

Abstract
Background
In plants the hormone cytokinin is perceived by members of a small cytokinin receptor family, which are hybrid sensor histidine kinases. While the immediate downstream signaling pathway is well characterized, the domain of the receptor responsible for ligand binding and which residues are involved in this process has not been determined experimentally.
Results
Using a live cell hormone-binding assay, we show that cytokinin is bound by a receptor domain predicted to be extracellular, the so called CHASE (cyclases, histidine kinase associated sensory extracellular) domain. The CHASE domain occurs not only in plant cytokinin receptors but also in numerous orphan receptors in lower eukaryotes and bacteria. Taking advantage of this fact, we used an evolutionary proteomics approach to identify amino acids important for cytokinin binding by looking for residues conserved in cytokinin receptors, but not in other receptors. By comparing differences in evolutionary rates, we predicted five amino acids within the plant CHASE domains to be crucial for cytokinin binding. Mutagenesis of the predicted sites and subsequent binding assays confirmed the relevance of four of the selected amino acids, showing the biological significance of site-specific evolutionary rate differences.
Conclusion
This work demonstrates the use of a bioinformatic analysis to mine the huge set of genomic data from different taxa in order to generate a testable hypothesis. We verified the hypothesis experimentally and identified four amino acids which are to a different degree required for ligand-binding of a plant hormone receptor.
Background
The plant hormone cytokinin is required for many fundamental processes and developmental programmes such as cell division, shoot branching, root development and senescence [1]. For the model plant Arabidopsis thaliana it has been shown that the cytokinin signal is perceived by members of the cytokinin receptor family, which are sensor histidine kinases [2-4]. Mutational analysis of the three cytokinin receptors (AHK2, AHK3, CRE1/AHK4) revealed that they act redundantly, but are absolutely required for normal cytokinin perception and plant growth [5-8]. In the current model, it is predicted that the hormone binds to the Arabidopsis histidine kinase receptors (AHKs) via an extracellular ligand binding domain, the so-called CHASE (cyclases/histidine kinases associated sensory extracellular) domain [9,10]. The CHASE domain, about 250 amino acids long, is exclusively found between two transmembrane regions as the N-terminal part of adenylyl cyclases, diguanylate cyclases or histidine kinases in a number of eukaryotes and numerous bacteria. It includes, for example, the spore differentiation factor, DhkA, and the osmosensing receptor-adenylyl cyclase ACG, which regulates spore dormancy, from the slime mold Dictyostelium discoideum. DhkA recognizes a small peptide, SDF-2 [11], and it was proposed that ACG binds discadenine [12]. Thus the CHASE domain is believed to bind diverse low molecular weight ligands. However, the ligand and its cognate receptor are only known in a few cases. Among higher eukaryotes the domain is found only in plants as part of specific sensor histidine kinases, the cytokinin receptors. It was proposed that plants acquired the CHASE domain through their chloroplasts, which have a cyanobacterial ancestry [9,10].
The binding of cytokinin to the receptor is thought to cause a conformational change leading to the autophosphorylation of a conserved histidine residue in the cytosolic part of the receptor. Subsequently, the signal is transferred to a canonical aspartate within the C-terminal part of the protein and transduced further by a multi-step two-component signaling system (for recent reviews see [13-15]).
The cytokinin binding activity of full-length CRE1/AHK4 was shown before by several different types of assays [3,4,16,17]. Although the CHASE domain is suspected of being the ligand binding domain, no systematic approach has been made with any cytokinin receptor to identify the binding domain unequivocally. Once a binding domain has been determined, the next step of the characterization is to identify functional amino acid residues. In this report we describe a novel knowledge-based approach that uses sequence information from distantly related organisms to predict putative functionally relevant sites in the ligand binding domain. The bioinformatics method was based on detecting differences in the evolution of individual amino acid sites between the CHASE domains of the different protein subclasses. The underlying premise was that a slower evolutionary rate of a given amino acid position, e.g. the conservation of a different amino acid in plants versus other organisms, would identify important positions for receptor function. These positions are putatively important in binding the plant-specific ligand, which is thought to be different from the other subgroups.
