

Abstract
Poly-N-acetylglucosamine (PNAG) is a surface polysaccharide produced by Staphylococcus aureus and Staphyloccus epidermidis and is an effective target for opsonic and protective Ab for these two organisms. Recently, it has been found that Escherichia coli produces an exo-polysaccharide, designated polyglucosamine, that is biochemically indistinguishable from PNAG. We analyzed 30 E. coli strains isolated from urinary tract and neonatal bloodstream infections for the pga locus, PNAG antigen production, and susceptibility to opsonic killing and protection from lethal infection by Ab to PNAG. Twenty-six of 30 strains carried the pga locus, 25 of 30 expressed immunologically detectable PNAG, and 21 of 30 could be killed by rabbit IgG specific for the deacetylated form of the staphylococcal PNAG. Ab to staphylococcal PNAG protected mice against lethality from five different E. coli strains expressing PNAG. PNAG expression by both Gram-negative and Gram-positive organisms could make this antigen a conserved vaccine target for multiple pathogenic species of bacteria.
Keywords: vaccine, polyglucosamine, pga locus, ica locus, biofilms
Staphylococcus aureus and coagulase negative staphylococci are the most frequent causes of nosocomial bloodstream infections (1). One virulence factor in many such infections is the ability to form a biofilm on synthetic materials (2–4). Most biofilm-forming strains of S. aureus secrete a large exo-polysaccharide, poly-N-acetylglucosamine (PNAG) that is involved in intercellular adhesion and is sometimes referred to as polysaccharide intercellular adhesin or PIA (5–7). PNAG also protects planktonic cells of S. aureus and Staphylococcus epidermidis from Ab-independent phagocytic killing by the host immune system (8–10). PNAG is synthesized by proteins encoded by the icaADBC genes of the intercellular adhesin locus (ica) (6, 11, 12). PNAG is also a promising vaccine candidate for staphylococcal infections (5, 13–17).
Wang et al. (18) recently found that Escherichia coli has a genetic locus homologous to the staphylococcal ica locus, which they termed pgaABCD, although it was originally designated as the ycdSRQP locus in the annotated E. coli K12 genome. pgaABCD encodes proteins able to synthesize an exo-polysaccharide that they designated polyglucosamine (PGA), which they found to be chemically nearly identical to PNAG (18). They also reported that homologues of the Gram-negative bacterial pga locus have been identified in a number of other important pathogens, including Yersinia pestis, Yersinia pseudotuberculosis, Yersinia enterocolitica, Bordetella pertussis, Bordetella parapertussis, and Bordetella bronchiseptica (18), but they did not demonstrate PNAG production by these species. However, Kaplan et al. (19) showed that Actinobacillus actinomycetemcomitans and Actinobacillus pleuropneumoniae have a pga-homologous locus and synthesize PNAG, as do Bordetella spp. (20). Thus, there is increasing evidence that PNAG can be synthesized as a surface polysaccharide by a variety of pathogenic bacteria.
In this work, we analyzed whether 30 clinical isolates of E. coli from urinary tract and neonatal blood stream infections contained the pga locus, expressed an antigen reactive with antibodies raised to staphylococcal PNAG, could be killed in an opsonophagocytosis assay with the antibodies to the staphylococcal PNAG and whether mice could be protected by passively administering Ab to PNAG when challenged with lethal doses of E. coli by i.p. injection.
Results
Presence of pga Locus and Production of PNAG by E. coli Clinical Strains.
Table 1 summarizes the characteristics of the 30 E. coli clinical isolates, comprising 18 urinary tract infection (UTI) isolates and 12 isolates from neonatal bacteremia for the presence of the pgaA and pgaC loci, which are not always present in the same operon in Gram-negative bacteria (18). We detected both of these pga-associated genes in 26 of the 30 clinical E. coli isolates. None had only one of the genes.
Table 1.
Molecular and immunological characterization of all E. coli clinical isolate strains (n = 30)
| Group | No. of strains | DNA* |
PNAG blot† |
Opsonophagocytic killing‡ |
|||||
|---|---|---|---|---|---|---|---|---|---|
| − | + | − | + | ++ | <10% | 11–30% | >31% | ||
| UTI | 18 | 2§ | 16 | 3§ | 6 | 9 | 4 (13%) | 5 (17%) | 9 (30%) |
| NICU | 12 | 2§ | 10 | 2§ | 2 | 8 | 5 (17%) | 0 (0%) | 7 (23%) |
*No. of strains without (−) or with (+) pga locus, determined by PCR amplification of pgaA and pgaC genes.
†No. of strains producing PNAG detected by immunoblot: −, no signal detected; +, weak signal detected; ++, strong signal detected.
‡No. of strains falling into each category; the percentage of all strains (UTI plus NICU groups) is shown in parentheses.
§All four strains negative for the pga locus were also negative for PNAG by immunoblotting.
