AGA GAA GTC TCC TCA GGA AAA ACT GG-3′; Cys-218 → Ser, 5′-CGA CGG GTA CTC CGG CCT CGG GG-3′; and Cys-227 → Ser, 5′-GCA CCG GAG TGT CTT CTC CCC TGG GTA-3′.
Four-liter fermentation cultures of Epicurian Coli BL21-Gold(DE3) cells (Stratagene) containing the expression plasmid were grown in LB-kanamycin broth (100 mg/liter) at 37°C, 750 rpm, and 10–15 liters of air per min. Expression was induced by the addition of isopropyl β-d-thiogalactoside (IPTG) to 0.1 mM at 1.0 OD600 and continuing growth for 4 h at 25°C and 250 rpm. The cells (15–30 g) were suspended in 100 ml of +T/G buffer [50 mM Hepes (pH 7.9)/10% glycerol/0.1% Triton X-100/0.5 M KCl/40 μg/ml DNase/1 mM MgCl2/15 mM methionine/5 mM imidazole/2 Complete/EDTA-free (Boehringer-Mannheim) inhibitor tablets] and lysed by passage through a French press cell. The resulting slurry was centrifuged at 40,000 × g for 30 min. The supernatant was loaded onto a 10-ml NTA-agarose column (Qiagen, Valencia, CA) equilibrated with +T/G buffer at 4°C. After washing with +T/G and −T/G buffer (+T/G buffer without glycerol, Triton X-100, and inhibitor mixture), bMsrA was eluted with a 500-ml gradient from 5 to 250 mM imidazole in −T/G buffer. The fractions corresponding to bMsrA, determined by SDS/PAGE analysis, were pooled. EDTA and DTT were added to give a final concentration of 5 mM and 15 mM, respectively. The protein solution was incubated on ice for 1 h and then dialyzed against 4 liters of 20 mM Mes (pH 6.5)/20 mM NaCl/0.1 mM EDTA (buffer A) at 4°C. The dialyzed protein was loaded in several batches onto a POROS SP/M column (BioCAD system; PerSeptive Biosystems, Foster City, CA) equilibrated with 20 mM Mes (pH 6.5)/0.3 M NaCl/0.1 mM EDTA. Protein was eluted at 10 ml/min with a 10-column volume gradient from 0.3 to 0.7 M NaCl. After dialysis overnight against 4 liters of buffer A, the protein was concentrated with an Amicon Centriprep device (10-kDa cutoff). Protein concentrations were determined by absorption at 280 nm with the theoretical extinction coefficient of each variant calculated by using the expasy protparam tool [www.expasy.ch, Swiss Institute of Bioinformatics; wild-type, 31,390 M-1⋅cm-1; single and double mutants, 31,270 M-1⋅cm-1; triple mutants, 31,150 M-1⋅cm-1]. The protein concentrations were in good agreement with those values obtained by using the Bradford method of protein determination. Typical yields were 5–25 mg/liter culture.
Analysis of the recombinant E. coli and bovine MsrAs by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) and electronic absorption spectroscopy showed no indication that either enzyme contains bound metals or cofactors (data not shown).
MsrA Activity Assay.
The enzymatic activity of bMsrA (2–4 μg) was assayed at 37°C in 25 mM Tris (pH 7.4) essentially as previously described by using N-acetyl-l-[3H]methionine sulfoxide as substrate (165 μM) in a total reaction volume of 30 μl (20). The incubation contained either 15 mM DTT or a thioredoxin-regenerating system (180 pmol of E. coli thioredoxin, 3 μg of E. coli thioredoxin reductase, and 50 nmol of NADPH). The reactions were stopped after 10 min by the addition of 0.5 M HCl (1 ml). The acidified solution was extracted with 3 ml of ethyl acetate, and the amount of product, N-Ac-l-[3H]Met, was determined by quantitating the amount of radioactivity in the ethyl acetate phase, corrected for the efficiency of extraction (50%).