Thus the aims of this study were twofold: (i) mapping of the ligand binding domain of CRE1/AHK4 and (ii) identifying amino acids crucial for the binding of cytokinin to the receptor. Using a binding assay we provide direct experimental evidence that cytokinin is bound via the CHASE domain of CRE1/AHK4. The substitution of four of the five amino acids, which were predicted by evolutionary analysis to be important for a functional ligand-binding domain, caused a clear change in the ligand binding, in this case a complete loss of, or strongly reduced, cytokinin binding. This demonstrates the power of combining bioinformatic predictions with experimental validation, which have been proven to be a very useful tool in other subjects in the past [18,19]. Thus the data further underpin the general potential of evolutionary proteomics to identify functionally relevant sites in proteins of known or even of unknown function.
Results
CRE1/AHK4 binds cytokinin via the CHASE domain
The cytokinin binding domain of CRE1/AHK4 was mapped by expressing the cytoplasmic part or the CHASE domain with the adjacent transmembrane domains as GST-fusion proteins in E. coli (Fig. 1A). The cytokinin binding capacity was tested in an in vivo binding assay [17] and compared to the binding capacity of the full-length protein in the same experimental setup. The full-length protein showed the highest relative trans-zeatin binding (Fig. 2). The binding capacity was slightly lower in the truncated version of CRE1/AHK4 consisting of the CHASE domain and the flanking transmembrane domains. The cytoplasmic domain and the empty vector control showed very weak or no binding (Fig. 2). The results of the binding assay indicate that the CHASE domain is the cytokinin binding domain of CRE1/AHK4.
Figure 1.
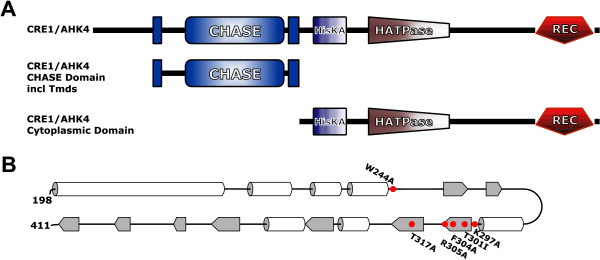
Domain structure of CRE1/AHK4 and secondary structure prediction of the CHASE domain. (A) The domain structure of the full-length protein and different truncated versions of CRE1/AHK4 used in this study. (B) The predicted secondary structure of the CHASE domain with the amino acid substitutions tested in the cytokinin binding assay marked in red. White tubes represent α-helices and grey areas β-sheets. Secondary structure prediction was done by PSIPred v2.4 [43]. Abbreviations: CHASE, cyclases, histidine kinase associated sensory extracellular; HisKA, histidine kinase A domain; HATPase, histidine kinase-like ATPase; Rec, receiver domain; Tmds, transmembrane domains.
Figure 2.
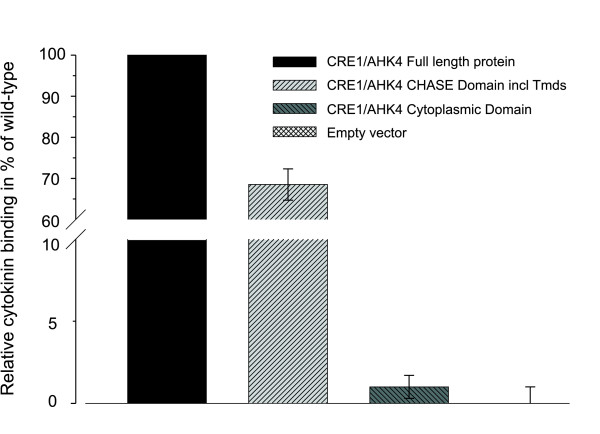
The CHASE domain of CRE1/AHK4 is necessary and sufficient for cytokinin binding. In vitro binding of trans-[2-3H]zeatin to full length CRE1/AHK4 protein or different domains of the receptor overexpressed in E. coli BL21. Bacterial cells were assayed for specific trans-[2-3H]zeatin binding. Data are mean ± s.d.; n = 4.