Fig. 1 Left shows the specificity of the PNAG immunoblot reagents when tested against S. aureus and S. epidermidis strains known to produce PNAG or deleted for the staphylococcal ica genes (Δica), as well as the sequenced E. coli UTI strain CFT073, known to have a pga locus (18, 21). We also found that the clinical E. coli UTI isolate UTI-J expressed detectable PNAG, whereas UTI-JΔpga did not; the UTI-JΔpga strain complemented in trans with pPGA reacquired the ability to produce PNAG. Identical results were obtained with the E. coli BW25113 parental, Δpga, and transcomplemented strains (data not shown).
Fig. 1.

PNAG expression in control strains and E. coli clinical isolates. Cell surface extracts were probed with affinity-purified goat Ab to PNAG. (Left) Control strains. Lanes: A1, S. aureus MN8; A2, S. aureus MN8Δica; A3, S. epidermidis M187; B1, E. coli CTF073; B2, E. coli UTI-J; B3, E. coli UTI-JΔpga; B4, E. coli UTI-JΔpga+pPGA. (Center) Clinical UTI isolates from different patients collected in the U.S. Lanes: C1, UTI-D; C2, UTI-E; C3, UTI-F; C4, UTI-G; D1, UTI-H; D2, UTI-I; D3, UTI-J; D4, UTI-K; E1, UTI-L; E2, UTI-MI; E3, UTI-N; E4, UTI-O; F1, UTI-P; F2, UTI-Q; F3, UTI-R; F4, UTI-S; G1, UTI-T; G2, UTI-U. (Right) Neonatal isolates from different patients in Europe and the U.S. Lanes: H1, NICU-1; I1, NICU-2; J1, NICU-3; K1, NICU-4; L1, NICU-5; H2, NICU-6; I2, NICU-7; J2, NICU-8; K2, NICU-9; L2, NICU-10; H3, NICU-11; I3, NICU-12. Extracts from isolates in the first and last rows (rows 1 and 3) are from E. coli strains also positive for the K1 capsular antigen.
When EDTA extracts prepared from 18 E. coli UTI isolates and 12 E. coli isolates from neonatal bacteremia were probed for expression of PNAG, 25 of 30 (83%) produced detectable levels of PNAG (Fig. 1 Center and Right). All 7 of the E. coli isolates from neonates known to produce the K1 capsular polysaccharide also produced PNAG (Fig. 1 Right, lanes H–K, rows 1 and 3). A summary of the semiquantitative detection of PNAG production determined by immunoblot is given in Table 1. The 5 strains producing no detectable PNAG included the 4 strains negative for the pga locus. Lack of detectable PNAG in the other strain could be due to low-level synthesis of PNAG or mutations in the pga locus preventing production of functional biosynthetic genes.
Opsonic Activity of Ab Raised to S. aureus PNAG Glycoforms Against Clinical Isolates of E. coli.
Fig. 2 presents the opsonic killing activity against 4 different E. coli clinical isolates using dilutions of rabbit polyclonal antibodies raised to either a poorly acetylated glycoform of PNAG designated dPNAG (deacetylated PNAG) or raised to highly acetylated (>95% substitution) native PNAG. Prior results with Staphylococcus showed that antibodies raised to dPNAG had superior opsonic killing activity and protective efficacy in animals (14). Although many E. coli strains can be killed in a bactericidal assay using only Ab to capsular polysaccharides and complement, we found that after absorption of the complement source with E. coli cells to remove any antibody from this reagent, the Ab to PNAG could not mediate bacterial killing in the absence of phagocytes. For all 4 of the strains at essentially all of the serum dilutions tested, dPNAG Ab mediated superior opsonic killing compared with Ab raised to native PNAG. Thus, as we found for S. aureus (14), Ab raised to the dPNAG antigen mediated better killing than Ab raised to then native PNAG antigen. Superior opsonic killing activity correlates with a better ability to deposit opsonically active fragments of the third component of complement on the bacterial surface (22).
Fig. 2.

Opsonophagocytic killing activity of goat Ab raised to either dPNAG (black diamonds) or PNAG (gray squares) against E. coli strains UTI-J, UTI-P, UTI-U, and NICU10. Points represent means of triplicates; SE bars are <5% and are contained within each point.
When we evaluated the opsonic killing of the 30 E. coli clinical isolates by the rabbit polyclonal Ab raised to dPNAG, we found that 9 of the pga-positive strains were poorly killed in vitro (<10%), 5 (all UTI isolates) were modestly killed (11–30%) and 16 were killed fairly well (>30%) (Fig. 3). Results are also summarized in Table 1. None of the E. coli strains lacking the pga locus was killed >10%. Several of the strains (indicated on the graph in Fig. 3) were then used in protection studies.
Fig. 3.

Opsonophagocytosis of the E. coli clinical isolates. Circles, UTI isolates. Squares, isolates from neonates in the NICU. A symbol indicates mean of triplicates, error bars the SD. Six isolates used in the protection studies are noted, as is E. coli UTI-JΔpga, for which only background levels of killing of were achieved. UTI strain H lacks the pga locus.