The oligomeric state of the bMsrA variants [2.5 mg/ml, 25 mM Tris (pH 7.4)] was determined either in the absence or in the presence of 0.9 mM Met(O) (10-fold molar excess) without the addition of an exogenous reducing system. After incubation for 1 h at 37°C, sample loading buffer [62.5 mM Tris (pH 6.8)/2% SDS/0.01% bromophenol blue] with and without DTT (100 mM) was added. The samples were heated at 100°C for 10 min and analyzed by SDS/PAGE using 10–15% gradient gels on a Pharmacia PhastSystem.
Mass Spectroscopic Determination of Free Thiols.
The number of free sulfhydryl groups of monomeric bMsrA was determined by derivatization with methyl methanethiosulfonate (MMTS, Sigma-Aldrich, ref. 21) and delayed-extraction matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry (PerSeptive Biosystems; Voyager-DE Biospectrometry Workstation). Stock enzyme solutions were diluted just before analysis to 100 μM in 25 mM Tris (pH 7.4) and 15 mM DTT and incubated for 10 min at 37°C. DTT was removed by using Micro Bio-Spin 6 columns (Bio-Rad). The enzyme was then incubated with 1 mM of either l-Met(O) or l-Met in 25 mM Tris (pH 7.4) for 1 h at 37°C. Substrate and product were removed by spin columns. The sample was then split into three parts for mass analysis: (i) no further treatment, (ii) addition of MMTS [10 mM Tris (pH 8.0), 233 μM MMTS (4.5 equivalents)] and incubation on ice for 30 min, or (iii) the addition of 10 mM Tris (pH 8.0) and 15 mM DTT and incubation at 37°C for 15 min. The DTT-treated samples were subsequently passed through spin columns to remove DTT and treated with MMTS as described above. The samples were purified and desalted for mass analysis by adding 1 μl of 25% trifluoroacetic acid (TFA) and passing the protein over miniature C18 columns (ZipTip, Millipore) using 0.1% TFA and 50% acetonitrile as wash and elution buffers, respectively. The samples were spotted onto the sample plate in the following manner: 0.5 μl of internal standards [0.1 pmol/μl in 10 mg/ml 3,5-dimethoxy-4-hydroxycinnamic acid (sinnapinic acid, Sigma-Aldrich), 30% acetonitrile, 0.1% TFA; E. coli thioredoxin (11,674 Da), horse apomyoglobin (16,952 Da) (PerSeptive Biosystems; Sequazyme Kit Mix 3)], 0.5 μl of sample, and 0.5 μl of sinnapinic acid stock. Because of the inherent formation of higher-order oligomers during the evaporation of the samples, only the mass of the monomeric form of each bMsrA variant was used in subsequent comparisons.
Results
MsrAs Show High Sequence Homology.
Sequence identity between bMsrA and most members of the family ranges from 34 to 88% (Fig. 1). During the construction of the alignment, it appeared that the MsrA from Neisseria gonorrhoeae (NEIGO) was unrelated to any other MsrA in the region around Cys-72 in bMsrA. However, reanalysis of the NEIGO MsrA DNA (22) with modern fluorescence dye terminators and cycle-sequencing methods revealed an error in the published sequence and confirmed the presence of the “GCFWG” motif (Fig. 1). Therefore, all MsrAs that have been sequenced contain an equivalent to Cys-72 in bMsrA. Cys-107 and Cys-218 are conserved within 70% of the proteins. Toward the C terminus of bMsrA, Cys-218 and Cys-227 bracket a glycine-rich region. Some other MsrAs also contain C-terminal cysteines, although in variable locations.
Cys-72 Is Essential for Activity.
Similar to previous reports for E. coli and bovine MsrA, it was found that the His-tag form of bMsrA used in these experiments was more active with thioredoxin than with DTT as the reducing system (12, 17).
The site-directed, Cys to Ser mutations had different effects on the enzymatic activity (Table 1). All variants containing the Cys-72 → Ser mutation were inactive. The mutation of Cys-107 had no significant effect on activity. A low level of residual activity was seen for the Cys-218 variants with both reducing systems. The mutation of Cys-227 caused a small reduction in activity in the DTT-based assay but had a much greater effect in the physiologically relevant, thioredoxin-based assay.