Evolutionary analysis of CHASE-subclasses reveals putative cytokinin-binding residues
Sequences containing the CHASE domain were identified in plants, slime-molds, cyanobacteria and proteobacteria and a multiple sequence alignment was generated for the region spanning the domain (Figure 3B and Additional file 1; for details see Material and Methods). The phylogenetic tree reveals several distinct subgroups, one of which includes all plant sequences (Figure 3A). Our computational approach to identify functional important positions is based on the assumption that CHASE domains found in plants recognize a different class of ligands (cytokinin) than bacterial domains and that these functional differences can be detected by a change in evolutionary rates of amino acid substitutions. A candidate position should have a slow evolutionary rate in plants and a fast evolutionary rate in other subgroups or a slow evolutionary rate in all subgroups, but then the subgroups should be conserved in a different amino acid to distinguish the plant sequences from bacterial sequences. The slime-mold sequences were not included in this analysis because their subgroup contained only two sequences and was not supported by a high bootstrap value. Among the positions that were derived from the analysis of bacterial and plant sequences and fulfilled the above criteria we selected five positions, which show the strongest evidence for functional divergence and thus, are likely to be important in cytokinin binding: Position T317 of CRE1/AHK4 is not only slowly evolving among plant sequences but also among all investigated bacterial subgroups (evolutionary site rate category 1; Fig. 3B). However, in each subgroup, this site is occupied by a different amino acid with varying biochemical properties. Only in the plant subgroup, this position is occupied by an amino acid with a hydroxyl containing side chain (threonine). The evolutionary rate of CRE1/AHK4 positions W244 and K297 is very low in the plant CHASE sequences compared to that of the CHASE domains of the other subgroups, indicating that these positions might be important in plants. The amino acids at positions 304 and 305 have different biochemical properties and evolutionary rates in the different subgroups (Fig. 3B). Position 304 is occupied by phenylalanine only in the plant sequences while the bacterial subgroups have mostly aliphatic amino acids as a residue in this position. At position 305, the plant subgroup has either a basic amino acid or a proline, while in the other subgroups the class of amino acids is not conserved. It should be noted that position 301, a functional important residue of the CRE1/AHK4 CHASE domain, did not fulfil our criteria, because some bacterial sequences are conserved in the same or a similar amino acid. In fact, an allele mutated in this position, which leads to an amino acid change to isoleucine, is known as wooden leg (wol) and was discovered in a screen for altered root morphology [20]. Subsequent analysis of this only known mutation of the CHASE domain in plants revealed the complete loss of cytokinin binding of the mutant protein [4]. We included this mutation as a positive control in our analysis.
Figure 3.

Phylogenetic tree and alignment of CHASE domains. (A) Phylogenetic tree of CHASE domains from five different subgroups (see Additional file 1 for all subgroups). Sequences of CHASE domains used for tree building are labelled with gi numbers and the start and end position of the respective CHASE domain. (B) Section of CHASE family alignment containing sequences used for the evolutionary rate analysis of the individual amino acids. The evolutionary site rate categories for each subgroup are given below the alignment, ranging from 1 (slow rate of evolution) to 8 (fast rate of evolution). Amino acid positions selected for experimental analysis (W244, K297, F304, R305, T317) are highlighted in orange, positions that are conserved throughout all CHASE sequences in grey. Interruptions of the alignment are indicated by two dots, gaps in the alignment by dashes. (See Additional file 1 for full sequence alignment). The selected blocks relate to the CRE1/AHK4 sequence 238–249, 292–308 and 313–321, respectively.
Specific amino acid substitutions lead to significant changes in cytokinin binding
The selected amino acids were substituted by alanine. Subsequently the ability of the mutated CHASE domains as part of the full length protein was tested for cytokinin binding activity. As controls, the wild-type CRE1/AHK4 protein showed strong cytokinin binding, while the empty vector did not show any cytokinin binding in the assay (Fig. 4A). We also confirmed that the known mutation (T301I) in the CHASE domain causes loss of cytokinin-binding activity [4] (Fig. 4A). In contrast and as expected, the substitution of the canonical histidine of the cytoplasmic histidine kinase domain by glutamine (H482Q), which abolishes the receptor's signaling capacity [21], does not alter the cytokinin binding capacity of the receptor (Fig. 4A).
Figure 4.