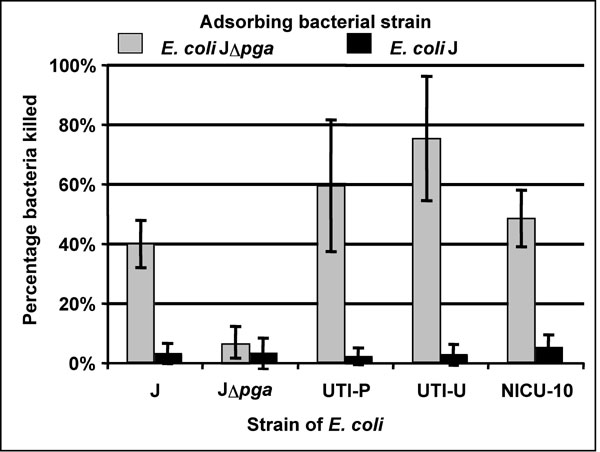
The specificity for the PNAG antigen of the opsonically active Ab in antisera raised to dPNAG conjugated to diphtheria toxin (dPNAG-DT) was shown by the loss of killing activity in the immune sera, initially diluted to have 40–70% killing activity, after adsorption with E. coli J. When the sera were adsorbed with the Δpga strain, no diminution in opsonic killing was observed [supporting information (SI) Fig. 4].
Protection Studies.
To determine the specificity of the goat antisera used in the protection studies, we first evaluated binding of pre- and postimmunization antisera to E. coli J and E. coli JΔpga whole cells by ELISA. Although there was binding of both the preimmune and immune goat antisera to both strains, there was no difference in binding of pre or postimmune sera when using the E. coli JΔpga strain (SI Fig. 5), whereas the PGA-positive E. coli J strain bound the immune serum noticeably better. Thus, if the preexisting Ab could protect against E. coli infection, it should be manifest in mice given preimmune serum. Table 2 summarizes the protective activity in a murine i.p. infection model of these goat antisera against a selection of E. coli strains. For each E. coli strain, we selected a challenge dose that would kill at least 75% of the mice. Goat Ab was used because of requirements for significant volumes of sera in these experiments, but this serum has been shown to have opsonic killing activity to S. aureus strains comparable with that of the rabbit antiserum used in the opsonic assays (Fig. 3) (14). This conclusion was verified by using 3 strains of E. coli (data not shown). As can be seen (Table 2), for the 5 E. coli strains expressing PNAG, there was significant protection ranging from 58% to 88% of animals given dPNAG-specific immune serum, whereas animals given preimmune goat serum had survival rates of 0–25% despite the presence of non-PNAG-specific Ab to E. coli in the preimmune serum. Furthermore, although both the preimmune and immune antisera bound to the E. coli JΔpga cells, there was no protective efficacy against this strain or against the E. coli UTI-H strain, which is negative for the pga locus.
Table 2.
Protection studies in an intraperitoneal murine model
| Strain | Dose per mouse, cfu* | Preimmune goat serum | Immune serum |
|---|---|---|---|
| Anti-dPNAG-DT goat | |||
| UTI-J | 2.5 × 108 | 0/8 (0%)† | 5/8 (63%)‡ |
| UTI-JΔpga§ | 1.0 × 109 | 2/8 (25%) | 2/8 (25%) |
| UTI-H§ | 5.0 × 108 | 0/8 (0%) | 0/8 (0%) |
| UTI-P | 2.5 × 108 | 0/8 (0%) | 7/8 (88%)‡ |
| UTI-U | 5.0 × 108 | 1/8 (13%) | 7/8 (88%)‡ |
| NICU-3 | 3.9 × 108 | 0/8 (0%) | 7/8 (88%)‡ |
| NICU-10 | 1.25 × 108 | 4/24 (17%) | 14/24 (58%)‡ |
| Anti-human IgG | |||
| UTI-J | 1.0 × 108 | 1/8 (13%)† | 2/8 (25%) |
| UTI-JΔpga§ | 2.0 × 109 | 1/8 (13%) | 1/8 (13%) |
*Minimal bacteria concentration necessary to kill at least 75% of the mice in each group.
†No. of surviving mice over the total mice used; percentage survival is in parentheses.
‡Survival ≥58% from immune goat serum significant at P ≤0.01, Fisher's exact test compared with normal goat serum giving survival of ≤25%.
§Lacks pga locus and PNAG production.
When an antiserum raised to an irrelevant antigen in conjunction with the same adjuvants as used with the dPNAG-DT conjugate was given to mice, no protection against either E. coli J or E. coli JΔpga was observed (Table 2). All dead mice had bacterial dissemination into their spleens, indicative of bacteremic spread from the initial focus of infection, whereas survivors had spleens lacking detectable bacteria as determined after the mice were killed.
Discussion
E. coli is an important cause of gastrointestinal diseases (23–25), UTIs (26–28), and neonatal meningitis and sepsis (29, 30), but the potential for vaccination against these strains has been deemed low because of the chemical and serologic variability of the most efficacious target antigens, lipopolysaccharide O antigens and capsular polysaccharides. Finding a conserved surface polysaccharide among diverse E. coli strains would raise the potential for use of such an antigen as a vaccine. Furthermore, finding that such an antigen was also expressed by other common but phylogenetically diverse pathogens, such as staphylococci, further raises interest in the vaccine potential of such an antigen. Given that vaccines injected into humans to prevent Streptococcus pneumoniae invasive disease, as well as Neiserria meningitidis, Haemophilus influenzae type b, and Salmonella enterica serovar Typhi infections are all composed of capsular polysaccharides as the major vaccine component, finding a broadly expressed surface polysaccharide such as PNAG among diverse pathogens that is a target for opsonic and protective Ab represents a potential means to have a vaccine effective against a wide number of important human bacterial pathogens.