Table 1.
The enzymatic activity of Cys to Ser mutants of bovine MsrA
| Cysteines replaced | Activity, %
|
|
|---|---|---|
| DTT | Thioredoxin | |
| None (WT) | 100 | 100 |
| 72 | 0 | 0 |
| 107 | 114 | 96 |
| 218 | 35 | 22 |
| 227 | 89 | 19 |
| 72, 218 | 0.7 | 0 |
| 107, 218 | 22 | 6 |
| 107, 227 | 96 | 14 |
| 218, 227 | 42 | 4 |
| 72, 107, 227 | 0 | 0 |
| 107, 218, 227 | 39 | 8 |
Activity of 100% represents 580 and 1,250 pmol of N-Ac-l-[3H]Met produced in the DTT and thioredoxin assays, respectively. Each value represents the average of three independent measurements at two enzyme concentrations (SD range 5–10%).
MsrA is inactivated by exposure to hydrogen peroxide. Pretreatment with MMTS modified all four cysteine residues, protecting them from oxidation by hydrogen peroxide (21). The addition of DTT reversed this covalent modification and restored enzymatic activity. Thus, in contrast to the unprotected protein, the addition of hydrogen peroxide to the MMTS-blocked protein did not affect activity (data not shown). These results not only confirm the requirement for free cysteine thiols in the action of bMsrA but strongly suggest that there are no other oxidizable residues in the protein that are essential for activity.
Disulfide Bond Formation During the Reaction Cycle.
The observed lower specific activity of the Cys-218 → Ser and the Cys-227 → Ser mutants in the presence of thioredoxin suggested that these residues may facilitate catalysis by relaying the reducing equivalents from thioredoxin through thiol–disulfide exchange. To investigate this possibility, the free sulfhydryl state of each variant was determined by derivatization with MMTS (21) after preincubation with substrate or product (Table 2). MMTS was used instead of the more traditional Ellman's reagent [5,5′-dithiobis(2-nitrobenzoic acid), DTNB] because derivatization of all cysteines by the smaller MMTS molecule occurred rapidly when the protein was in its native, folded state (data not shown). MMTS treatment resulted in the covalent attachment of a methylthiol group (—SCH3, 47 Da) to each free thiol. This addition to the monomeric form of each variant (see Materials and Methods) was confirmed by delayed-extraction matrix-assisted laser desorption ionization-time of flight mass spectrometry. The loss of two free sulfhydryls for the wild-type enzyme, only upon treatment with substrate, indicated the formation of a disulfide bond. The inactive Cys-72 mutant showed no loss in free thiols. Each of the other single mutants, however, did show disulfide bond formation in the presence of substrate. Further analysis of all potential cysteine mutant pairs revealed that disulfide bond formation could occur between either Cys-72 and Cys-218 or Cys-72 and Cys-227. The disulfide bonds were reduced by DTT (data not shown).
Table 2.
Free sulfhydryl content of bMsrA
| Cysteines replaced | No. of Cys | Mass increase of monomer, Da*
|
Decrease in free thiols† | |
|---|---|---|---|---|
| l-Met(O) | l-Met | |||
| None (WT) | 4 | 81 /86 | 201 /197 | 2 |
| 72 | 3 | 138 /142 | 140 /144 | 0 |
| 107 | 3 | 35 /38 | 145 /153 | 2 |
| 218 | 3 | 44 /51 | 160 /157 | 2 |
| 227 | 3 | 49 /50 | 154 /158 | 2 |
| 72, 218 | 2 | 91 /92 | 90 /92 | 0 |
| 107, 218 | 2 | 1 /0 | 104 /100 | 2 |
| 107, 227 | 2 | 7 /0 | 102 /106 | 2 |
| 218, 227 | 2 | 78 /75 | 101 /100 | 0 |
| 72, 107, 227 | 1 | 48 /52 | 50 /53 | 0 |
| 107, 218, 227 | 1 | 45 /39 | 66 /72 | 0 |
The values indicate the observed increase in mass of the monomer of each variant upon treatment with MMTS from two independent experiments as described in Materials and Methods. The mass of each variant without pretreatment was used as the reference. The theoretical mass increases for the covalent addition of one to four methylthiol groups (—SCH3) from MMTS to cysteine are 47, 94, 141, and 188 Da, respectively.