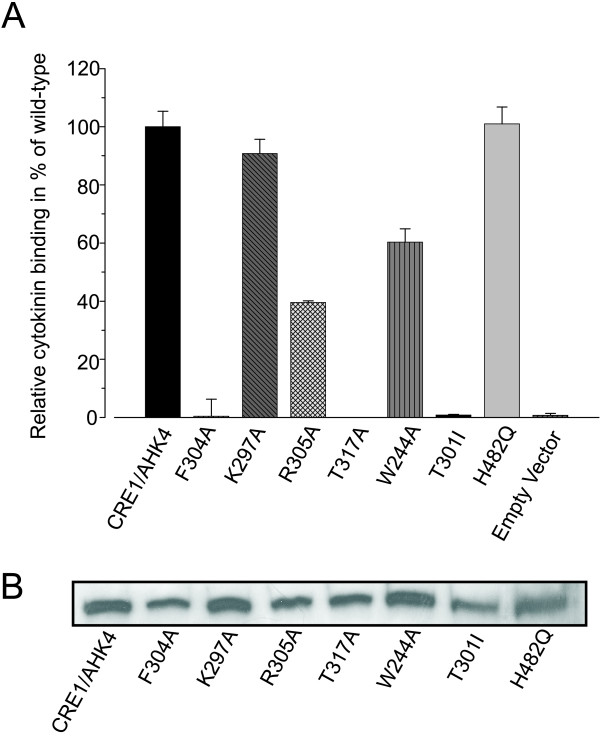
Identification of amino acid residues of the CRE1/AHK4 CHASE domain important for ligand binding. (A) Effect of point mutations in the CRE1/AHK4 CHASE domain on the specific binding of trans-[2-3H]zeatin. For the localization of the mutated sites see Fig. 1B. H482Q is a control carrying a mutated histidine residue of the cytoplasmic domain. Data are means ± S.D. from measurements with two different E. coli clones for each construct. (B) Western blot with whole protein extracts of an aliquot of E. coli cells used for the binding assay shown in (A). For protein detection a mouse-anti-GST antibody (GST B-14, Santa Cruz Biotechnology) was used.
Two of the candidate residues, namely F304A and T317A led to a complete abolishment of ligand binding (Fig. 4A). Two other amino acid substitutions – W244A and R305A – resulted in strongly reduced binding capacity, approximately 60% and 40% of the CRE1/AHK4 wild-type control, respectively (Fig. 4A). In contrast, the substitution of K297A caused only a slight decrease of the cytokinin binding of the mutant protein compared to the wild-type CRE1/AHK4. The protein level of all GST-fusion proteins was checked by Western blot and did not show significant differences (Fig. 4B).
Discussion
CRE1/AHK4 binds cytokinin via the CHASE domain
In this study we investigated how the plant hormone cytokinin is recognized by its receptor. It has been hypothesized that cytokinin is bound by the CHASE domain of the receptors [2-4,9,10], but no experiment has been performed to prove this hypothesis. Testing truncated versions of a protein for activity is a first step to delineate those parts of the protein important for the investigated function [22-24]. The binding assays using the full-length and several truncated versions of CRE1/AHK4 (Fig. 2) confirm the hypothesis that trans-zeatin is bound by CRE1/AHK4 via the CHASE domain. This is relevant as cytokinin occurs also inside the plant cell and it could be, therefore, possible that cytokinin is bound also via the cytoplasmic part of the cytokinin receptors. However, our data show that this possibility is not realized in CRE1/AHK4.
Bioinformatic analysis identifies residues that are crucial for a functional ligand binding domain
Functional important amino acid positions are often predicted by combining evolutionary information of a protein family with 3D structures. The underlying assumption is, if a position adopts a new beneficial function, it will be subject to stronger selective constraint, which will be reflected in the evolutionary rates of amino acid replacement [25]. Several methods have been described in the past that search for spatial clusters of subfamily conserved residues or that search for shifts in the evolutionary rates of protein subfamilies [26-33]. The accuracy of computer predictions has been confirmed in several cases through mutagenesis experiments [18,34,35]. In the lack of a 3D structure of the CHASE domain, we focused on identifying sites that stand out by smaller evolutionary rates in the plant subfamily or, at sites that evolve slowly in all subfamilies, by biochemical different amino acids that are conserved in the different subfamilies.
Experimental evaluation of the roles of the selected amino acids in cytokinin binding underpinned the validity of the approach. Substitutions to alanine of four of the five selected amino acids in CRE1/AHK4 showed a dramatic alteration of the cytokinin binding, in this case a strong reduction to total abolishment of the trans-zeatin binding (Fig. 4A), thus confirming their importance in cytokinin binding.