As a first step in determining whether PNAG has such broad vaccine potential, we followed up on the recent findings of Wang et al. (18), who showed that strains of E. coli for which a sequenced genome has been produced all contain a genetic locus homologous to the staphylococcal icaADBC locus, designated pgaABCD by them (18). The presence of a pga locus is associated with the production of PNAG. In both staphylococci and E. coli, the PNAG polymer promotes biofilm formation and is cell surface-associated (5, 6, 18). In staphylococci, PNAG has been found to be a virulence factor (8, 31–33) and target for protective Ab (14–16). In this work, we report that a high percentage of E. coli UTI and neonatal bacteremia isolates contain the ica-homologous pga genetic locus and synthesize PNAG that is immunologically cross-reactive with PNAG isolated from S. aureus, and the production of PNAG by E. coli allows opsonically active antibodies raised to the S. aureus dPNAG antigen to mediate in vitro killing of these organisms. When the pga locus was deleted from E. coli strains BW25113 and clinical UTI isolate strain J, they no longer synthesized PNAG, but when the pga locus was added back in trans, PNAG synthesis was restored. In addition, clinical isolates lacking detectable pgaABCD also failed to produce PNAG. One isolate contained the pgaA and pgaC genes but nonetheless failed to produce detectable PNAG, potentially as a result of a mutation or gene repression. Overall, production of immunologically cross-reactive PNAG antigen was detected in 83% of 30 clinical isolates of E. coli.
To explore the potential of PNAG to vaccinate against E. coli infections, we initially compared the opsonic killing activity of antibodies raised with a mostly deacetylated form of PNAG (dPNAG, ≈15% residual N-acetyl groups) with that of antibodies raised to native PNAG (>95% N-acetyl groups). Prior results with staphylococci have shown that the Ab to the dPNAG antigen has greater activity in opsonizing S. aureus and S. epidermidis for phagocytic killing (14, 22, 34) and in mediating protective efficacy in experimental animal infections (14, 34). Ab to dPNAG did not mediate killing of E. coli in the absence of phagocytes. As we found with staphylococci, Ab raised to the dPNAG had superior opsonic activity against E. coli. The molecular basis for this phenomenon has been elucidated by the studies of Vuong et al. (10) by using S. epidermidis, wherein partial deacetylation of PNAG by the IcaB protein is needed to retain the antigen on the bacterial cell surface. We have found that the IcaB protein in S. aureus behaves identically (N.C., K.K.J., T.M.-L., D. B. Pier, C. Kelly-Quintos, D.A.G., J. Azeredo, and G.B.P, unpublished observations). Therefore, the epitopes recognized by the antibodies raised to dPNAG are present at a higher density on the bacterial cell surface, which is needed for efficient opsonic killing.
We next analyzed the protective efficacy of Ab raised to dPNAG conjugated to DT (14) against several E. coli strains expressing high or moderate levels of PNAG, as well as some negative controls comprising E. coli strains lacking the pga locus. Compared with normal goat serum, the goat serum raised to dPNAG-DT effectively protected against lethal sepsis after i.p. infection, except with the pga-negative controls, as expected. Although the i.p. infection model might be considered to mimic aspects of E. coli peritonitis, including bacteremic spread as evidenced by the presence of bacteria in the spleens of nonprotected animals, it clearly does not mimic the various manifestations of E. coli UTI or neonatal bacteremia. Nonetheless, a high-dose challenge model is a robust initial test of a vaccine's potential efficacy, primarily evaluating the in vivo activities of the antibodies and expression of the antigen. Although additional studies are needed to determine whether Ab to dPNAG can protect against experimental E. coli bladder infections (35, 36) or bacteremia emanating from the gastrointestinal tract (37, 38), these initial studies validate the potential of dPNAG as a protective vaccine for diverse and common pathogens such as E. coli, S. aureus, and coagulase-negative staphylococci. In addition, if pathogens such as Bordetella spp., Yersinia spp., A. actinomycetemcomitans, and others known to have a pga-homologous genetic locus (18, 19) also produce PNAG in a manner and form rendering antibodies to the antigen opsonic and protective, then a properly constructed PNAG-based vaccine could be protective against a broad array of human and animal pathogens. Currently, no such broadly expressed, protective surface polysaccharide antigen, the basis for most successful vaccines used in humans to elicit antibodies that kill bacterial pathogens, has been identified.
Materials and Methods
Strains and Media.