† The difference in the number of free cysteine thiols upon treatment with l-Met(O) vs. l-Met. A value of 2 represents the formation of a disulfide bond. The disulfide bonds formed could be reduced with the addition of DTT (data not shown).
Intermolecular Disulfide Bonds Formed in the Absence of DTT or Thioredoxin.
The complete loss of activity for the Cys-72 mutants clearly shows the primary importance of Cys-72 in catalysis (Table 1). The residual activity of the triple mutant, with Cys-107, Cys-218, and Cys-227 replaced and only Cys-72 retained, was surprising, however, given the apparent involvement of a disulfide bond between Cys-72 and either Cys-218 or Cys-227. Single-turnover experiments performed with the same mutant without the addition of reducing agent also unexpectedly showed the formation of product (data not shown). SDS/PAGE analysis of the bMsrA variants (Fig. 2) indicated that, under nonreducing conditions, incubation with Met(O) could stimulate the formation of intermolecular, mixed disulfide bonds. These interactions were not seen after the addition of DTT. For example, both the wild-type enzyme and the Cys-107 mutant were able to oligomerize up to a tetramer, but only in the presence of substrate. All Cys-72 → Ser mutants showed no change upon treatment with substrate. Some variants showed dimer formation, but the formation of trimers depended on the presence of either Cys-107 or Cys-218. The triple mutant containing only Cys-72 clearly formed dimer with the addition of substrate. Even though the enzyme concentrations used in these experiments were artificially high, the specific formation of intermolecular disulfide bonds upon the addition of substrate suggests that the release of product, after the action of Cys-72, is facilitated by an additional thiol interaction. Moreover, the source of the thiol can be either intramolecular, i.e., protein thiols, or intermolecular, i.e., DTT, thioredoxin or another molecule of bMsrA.
Figure 2.

SDS/PAGE analysis of the oligomeric state of bMsrA upon treatment with Met(O). The Coomassie-stained gels show the formation of intermolecular disulfide bonds between some of the variants of bMsrA after incubation with substrate under nonreducing conditions (see Materials and Methods). The addition of DTT reduced the polymeric forms [e.g., dimer (D), trimer (TR), and tetramer (TE)] to the monomeric form (M), which migrated just below the 31-kDa molecular mass standard. Lane 1, molecular mass standards; lane 2, protein incubated in the absence of l-Met(O) and analyzed under nonreducing conditions; lane 3, protein incubated in the presence of l-Met(O) and analyzed under nonreducing conditions; lane 4, protein incubated with l-Met(O) and analyzed after treatment with DTT.
Discussion
The wide tissue distribution and high sequence homology (Fig. 1) of MsrAs supports their pivotal role in the repair of oxidative damage to proteins and free methionine (9, 12–15). Potential pathophysiological manifestations of increased oxidation of methionine in proteins include smokers' emphysema, adult respiratory distress syndrome, rheumatoid arthritis, and Alzheimer's disease (11, 23–25). Moreover, MsrAs appear to be important for the infection of eukaryotic and plant cells by pathogenic bacteria (5, 7).
Mechanism of Catalysis.
The reactions of small molecule sulfoxides with thiols have been extensively studied (26–29). The slow step (step 1) is considered to be the attack on the sulfur atom of the sulfoxide by the thiol.
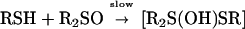 |
1 |
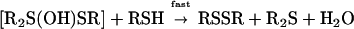 |
2 |
Integral components of this step are thought to include the protonation of the sulfoxide oxygen, the formation of a sulfonium ion, and the deprotonation of the thiol to generate a nucleophilic thiolate ion. The importance of forming a thiolate ion is supported by the observed rate acceleration with thiols that exhibit lower pKa values. Breakdown of the trigonal-bipyramidal intermediate (30–35) occurs with the addition of a second molecule of thiol (step 2).