Interestingly, the three amino acids which show the strongest effect and also the positive control T301 are located in close vicinity in two predicted β-sheets in the center part of the CHASE domain (Fig. 1B). The only exception in this study, position K297, which, while completely conserved among plant sequences in contrast to bacterial sequences, did not show any significant reduction in cytokinin binding (Fig. 3A), is located just outside the first of these two central β-sheets. We hypothesize that these β-sheets are part of a binding pocket for cytokinins. It is understood that only the experimental determination of the structure will provide conclusive evidence. The identification of important amino acids presented here will be an important contribution to understand the functioning of the CHASE domain, once structural data become available.
Future applications
Plant tissues typically contain a mixture of different biologically active cytokinin metabolites. The specificity of ligand recognition was investigated for Arabidopsis and maize cytokinin receptors and it was shown that their relative affinities for different cytokinins varies [4,16,17,36]. Analysis of the mutant receptors generated in this study has shown that the mutations do not affect the recognition of different cytokinins in a distinct way (Romanov et al., unpublished result). Thus these amino acids appear to be of general relevance. As they are conserved among all plant CHASE domains they might present the key residues for binding the hormonal core. Fine-tuning for specific cytokinins could be achieved by additional, less conserved positions. It will be interesting to see whether our bioinformatical design also enables the identification of amino acid residues which are relevant to the detection of different types of cytokinins by receptors of the same species. However, such an analysis will require a greater number of different cytokinin receptors to be studied with respect to their cytokinin binding preference.
Conclusion
Using truncated versions of the cytokinin receptor CRE1/AHK4 in a binding assay, we have experimentally defined the CHASE domain as the ligand binding domain of this class of receptor. Based on our bioinformatical approach, combined with experimental validation, we successfully identified functionally important amino acids in this domain. The experimental confirmation of these residues highlights the significance of evolutionary proteomics in the post genomic era and demonstrates its potential for the characterization of protein functioning. In addition to the analysis of proteins with known functions, the approach can be envisioned as an aid in the enormous task of functionally annotating the vastly increasing amount of sequence information provided by the numerous genome sequencing projects.
Methods
Construction of the different CRE1/AHK4 variants
The truncated variants of CRE1/AHK4 (At2g01830.2) were generated by PCR using the respective primers (Additional file 2) and cloned into the entry vector pDONR221 of the Gateway™ cloning system (Invitrogen, Carlsbad). For the amino acid substitutions the HindIII fragment of CRE1/AHK4 containing the entire CHASE domain was cloned into pBluescript II KS (Stratagene, La Jolla). Site-directed mutagenesis was carried out with the QuikChange® II Site-Directed Mutagenesis Kit (Stratagene, La Jolla) using the respective primers (Additional file 2). The mutated fragments were sequenced and recloned into a CRE1/AHK4 clone in the pDONR221 vector missing the HindIII fragment in the cDNA. Subsequently all clones were shuttled into the pDEST15 vector (Invitrogen, Carlsbad) and transformed into the E. coli strain BL21DE3pLys (Novagen, San Diego).
Cytokinin binding assay and immunoblotting
For in vivo cytokinin binding the assays were performed as published [17]. Pellets of 1 ml of the respective E. coli culture (used in the binding assay) were resuspended in 50 μL reducing 1x Laemmli buffer [37] and heated to 95°C for 5 min before separating 25 μL of the protein extract on a 10% SDS-PAGE (Bio-Rad Laboratories, München, Germany). Proteins were transferred to PVDF membrane (Bio-Rad) using a tank transfer system (Bio-Rad) with Towbin buffer without methanol [38]. After blocking for 2 h at room temperature with Tris buffered saline (TBS) including 5% low fat dried milk powder, the membrane was incubated with anti GST primary antibody (B-14) (1:500 in blocking buffer; Santa Cruz Biotechnology, Heidelberg, Germany) 3 h at room temperature. Horseradish peroxidase-conjugated goat anti mouse secondary antibody (1:4000 in blocking buffer; Santa Cruz Biotechnology, Heidelberg, Germany) and enhanced chemiluminescence reagent (Pierce SuperSignal West Pico; Perbio Science, Bonn, Germany) were used for detection. Blots were exposed to Pierce CL-Xposure films (Perbio Science).