To evaluate the expression of PNAG, we used several strains of staphylococci, including PNAG-positive S. aureus MN8 (15) and S. epidermidis M187 (39) and PNAG-negative S. aureus MN8Δica (8). E. coli strains used included CFT073, a urinary tract isolate previously shown to contain the pga locus (18), and 30 clinical isolates of E. coli collected over different periods of time and also geographically distant. These isolates included 18 obtained from 18 different patients hospitalized at Brigham and Women's Hospital, Boston, MA, and were provided by the Clinical Microbiology Laboratory, courtesy of Andrew Onderdonk, and 12 blood isolates from patients in neonatal intensive care units (NICU) in Europe and the U.S. which were kindly provided by Kwang-Sik Kim, Baltimore, MD, or Peter W. Taylor, London, U.K. Seven of the E. coli isolates from neonates were known to express the K1 capsular polysaccharide frequently found among isolates from such patients (40). S. epidermidis, S. aureus, and E. coli strains were grown for 16 h at 37°C in tryptic soy broth/1% (gram percent) glucose (TSBG).
Construction of E. coli pga-Deleted and Complemented Strains.
The pga locus was replaced with a chloramphenicol acetyltransferase cassette by the PCR-mediated one-step method of gene inactivation described by Datsenko and Wanner (41). To accomplish this goal, E. coli BW25113 and the clinical UTI isolate strain J were first transformed with the red recombinase expression plasmid pKD46 then grown overnight at 25°C in SOC medium (2 × 10−2 g/ml Bacto-tryptone/5 × 10−3 g/ml yeast extract/5.84 × 10−4 g/ml NaCl/1.86 × 10−4 g/ml KCl/20 mM MgCl2/20 mM Glc, pH 7.0) containing 100 μg of ampicillin per ml and 10 mM l-arabinose. The next day, the cells were collected by centrifugation, washed three times with deionized H2O containing 10% glycerol, and resuspended at a 50-fold concentration over the original volume. A PCR product of the chloramphenicol acetyltransferase cassette was generated from plasmid pKD3 by using the primer pair pgadeleteFWD (5′-CCGGACTAGCGCTTTTTCTGAAACCACCATTTTTATTTGCCCCT G G C T G G GTGTAGGCTGGAGCTGCTTC-3′) and pgadeleteREV (5′-AAGTAGCAGAAAAAGGTGCCCGAAAACCAA A T G GGCTTTGAAACTTCTTACATATGAATATCC T C C TTAG-3′). The PCR product had 5′ and 3′ ends homologous to the 5′ and 3′ ends of the E. coli pga locus and was gel-purified by using the QIAquick gel extraction kit (Qiagen, Valencia, CA). Electrocompetent E. coli cells were transformed with the purified PCR product by electroporation, and transformants were selected on LB agar containing 30 μg of chloramphenicol per ml. Chloramphenicol-resistant bacteria were analyzed by PCR to ascertain that the chromosomal pga locus was deleted (Δpga strains) and were then cured of the pKD46 plasmid by two rounds of growth at 42°C in the absence of ampicillin. The E. coli BW25113 parental and Δpga strains were used to confirm the specificity of the immunoblot, and the Δpga strain was used to absorb antisera used in the opsonic killing assay.
To transcomplement the pga-deletion mutants, the entire pga locus was amplified from E. coli BW25311 DNA by PCR using primers pgaexpressFWD (5′-ATGTATTCAAGTAGCAGAAAAAGGTGC-3′) and pgaexpressREV (5′-GTGTTTACGCCCGGACTAGC-3′) and cloned into the arabinose-inducible pBAD-TOPO vector (Invitrogen, Carlsbad, CA). The DNA sequence of the insert in the resulting construct, pPGA, was determined to confirm identity to the known pga DNA sequence and then used to transform the pga-deletion mutant strains by electroporation. Transformants were selected on LB agar containing 100 μg ampicillin per ml. Addition of arabinose to the growth medium was not necessary for sufficient expression levels in pPGA-complemented strains.
Detection of PNAG Production by Bacteria.
Immunoblots for production of PNAG were performed essentially as described previously (7, 42) with minor modifications. Bacteria were grown to stationary phase in TSBG, and the cultures were diluted in this growth medium to produce an A600 = 1.5. Bacterial cells were pelleted by centrifugation, and equivalent amounts of each of the different bacterial preparations were resuspended in 300 μl of 0.5 M EDTA (pH 8.0) and incubated for 5 min at 100°C. Cells were removed by centrifugation at 10,500 × g for 6 min, and 100 μl of the supernatant was then incubated with 10 μl of proteinase K (20 mg/ml; Qiagen) for 60 min at 60°C. Proteinase K was heat-inactivated by incubating for 30 min at 80°C. This solution was diluted 3-fold in Tris-buffered saline (20 mM Tris·HCl/150 mM NaCl, pH 7.4), except for the extract from S. epidermidis M187, which was diluted 1,000-fold because of hyperproduction of PNAG by this strain, and 200 μl of each dilution was immobilized on a nitrocellulose filter attached to a slot-blot vacuum manifold. The filter was blocked with a solution of 1% BSA, then incubated for 2 h with a rabbit Ab raised to S. aureus PNAG conjugated to DT, produced as described previously (14) and further purified on an affinity column, also as described previously (22). The secondary Ab used was a horseradish peroxidase-conjugated anti-rabbit IgG Ab (Southern Biotech, Birmingham, AL) diluted 1:6,000, and detection of the PNAG antigen used the enhanced chemiluminescence (Amersham, Piscataway, NJ) Western blotting system.