Based on the available data, a mechanism for the reduction of Met(O) by MsrA is proposed in Fig. 3. It involves the overall transfer of two protons and two electrons. The loss of activity for the Cys-72 mutants (Fig. 1, Table 1) strongly suggests that Cys-72 acts as the nucleophile in the reaction. Proton transfer from either Cys-72 or a specific active site donor generates a sulfonium ion (II). An attack, perhaps simultaneously with protonation of the sulfoxide, by the thiolate form of Cys-72 leads to the formation of a tetravalent sulfur intermediate (III). Collapse of the intermediate is facilitated by the concomitant proton transfer from solvent, Cys-218, or an active site donor and the attack of Cys-218 on Cys-72 (IV). Hence, one turnover of the reaction results in the release of water and product and the formation of a disulfide bond. Return of the active site to a fully reduced state (I) is facilitated by thiol—disulfide exchange via Cys-227 (V) and either DTT or a thioredoxin regeneration system (VI). The formation of a disulfide bond between Cys-72 and Cys-218 is supported by the analysis of the double mutants of bMsrA by mass spectrometry (Table 2). The inability of DTT to rescue the lower activity of the Cys-218 variants, in contrast to the Cys-227 variants (Table 1), also suggests that Cys-218 is the preferred thiol for release of product. Moreover, the observed increase in activity with the use of DTT as compared with thioredoxin for the Cys-227 variants indicates that the role of Cys-227 is to facilitate the transfer of reducing equivalents from thioredoxin.
Figure 3.

Proposed reaction mechanism for MsrA catalysis. Protonation of Met(O) (I) leads to the formation of a sulfonium ion (II). Possibly concomitant with sulfoxide protonation, Cys-72 attacks the sulfur atom of the sulfonium ion, leading to the formation of a covalent intermediate (III). Breakdown of the complex is facilitated by proton transfer and the attack on Cys-72 by Cys-218 (IV). Return of the active site to a fully reduced state (I) is facilitated by thiol—disulfide exchange via Cys-227 (V) and either DTT or a thioredoxin regeneration system (VI) (see Discussion for details). Abbreviations: TR, thioredoxin; TRR, thioredoxin reductase.
Cys-218 and Cys-227 of bMsrA are located within a glycine-rich “C-terminal-tail.” This tail could be either in close proximity to the active site or may be able to “flip in and out” providing reducing equivalents. It is clear from the enzyme activities of the cysteine mutants that, if Cys-218 is removed, for example, a cysteine in the vicinity can compensate and restore activity. This thiol can be either from the protein, i.e., Cys-227, or from an exogenous source, i.e., DTT, thioredoxin, or another molecule of bMsrA. These observations also suggest that the active site cysteines are probably near the surface, enabling an interaction with thioredoxin, which is quite large (≈12 kDa). Because the reducing equivalents can come from a variety of sources, the apparent diversity (Fig. 1) in the location and number of C-terminal residues is reasonable. As long as there is a source of thiols to shuttle electrons to Cys-72, the reduction of Met(O) can occur. It is interesting to note, however, that activity with 2-mercaptoethanol or glutathione is not observed (12, 17). This lack of activity is presumably a consequence of decreased reducing power and the inability to form a more stable intermolecular ring structure like DTT or the formation of a stable disulfide bond in thioredoxin (36–41).
The proposed mechanism indicates the need for proton donors and acceptors to facilitate the formation of the intermediate and the ultimate release of water and methionine. Several other residues besides Cys-72 are strictly conserved in MsrAs (Fig. 1; e.g., Glu-115, Tyr-155, Glu-203, and His-206 in bMsrA). These residues could provide essential hydrogen-bonding or ion-pairing interactions to activate Cys-72, Cys-218, and Cys-227 for the initial nucleophilic attack and subsequent thiol–disulfide exchange steps (29, 42, 43). In addition, these conserved residues may mediate the formation and stabilization of the covalent intermediate.