Sequence analysis
Sequences containing the CHASE domain were retrieved from Genbank's non-redundant database using a Hidden Markov Model, which was built from a representative multiple sequence alignment [10] obtained from the Pfam database (HMMER package)[39]. To identify additional plant sequences, Genbank's EST database was searched with the CHASE domain of CRE1/AHK4. EST sequences were translated into proteins. Protein regions containing the CHASE domain were aligned with hmmalign (HMMER). The alignment (Additional file 1) was manually optimized to minimise gaps in loop regions and used to calculate a phylogenetic tree with CLUSTAL W [40]. Five stable subtrees with more than 90% bootstrap support were chosen for the analysis of evolutionary rates, of which one represents all plant sequences and four subtrees represent bacterial sequences. In order to identify functionally diverged amino acid sites, we estimated evolutionary rates for each position in the five alignments and compared the rates between the plant and bacterial groups. This allows the identification of positions that are under selective pressure in the plant group, while they are free of any constraint in bacterial groups. In contrast to more simple amino acid conservation scores, evolutionary rates reflect the amino acid changes considering the phylogenetic distance of the sequences. Evolutionary site rates were estimated with the maximum likelihood method implemented in the TREE-PUZZLE program v5.1 [41,42]. For the estimation of the evolutionary site rates with TREE-PUZZLE, we have chosen a heterogeneity rate model that was compared with the null model, which assumes a uniform rate among sites. The two models were compared in a log likelihood ratio test and in four out of five cases the null model was rejected (p-value < 0.01). We used the Jones Taylor amino acid substitution model and assumed an eight category discretized gamma model for the variation of substitution rates among sites. We searched for sites that are conserved in the plant group but fast evolving in bacterial groups, or that are slow evolving in all groups, but conserved in different amino acids.
Authors' contributions
AH, KW, NN and GAR carried out the molecular studies. BP did the bioinformatic analysis. AH, BP and TS conceived the study and wrote the manuscript. KW and GAR contributed to draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Full sequence alignment of the CHASE domain. The sequences are labelled with gi numbers and the start and end position of the subset of the sequences used for the alignment is given. In case of EST data, the complete translated sequences were used. Evolutionary site rate categories for the 5 subclasses depicted in Fig. 3 are given below the alignment.
Primers used for cloning in the different amino acid substitutions and truncation experiments.
Acknowledgments
Acknowledgements
This work was supported by a grant of the DFG in the frame of Sfb 449 (Structure and function of membrane receptors) and RFBR grant N 07-04-00331.
Contributor Information
Alexander Heyl, Email: heyl@zedat.fu-berlin.de.
Klaas Wulfetange, Email: kwulfe@zedat.fu-berlin.de.
Birgit Pils, Email: pils@well.ox.ac.uk.
Nicola Nielsen, Email: n.nielsen@gmx.de.
Georgy A Romanov, Email: gar@ippras.ru.
Thomas Schmülling, Email: tschmue@zedat.fu-berlin.de.
References
- Mok DWS, Mok MC. Cytokinin metabolism and action. Annu Rev Plant Physiol Mol Biol. 2001;52:89–118. doi: 10.1146/annurev.arplant.52.1.89. [DOI] [PubMed] [Google Scholar]
- Inoue T, Higuchi M, Hashimoto Y, Seki M, Kobayashi M, Kato T, Tabata S, Shinozaki K, Kakimoto T. Identification of CRE1 as a cytokinin receptor from Arabidopsis. Nature. 2001;409:1060–1063. doi: 10.1038/35059117. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Miwa K, Ishikawa K, Yamada H, Aiba H, Mizuno T. The Arabidopsis sensor His-kinase, AHK4, can respond to cytokinins. Plant Cell Physiol. 2001;42:107–113. doi: 10.1093/pcp/pce037. [DOI] [PubMed] [Google Scholar]