Detection of the pga Locus in E. coli Clinical Isolates by PCR.
Genomic DNA from E. coli strains was extracted by using a DNeasy tissue kit (Qiagen). Primers used for the detection of the pgaA (5′-GGCTTTGAAACTTCTTACTGC-3′ and 5′-CCTGTTTATCTTGCCCGGCC-3′) and pgaC (5′-ATGATTAATCGCATCGTATCG-3′ and 5′-CATCGGTTCCACAATATATGC-3′) were custom synthesized by Qiagen Operon (Alameda, CA). For the PCR we added 25 μl of PCR supermix high-fidelity (Invitrogen), 0.5 μl of each primer, and 2 μl of DNA from the bacterial strains. PCR conditions comprised an initial 5-min denaturation step at 94°C followed by 32 cycles of 30 s at 94°C, 30 s at 50°C, and 45 s at 72°C, and a final extension step of 5 min at 72°C.
Antisera.
We used antibodies raised to either native S. aureus PNAG, wherein the glucosamine monosaccharides contained ≈95% acetate groups, or dPNAG, wherein the level of substitution with acetate groups was ≈15% (29). Both polysaccharides were conjugated to DT as described (14) and used to raise antibodies in rabbits, also as described previously (14). The dPNAG-DT conjugate was also used to immunize a goat as described previously (20). Specificity of the rabbit antiserum used in the opsonophagocytic assay was evaluated by adsorbing the sera with either PNAG-expressing E. coli J or the isogenic PNAG-negative E. coli Δpga strain. Specificity of the goat antisera for PNAG expression by E. coli was evaluated by whole-cell ELISA using the E. coli J and JΔpga strains. For some control experiments, we used a goat antiserum to human IgG raised by using the same adjuvants and immunization schedule as was used for the dPNAG-DT conjugate vaccine and was supplied by Lampire Biological Laboratories, Pipersville, PA. We have shown previously that the goat antiserum has comparable binding and opsonic activity against PNAG-producing S. aureus as does the rabbit antiserum raised to the dPNAG-DT conjugate (14).
Opsonic Killing of E. coli Clinical Isolates.
WBC were prepared from fresh human blood collected from healthy adult volunteers. Twenty-five milliliters was mixed with an equal volume of dextran/heparin buffer and incubated at 37°C for 1 h. The upper layer containing the leukocytes was collected, the cells were pelleted by centrifugation, and the remaining erythrocytes were removed by hypotonic lysis after resuspension of the cell pellet in 1% NH4Cl with incubation for 10 min at room temperature. WBC were then washed three times and resuspended with RPMI containing 15% heat-inactivated (56°C, 30 min) FBS (RPMI/FBS). Trypan blue staining was used to determine the number of viable leukocytes, then the final WBC count was adjusted to 2.5 × 107 WBC per ml. The complement source (1 ml of baby rabbit serum diluted 1:10 in RPMI/FBS) was adsorbed twice at 4°C for 45 min with continual mixing using bacteria resuspended from a pellet containing ≈109 cfu of E. coli BW25113 to remove natural Ab that mediated bactericidal killing of some of the target E. coli strains. The rabbit antisera were diluted 1:10 in RPMI/FBS and absorbed twice at 4°C for 45 min with continual mixing using bacteria resuspended from a pellet containing ≈109 cfu of E. coli BW25113Δpga. After absorption, the complement solution and the Ab were centrifuged and filter-sterilized. The bacterial strains to be evaluated for phagocyte-dependent killing activity of the rabbit Ab to PNAG were grown overnight in TSBG and then adjusted to an A650 = 0.4. A 1:100 dilution (3–6 × 107 cfu/ml) was then made in RPMI/FBS for use in the killing assay.
The opsonophagocytic assay was performed with 100 μl of leukocytes/100 μl of bacteria/100 μl of the complement solution/100 μl of a 1:10 or higher dilution of the Ab. Several controls were used: each component individually and all of the possible combinations of the individual components with one missing. The missing volume was made up with 100 μl of RPMI/FBS. The reaction mixture was incubated on a rotor rack at 37°C for 90 min. The tubes were vortexed for 15 s, and then dilutions were made in TSB with 0.025% Tween (to prevent bacterial sticking to pipette tips and the walls of dilution vessels); samples were than plated onto tryptic soy agar plates. The percentage of killing was calculated by determining the ratio of the cfu surviving in the tubes with bacteria, leukocytes, complement and Ab, to the cfu surviving in the tubes with bacteria, complement, leukocytes, and RPMI/FBS. Each assay was done in triplicate and repeated two or three times.
Murine Lethality Model.