Comparison to Other Reductases.
A comparison of the proposed mechanism of MsrAs to that of other reductases that exhibit similar characteristics suggests that the reduction of Met(O) by MsrAs is distinctly different. For example, the use of a cysteine-rich, “C-terminal-tail” to reduce the active site has also been described for the class I ribonucleotide reductase from E. coli (44–47). Despite this apparent resemblance, the proposed reaction mechanisms for this class of enzymes and MsrAs are very different. Ribonucleotide reductase relies on the formation of a stable tyrosine radical adjacent to a diferric metal cluster in the R1 subunit. The radical is subsequently transferred to the R2 subunit to form a cysteine thiyl radical. The movement of the radical through the substrate results in the production of deoxynucleotides and the formation of a disulfide bond that must be reduced for additional catalytic cycles. In contrast, MsrAs show no metal dependence and thus probably do not use a free radical in catalysis. Because MsrAs can reduce dimethyl sulfoxide (DMSO, ref. 17), one might also expect that MsrAs are similar to DMSO and biotin sulfoxide reductases. These reductases, however, employ a sulfur-rich, molybdopterin guanine dinucleotide cofactor for catalysis (48–50).
The alkyl hydroperoxide reductase (AhpR) system exhibits a dependence on thiol–disulfide exchange between partners of a two-component system, similar to MsrA and thioredoxin. This system is made up of a complex between a dimer of the AhpC protein and a flavoprotein reductase, AhpF (51, 52). During the reduction reaction, however, a cysteine sulfenic acid is formed. A recent review of AhpR and other reductases, peroxiredoxins, and phosphatases that use this unusual cysteine modification illustrates that the intermediate is formed by an attack on the oxygen atom of hydrogen peroxide and organic hydroperoxides (53). Because the reactions of thiols with sulfoxides are so well characterized and involve an attack at the sulfur atom, it is unlikely that the reaction catalyzed by MsrAs utilizes a sulfenic acid. Nonetheless, this possibility cannot be excluded at this time.
Acknowledgments
We acknowledge the technical assistance of Leslie S. Gay, Mei Wang, Debra A. McMillen, and Yanling Wang and thank Dr. Bruce Branchaud for reading the manuscript. This work was supported in part by National Research Service Award F32-GM17536 (W.T.L.) and Grant GM20066 (B.W.M.) from the National Institutes of Health. N.B. and H.W. were supported by funds from Hoffmann–La Roche. J.F.H. was supported by the Natural Sciences and Engineering Research Council (Canada).
Abbreviations
- Met(O)
methionine sulfoxide
- MMTS
methyl methanethiosulfonate
- MsrA
peptide methionine sulfoxide reductase
- bMsrA
bovine MsrA
- TFA
trifluoroacetic acid
Note Added in Proof
A recent report has also demonstrated the importance of Cys-72 in yeast MsrA (54).