- Yamada H, Suzuki T, Terada K, Takei K, Ishikawa K, Miwa K, Yamashino T, Mizuno T. The Arabidopsis AHK4 histidine kinase is a cytokinin-binding receptor that transduces cytokinin signals across the membrane. Plant Cell Physiol. 2001;42:1017–1023. doi: 10.1093/pcp/pce127. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Pischke MS, Mähönen AP, Miyawaki K, Hashimoto Y, Seki M, Kobayashi M, Shinozaki K, Kato T, Tabata S, et al. In planta functions of the Arabidopsis cytokinin receptor family. Proc Natl Acad Sci USA. 2004;101:8821–8826. doi: 10.1073/pnas.0402887101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Ryu H, Hong SH, Woo HR, Lim PO, Lee IC, Sheen J, Nam HG, Hwang I. Cytokinin-mediated control of leaf longevity by AHK3 through phosphorylation of ARR2 in Arabidopsis. Proc Natl Acad Sci USA. 2006;103:814–819. doi: 10.1073/pnas.0505150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura C, Ohashi Y, Sato S, Kato T, Tabata S, Ueguchi C. Histidine kinase homologs that act as cytokinin receptors possess overlapping functions in the regulation of shoot and root growth in Arabidopsis. Plant Cell. 2004;16:1365–1377. doi: 10.1105/tpc.021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riefler M, Novak O, Strnad M, Schmülling T. Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development and cytokinin metabolism. Plant Cell. 2006;18:40–54. doi: 10.1105/tpc.105.037796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougel C, Zhulin IB. CHASE: an extracellular sensing domain common to transmembrane receptors from prokaryotes, lower eukaryotes and plants. Trends Biochem Sci. 2001;26:582–584. doi: 10.1016/S0968-0004(01)01969-7. [DOI] [PubMed] [Google Scholar]
- Anantharaman V, Aravind L. The CHASE domain: a predicted ligand-binding module in plant cytokinin receptors and other eukaryotic and bacterial receptors. Trends Biochem Sci. 2001;26:579–582. doi: 10.1016/S0968-0004(01)01968-5. [DOI] [PubMed] [Google Scholar]
- Wang N, Soderbom F, Anjard C, Shaulsky G, Loomis WF. SDF-2 induction of terminal differentiation in Dictyostelium discoideum is mediated by the membrane-spanning sensor kinase DhkA. Mol Cell Biol. 1999;19:4750–4756. doi: 10.1128/mcb.19.7.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter DA, Dunbar AJ, Buconjic SD, Wheldrake JF. Ammonium phosphate in sori of Dictyostelium discoideum promotes spore dormancy through stimulation of the osmosensor ACG. Microbiology. 1999;145:1891–1901. doi: 10.1099/13500872-145-8-1891. [DOI] [PubMed] [Google Scholar]
- Ferreira FJ, Kieber JJ. Cytokinin signaling. Curr Opin Plant Biol. 2005;8:518–525. doi: 10.1016/j.pbi.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Heyl A, Werner T, Schmülling T. Cytokinin metabolism and signal transduction. In: Hedden P, Thomas S, editor. Plant Hormone signaling. Oxford: Blackwell Publishing; 2006. pp. 93–123. [Google Scholar]
- Mizuno T. Two-component phosphorelay signal transduction systems in plants: from hormone responses to circadian rhythms. Biosci Biotechnol Biochem. 2005;69:2263–2276. doi: 10.1271/bbb.69.2263. [DOI] [PubMed] [Google Scholar]
- Spichal L, Rakova NY, Riefler M, Mizuno T, Romanov GA, Strnad M, Schmülling T. Two cytokinin receptors of Arabidopsis thaliana, CRE1/AHK4 and AHK3, differ in their ligand specificity in a bacterial assay. Plant Cell Physiol. 2004;45:1299–1305. doi: 10.1093/pcp/pch132. [DOI] [PubMed] [Google Scholar]
- Romanov GA, Spichal L, Lomin SN, Strnad M, Schmülling T. A live cell hormone-binding assay on transgenic bacteria expressing a eukaryotic receptor protein. Anal Biochem. 2005;347:129–134. doi: 10.1016/j.ab.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Sowa ME, He W, Slep KC, Kercher MA, Lichtarge O, Wensel TG. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat Struct Biol. 2001;8:234–237. doi: 10.1038/84974. [DOI] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal microRNAs confer robustness to gene expression and have a significant impact on 3'UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Mähönen AP, Bonke M, Kauppinen L, Riikonen M, Benfey PN, Helariutta Y. A novel two-component hybrid molecule regulates vascular morphogenesis of the Arabidopsis root. Genes Dev. 2000;14:2938–2943. doi: 10.1101/gad.189200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang I, Sheen J. Two-component circuitry in Arabidopsis cytokinin signal transduction. Nature. 2001;413:383–389. doi: 10.1038/35096500. [DOI] [PubMed] [Google Scholar]