To evaluate the in vivo protective efficacy of Ab to PNAG, we used an i.p. infection model in mice, as described previously (8, 14). Briefly, groups of eight mice (FVB, female, 6–8 weeks of age) were immunized i.p. 24 h before and 4 h after infection with 0.3 ml of normal goat serum or immune goat serum raised against staphylococcal dPNAG conjugated to DT. Some mice received a heterologous immune serum raised to human IgG with the same adjuvants and schedule as used with the dPNAG-DT conjugate. Bacteria were grown overnight in TSBG and then resuspended in sterile PBS to ≈5 × 108 to 5 × 109 cfu/ml, depending on the strain. For each strain, we selected a minimal dose that would kill at least 75% of the mice. Mice were challenged i.p. with a dose of 1.25 × 108 to 1 × 109 cfu in 0.2 ml of PBS and monitored at least twice daily. When mice became moribund, as defined by hunched appearance, markedly increased respiration rate, piloerection, and lack of an ability to move in response to being touched, they were considered moribund; they were killed and counted as dead for these experimental outcomes. After death, the spleens of the mice were removed, homogenized in RPMI, and cultured on tryptic soy agar and MacConkey agar to determine that the E. coli had disseminated from the initial site of infection.
Statistical Analysis.
Differences in outcomes in the protection studies were determined by Fisher's exact test.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants AI46706 (to G.B.P.) and R21AI61590 (to K.K.J.). N.C. was supported by Portuguese Fundação para a Ciência e Tecnologia Grant SFRH/BD/8676/2002 and by the Portuguese Fulbright Commission.
Abbreviations
- dPNAG
deacetylated PNAG
- DT
diphtheria toxoid
- ica
intercellular adhesin
- NICU
neonatal intensive care unit
- PGA
polyglucosamine
- PNAG
poly-N-acetylglucosamine
- UTI
urinary tract infection.
Footnotes
Conflict of interest statement: T.M.-L., K.K.J., and G.B.P. have filed patents and received royalty/licensing income for development of PNAG as a vaccine.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0700630104/DC1.
References
- 1.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Clin. Infect. Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 2.Mack D, Becker P, Chatterjee I, Dobinsky S, Knobloch JK, Peters G, Rohde H, Herrmann M. Int J Med Microbiol. 2004;294:203–212. doi: 10.1016/j.ijmm.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 3.Costerton JW, Montanaro L, Arciola CR. Int J Artif Organs. 2005;28:1062–1068. doi: 10.1177/039139880502801103. [DOI] [PubMed] [Google Scholar]
- 4.Vuong C, Otto M. Microbes Infect. 2002;4:481–489. doi: 10.1016/s1286-4579(02)01563-0. [DOI] [PubMed] [Google Scholar]
- 5.Maira-Litran T, Kropec A, Abeygunawardana C, Joyce J, Mark G, III, Goldmann DA, Pier GB. Infect Immun. 2002;70:4433–4440. doi: 10.1128/IAI.70.8.4433-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cramton SE, Gerke C, Schnell NF, Nichols WW, Gotz F. Infect Immun. 1999;67:5427–5433. doi: 10.1128/iai.67.10.5427-5433.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cramton SE, Ulrich M, Gotz F, Doring G. Infect Immun. 2001;69:4079–4085. doi: 10.1128/IAI.69.6.4079-4085.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kropec A, Maira-Litran T, Jefferson KK, Grout M, Cramton SE, Gotz F, Goldmann DA, Pier GB. Infect Immun. 2005;73:6868–6876. doi: 10.1128/IAI.73.10.6868-6876.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vuong C, Voyich JM, Fischer ER, Braughton KR, Whitney AR, DeLeo FR, Otto M. Cell Microbiol. 2004;6:269–275. doi: 10.1046/j.1462-5822.2004.00367.x. [DOI] [PubMed] [Google Scholar]
- 10.Vuong C, Kocianova S, Voyich JM, Yao Y, Fischer ER, DeLeo FR, Otto M. J Biol Chem. 2004;279:54881–54886. doi: 10.1074/jbc.M411374200. [DOI] [PubMed] [Google Scholar]
- 11.Heilmann C, Schweitzer O, Gerke C, Vanittanakom N, Mack D, Gotz F. Mol Microbiol. 1996;20:1083–1091. doi: 10.1111/j.1365-2958.1996.tb02548.x. [DOI] [PubMed] [Google Scholar]
- 12.McKenney D, Hubner J, Muller E, Wang Y, Goldmann DA, Pier GB. Infect Immun. 1998;66:4711–4720. doi: 10.1128/iai.66.10.4711-4720.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maira-Litran T, Kropec A, Goldmann D, Pier GB. Vaccine. 2004;22:872–879. doi: 10.1016/j.vaccine.2003.11.033. [DOI] [PubMed] [Google Scholar]
- 14.Maira-Litran T, Kropec A, Goldmann DA, Pier GB. Infect Immun. 2005;73:6752–6762. doi: 10.1128/IAI.73.10.6752-6762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenney D, Pouliot KL, Wang Y, Murthy V, Ulrich M, Doring G, Lee JC, Goldmann DA, Pier GB. Science. 1999;284:1523–1527. doi: 10.1126/science.284.5419.1523. [DOI] [PubMed] [Google Scholar]