References
- 1.Vogt W. Free Radical Biol Med. 1995;18:93–105. doi: 10.1016/0891-5849(94)00158-g. [DOI] [PubMed] [Google Scholar]
- 2.Brot N, Weissbach H. Biofactors. 1991;3:91–96. [PubMed] [Google Scholar]
- 3.Stadtman E R. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 4.Moskovitz J, Berlett B S, Poston J M, Stadtman E R. Methods Enzymol. 1999;300:239–244. doi: 10.1016/s0076-6879(99)00130-5. [DOI] [PubMed] [Google Scholar]
- 5.Wizemann T M, Moskovitz J, Pearce B J, Cundell D, Arvidson C G, So M, Weissbach H, Brot N, Masure H R. Proc Natl Acad Sci USA. 1996;93:7985–7990. doi: 10.1073/pnas.93.15.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciorba M A, Heinemann S H, Weissbach H, Brot N, Hoshi T. Proc Natl Acad Sci USA. 1997;94:9932–9937. doi: 10.1073/pnas.94.18.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hassouni M E, Chambost J P, Expert D, Van Gijsegem F, Barras F. Proc Natl Acad Sci USA. 1999;96:887–892. doi: 10.1073/pnas.96.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun H, Gao J, Ferrington D A, Biesiada H, Williams T D, Squier T C. Biochemistry. 1999;38:105–112. doi: 10.1021/bi981295k. [DOI] [PubMed] [Google Scholar]
- 9.Kuschel L, Hansel A, Schonherr R, Weissbach H, Brot N, Hoshi T, Heinemann S H. FEBS Lett. 1999;456:17–21. doi: 10.1016/s0014-5793(99)00917-5. [DOI] [PubMed] [Google Scholar]
- 10.Levine R L, Mosoni L, Berlett B S, Stadtman E R. Proc Natl Acad Sci USA. 1996;93:15036–15040. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabbita S P, Aksenov M Y, Lovell M A, Markesbery W R. J Neurochem. 1999;73:1660–1666. doi: 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- 12.Brot N, Weissbach L, Werth J, Weissbach H. Proc Natl Acad Sci USA. 1981;78:2155–2158. doi: 10.1073/pnas.78.4.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moskovitz J, Jenkins N A, Gilbert D J, Copeland N G, Jursky F, Weissbach H, Brot N. Proc Natl Acad Sci USA. 1996;93:3205–3208. doi: 10.1073/pnas.93.8.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moskovitz J, Rahman M A, Strassman J, Yancey S O, Kushner S R, Brot N, Weissbach H. J Bacteriol. 1995;177:502–507. doi: 10.1128/jb.177.3.502-507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moskovitz J, Berlett B S, Poston J M, Stadtman E R. Proc Natl Acad Sci USA. 1997;94:9585–9589. doi: 10.1073/pnas.94.18.9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moskovitz J, Flescher E, Berlett B S, Azare J, Poston J M, Stadtman E R. Proc Natl Acad Sci USA. 1998;95:14071–14075. doi: 10.1073/pnas.95.24.14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moskovitz J, Weissbach H, Brot N. Proc Natl Acad Sci USA. 1996;93:2095–2099. doi: 10.1073/pnas.93.5.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharov V S, Ferrington D A, Squier T C, Schöneich C. FEBS Lett. 1999;455:247–250. doi: 10.1016/s0014-5793(99)00888-1. [DOI] [PubMed] [Google Scholar]
- 19.Rosenberg A H, Goldman E, Dunn J J, Studier F W, Zubay G. J Bacteriol. 1993;175:716–722. doi: 10.1128/jb.175.3.716-722.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brot N, Werth J, Koster D, Weissbach H. Anal Biochem. 1982;122:291–294. doi: 10.1016/0003-2697(82)90283-4. [DOI] [PubMed] [Google Scholar]
- 21.Smith D J, Maggio E T, Kenyon G L. Biochemistry. 1975;14:766–771. doi: 10.1021/bi00675a019. [DOI] [PubMed] [Google Scholar]
- 22.Taha M K, So M, Seifert H S, Billyard E, Marchal C. EMBO J. 1988;7:4367–4378. doi: 10.1002/j.1460-2075.1988.tb03335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Travis J, Salvesen G S. Annu Rev Biochem. 1983;52:655–709. doi: 10.1146/annurev.bi.52.070183.003255. [DOI] [PubMed] [Google Scholar]