- Kinoshita T, Cano-Delgado A, Seto H, Hiranuma S, Fujioka S, Yoshida S, Chory J. Binding of brassinosteroids to the extracellular domain of plant receptor kinase BRI1. Nature. 2005;433:167–171. doi: 10.1038/nature03227. [DOI] [PubMed] [Google Scholar]
- Charpenteau M, Jaworski K, Ramirez BC, Tretyn A, Ranjeva R, Ranty B. A receptor-like kinase from Arabidopsis thaliana is a calmodulin-binding protein. Biochem J. 2004;379:841–848. doi: 10.1042/BJ20031045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Schaller GE. Requirement of the histidine kinase domain for signal transduction by the ethylene receptor ETR1. Plant Physiol. 2004;136:2961–2970. doi: 10.1104/pp.104.047126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge; New York: Cambridge University Press; 1983. [Google Scholar]
- Gu X, Vander Velden K. DIVERGE: phylogeny-based analysis for functional-structural divergence of a protein family. Bioinformatics. 2002;18:500–501. doi: 10.1093/bioinformatics/18.3.500. [DOI] [PubMed] [Google Scholar]
- Landgraf R, Fischer D, Eisenberg D. Analysis of heregulin symmetry by weighted evolutionary tracing. Protein Eng. 1999;12:943–951. doi: 10.1093/protein/12.11.943. [DOI] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- Armon A, Graur D, Ben-Tal N. ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J Mol Biol. 2001;307:447–463. doi: 10.1006/jmbi.2000.4474. [DOI] [PubMed] [Google Scholar]
- Casari G, Sander C, Valencia A. A method to predict functional residues in proteins. Nat Struct Biol. 1995;2:171–178. doi: 10.1038/nsb0295-171. [DOI] [PubMed] [Google Scholar]
- del Sol Mesa A, Pazos F, Valencia A. Automatic methods for predicting functionally important residues. J Mol Biol. 2003;326:1289–1302. doi: 10.1016/S0022-2836(02)01451-1. [DOI] [PubMed] [Google Scholar]
- Madabushi S, Yao H, Marsh M, Kristensen DM, Philippi A, Sowa ME, Lichtarge O. Structural clusters of evolutionary trace residues are statistically significant and common in proteins. J Mol Biol. 2002;316:139–154. doi: 10.1006/jmbi.2001.5327. [DOI] [PubMed] [Google Scholar]
- Gaucher EA, Gu X, Miyamoto MM, Benner SA. Predicting functional divergence in protein evolution by site-specific rate shifts. Trends Biochem Sci. 2002;27:315–321. doi: 10.1016/S0968-0004(02)02094-7. [DOI] [PubMed] [Google Scholar]
- Onrust R, Herzmark P, Chi P, Garcia PD, Lichtarge O, Kingsley C, Bourne HR. Receptor and betagamma binding sites in the alpha subunit of the retinal G protein transducin. Science. 1997;275:381–384. doi: 10.1126/science.275.5298.381. [DOI] [PubMed] [Google Scholar]
- Budovskaya YV, Stephan JS, Deminoff SJ, Herman PK. An evolutionary proteomics approach identifies substrates of the cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 2005;102:13933–13938. doi: 10.1073/pnas.0501046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Kojima M, Yamaya T, Sakakibara H. Molecular characterization of cytokinin-responsive histidine kinases in maize. Differential ligand preferences and response to cis-zeatin. Plant Physiol. 2004;134:1654–1661. doi: 10.1104/pp.103.037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://hmmer.wustl.edu
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 1994;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- Strimmer K, von Haeseler A. Quartet puzzling: A quartet maximum likelihood method for reconstructing tree topologies. Mol Biol Evol. 1996;13:964–969. [Google Scholar]
- Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005;33:W36–38. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full sequence alignment of the CHASE domain. The sequences are labelled with gi numbers and the start and end position of the subset of the sequences used for the alignment is given. In case of EST data, the complete translated sequences were used. Evolutionary site rate categories for the 5 subclasses depicted in Fig. 3 are given below the alignment.
Primers used for cloning in the different amino acid substitutions and truncation experiments.