- 16.Kojima Y, Tojo M, Goldmann DA, Tosteson TD, Pier GB. J Infect Dis. 1990;162:435–441. doi: 10.1093/infdis/162.2.435. [DOI] [PubMed] [Google Scholar]
- 17.Takeda S, Pier GB, Kojima Y, Tojo M, Muller E, Tosteson T, Goldmann DA. Circulation. 1991;84:2539–2546. doi: 10.1161/01.cir.84.6.2539. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Preston JF, III, Romeo T. J Bacteriol. 2004;186:2724–2734. doi: 10.1128/JB.186.9.2724-2734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan JB, Velliyagounder K, Ragunath C, Rohde H, Mack D, Knobloch JK, Ramasubbu N. J Bacteriol. 2004;186:8213–8220. doi: 10.1128/JB.186.24.8213-8220.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parise G, Mishra M, Itoh Y, Romeo T, Deora R. J Bacteriol. 2006;189:750–760. doi: 10.1128/JB.00953-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welch RA, Burland V, Plunkett G, III, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, et al. Proc Natl Acad Sci USA. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly-Quintos C, Kropec A, Briggs S, Ordonez C, Goldmann DA, Pier GB. J Infect Dis. 2005;192:2012–2019. doi: 10.1086/497604. [DOI] [PubMed] [Google Scholar]
- 23.Garmendia J, Frankel G, Crepin VF. Infect Immun. 2005;73:2573–2585. doi: 10.1128/IAI.73.5.2573-2585.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qadri F, Svennerholm AM, Faruque AS, Sack RB. Clin Microbiol Rev. 2005;18:465–483. doi: 10.1128/CMR.18.3.465-483.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DuPont HL. Curr Opin Infect Dis. 2005;18:407–412. doi: 10.1097/01.qco.0000182535.54081.68. [DOI] [PubMed] [Google Scholar]
- 26.Andreu A, Alos JI, Gobernado M, Marco F, de la Rosa M, Garcia-Rodriguez JA. Enferm Infecc Microbiol Clin. 2005;23:4–9. doi: 10.1157/13070401. [DOI] [PubMed] [Google Scholar]
- 27.Tal S, Guller V, Levi S, Bardenstein R, Berger D, Gurevich I, Gurevich A. J Infect. 2005;50:296–305. doi: 10.1016/j.jinf.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Wu CY, Chiu PC, Hsieh KS, Chiu CL, Shih CH, Chiou YH. Acta Paediatr Taiwan. 2004;45:328–333. [PubMed] [Google Scholar]
- 29.Jones B, Peake K, Morris AJ, McCowan LM, Battin MR. Aust NZ J Obstet Gynaecol. 2004;44:558–561. doi: 10.1111/j.1479-828X.2004.00304.x. [DOI] [PubMed] [Google Scholar]
- 30.Zaidi AK, Huskins WC, Thaver D, Bhutta ZA, Abbas Z, Goldmann DA. Lancet. 2005;365:1175–1188. doi: 10.1016/S0140-6736(05)71881-X. [DOI] [PubMed] [Google Scholar]
- 31.Shiro H, Muller E, Gutierrez N, Boisot S, Grout M, Tosteson TD, Goldmann D, Pier GB. J Infect Dis. 1994;169:1042–1049. doi: 10.1093/infdis/169.5.1042. [DOI] [PubMed] [Google Scholar]
- 32.Rupp ME, Ulphani JS, Fey PD, Mack D. Infect Immun. 1999;67:2656–2659. doi: 10.1128/iai.67.5.2656-2659.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rupp ME, Ulphani JS, Fey PD, Bartscht K, Mack D. Infect Immun. 1999;67:2627–2632. doi: 10.1128/iai.67.5.2627-2632.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly-Quintos C, Cavacini LA, Posner MR, Goldmann D, Pier GB. Infect Immun. 2006;74:2742–2750. doi: 10.1128/IAI.74.5.2742-2750.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts JA, Marklund BI, Ilver D, Haslam D, Kaack MB, Baskin G, Louis M, Mollby R, Winberg J, Normark S. Proc Natl Acad Sci USA. 1994;91:11889–11893. doi: 10.1073/pnas.91.25.11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langermann S, Palaszynski S, Barnhart M, Auguste G, Pinkner JS, Burlein J, Barren P, Koenig S, Leath S, Jones CH, et al. Science. 1997;276:607–611. doi: 10.1126/science.276.5312.607. [DOI] [PubMed] [Google Scholar]
- 37.Mushtaq N, Redpath MB, Luzio JP, Taylor PW. J Antimicrob Chemother. 2005;56:160–165. doi: 10.1093/jac/dki177. [DOI] [PubMed] [Google Scholar]
- 38.Mushtaq N, Redpath MB, Luzio JP, Taylor PW. Antimicrob Agents Chemother. 2004;48:1503–1508. doi: 10.1128/AAC.48.5.1503-1508.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller E, Huebner J, Gutierrez N, Takeda S, Goldmann DA, Pier GB. Infect Immun. 1993;61:551–558. doi: 10.1128/iai.61.2.551-558.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie Y, Kim KJ, Kim KS. FEMS Immunol Med Microbiol. 2004;42:271–279. doi: 10.1016/j.femsim.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Datsenko KA, Wanner BL. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jefferson KK, Cramton SE, Gotz F, Pier GB. Mol Microbiol. 2003;48:889–899. doi: 10.1046/j.1365-2958.2003.03482.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}