- 24.Cochrane C G, Spragg R, Revak S D. J Clin Invest. 1983;71:754–761. doi: 10.1172/JCI110823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong P S, Travis J. Biochem Biophys Res Commun. 1980;96:1449–1454. doi: 10.1016/0006-291x(80)90113-8. [DOI] [PubMed] [Google Scholar]
- 26.Yiannios C N, Karabinos J V. J Org Chem. 1963;28:3246–3248. [Google Scholar]
- 27.Wallace T J. J Am Chem Soc. 1964;86:2018–2021. [Google Scholar]
- 28.Wallace T J, Mahon J J. J Am Chem Soc. 1964;86:4099–4103. [Google Scholar]
- 29.Wallace T J, Mahon J J. J Org Chem. 1965;30:1502–1506. [Google Scholar]
- 30.Owsley D C, Helmkamp G K, Rettig M F. J Am Chem Soc. 1969;91:5239–5242. [Google Scholar]
- 31.Johnson C R, Rigau J J. J Am Chem Soc. 1969;91:5398–5399. [Google Scholar]
- 32.Kapovits I, Kalman A. Chem. Commun. 1971. 649–650. [Google Scholar]
- 33.Martin J C, Arhart R J. J Am Chem Soc. 1971;93:2339–2341. [Google Scholar]
- 34.Martin J C, Arhart R J. J Am Chem Soc. 1971;93:2341–2342. [Google Scholar]
- 35.Paul I C, Martin J C, Perozzi E F. J Am Chem Soc. 1972;94:5010–5017. [Google Scholar]
- 36.Shaked Z, Szajewski R P, Whitesides G M. Biochemistry. 1980;19:4156–4166. doi: 10.1021/bi00559a004. [DOI] [PubMed] [Google Scholar]
- 37.Szajewski R P, Whitesides G M. J Am Chem Soc. 1980;102:2011–2026. [Google Scholar]
- 38.Houk J, Whitesides G M. J Am Chem Soc. 1987;109:6825–6836. [Google Scholar]
- 39.Burns J A, Whitesides G M. J Am Chem Soc. 1990;112:6296–6303. [Google Scholar]
- 40.Jeng M F, Campbell A P, Begley T, Holmgren A, Case D A, Wright P E, Dyson H J. Structure. 1994;2:853–868. doi: 10.1016/s0969-2126(94)00086-7. [DOI] [PubMed] [Google Scholar]
- 41.Qin J, Clore G M, Gronenborn A M. Structure. 1994;2:503–522. doi: 10.1016/s0969-2126(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 42.Modena G. Int J Sulfur Chem Part C. 1972;7:95–102. [Google Scholar]
- 43.Torchinsky Y M. Sulfur in Proteins. New York: Pergamon; 1981. pp. 133–139. [Google Scholar]
- 44.Mao S S, Yu G X, Chalfoun D, Stubbe J. Biochemistry. 1992;31:9752–9759. doi: 10.1021/bi00155a031. [DOI] [PubMed] [Google Scholar]
- 45.Mao S S, Holler T P, Bollinger J M, Jr, Yu G X, Johnston M I, Stubbe J. Biochemistry. 1992;31:9744–9751. doi: 10.1021/bi00155a030. [DOI] [PubMed] [Google Scholar]
- 46.Mao S S, Holler T P, Yu G X, Bollinger J M, Jr, Booker S, Johnston M I, Stubbe J. Biochemistry. 1992;31:9733–9743. doi: 10.1021/bi00155a029. [DOI] [PubMed] [Google Scholar]
- 47.Stubbe J, van der Donk W A. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 48.Schindelin H, Kisker C, Hilton J, Rajagopalan K V, Rees D C. Science. 1996;272:1615–1621. doi: 10.1126/science.272.5268.1615. [DOI] [PubMed] [Google Scholar]
- 49.McAlpine A S, McEwan A G, Bailey S. J Mol Biol. 1998;275:613–623. doi: 10.1006/jmbi.1997.1513. [DOI] [PubMed] [Google Scholar]
- 50.Pollock V V, Barber M J. J Biol Chem. 1997;272:3355–3362. doi: 10.1074/jbc.272.6.3355. [DOI] [PubMed] [Google Scholar]
- 51.Ellis H R, Poole L B. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 52.Ellis H R, Poole L B. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 53.Claiborne A, Yeh J I, Mallett T C, Luba J, Crane E J, 3rd, Charrier V, Parsonage D. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 54.Moskovitz J, Poston J M, Berelett B S, Nosworthy N J, Szczepanowski R, Stadtman E R. J Biol Chem. 2000;275:14167–14172. doi: 10.1074/jbc.275.19.14167. [DOI] [PubMed] [Google Scholar]