

Abstract
Experimental data from protein engineering studies and NMR spectroscopy have been used by theoreticians to develop algorithms for helix propensity and to benchmark computer simulations of folding pathways and energy landscapes. Molecular dynamic simulations of the unfolding of chymotrypsin inhibitor 2 (CI2) have provided detailed structural models of the transition state ensemble for unfolding/folding of the protein. We now have used the simulated transition state structures to design faster folding mutants of CI2. The models pinpoint a number of unfavorable local interactions at the carboxyl terminus of the single α-helix and in the protease-binding loop region of CI2. By removing these interactions or replacing them with stabilizing ones, we have increased the rate of folding of the protein up to 40-fold (τ = 0.4 ms). This correspondence, and other examples of agreement between experiment and theory in general, Φ-values and molecular dynamics simulations, in particular, suggest that significant progress has been made toward describing complete folding pathways at atomic resolution by combining experiment and simulation.
Efforts are being made to describe the folding pathways of proteins at atomic resolution by experiment using Φ-value analysis (1–6) and NMR (7–10) and by simulation using molecular dynamics (MD) (11–20). Φ-value analysis infers structure of transition states and intermediates from measurements of changes in energetics of kinetics and equilibrium thermodynamics on mutation of structure (1–5, 21). NMR is particularly useful for obtaining information about denatured and partly folded states (22, 23). MD simulation, on the other hand, has the potential of describing the complete energy landscape during folding at the atomic level. MD is applied most readily to unfolding, from a known native structure, and the folding process inferred from microscopic reversibility (24, 25). There is uncertainty in the interpretation of MD simulations because of the extrapolation from high temperatures or other extreme conditions and the use of empirical energy functions and empirical methods for identifying transition states. The importance of Φ-value analysis is that it is the only experimental procedure that provides evidence about side chain interactions in transition states, and these can be used to benchmark MD simulation and check its reliability. Thus, simulation and experiment can complement one another to provide an in-depth and testable reconstruction of protein-folding pathways. We are using this approach to characterize the transition state of folding/unfolding of chymotrypsin inhibitor 2 (CI2) (6, 12–14, 27).
CI2 folds by simple two-state kinetics from a relatively expanded denatured state through only a single detectable transition state ensemble (26–28). Both experiment and simulation indicate that the transition state for folding of CI2 is an ensemble of structures with a nearly intact single α-helix (residues 12–24), and a disrupted β-sheet, except for a portion of the β-sheet that participates with the α-helix in the formation of the folding nucleus (6, 14, 19). Lattice simulations (29, 30), energy landscape methods (31), and MD simulations (12–14, 19) have shown good agreement with the experimental data. An analysis of a set of MD simulations by Lazaridis and Karplus (19) has led to a reconciliation of the “New View” of protein folding with the “Old” (19). If models generated by MD are able to capture the essential structural features of a protein-folding reaction, then we are on the path toward solving the protein-folding problem. This idea calls for experimental tests of predictions that emerge from the simulation of transition state structures. To this end, we have identified particular interactions in our four wild-type transition state ensembles, generated with MD (13, 14), that appear to destabilize the transition state.
Specifically, we have identified two regions of structure in which mutation may speed up folding. The first is at the C terminus of the single helix. Asp-23 stabilizes the native protein by making a salt bridge with Lys-2. But, the presence of an aspartate at the C terminus of an isolated helix is destabilizing. The Asp-23–Lys-2 salt bridge is broken in the simulated transition state, giving rise, effectively, to an isolated helix (Fig. 1). The second region involves a cluster of arginines—Arg-46, Arg-48, and Arg-62 (Fig. 2). The hydrophobic portions of these side chains stack, leading to close guanidinium groups and electrostatic repulsion. This unfavorable arrangement is partly relieved by a network of hydrogen bonds with the carboxylate of the C-terminal residue Gly-64. We have constructed site-specific mutants to disrupt interactions in these two regions, and the mutants do indeed fold faster than wild type. This is notable because most mutations result in slower folding proteins. Furthermore, denaturation simulations of the mutants were performed and transition state ensembles were identified. This completes the cycle and allows us to pinpoint the specific interactions responsible for stabilization of the mutant transition states. These combined studies provide a basis for rational design of faster folding proteins.
Figure 1.

Interactions between the α-helix and N-terminal loop of CI2. Lys-2, Glu-7, Lys-18, and Asp-23 are shown for the native protein, the four wild-type transition state models (14), and the DA23 and KR18/DR23 mutant transition state structures (at 125 and 560 ps, respectively). Lys-2, Glu-7, and Asp-23 comprise a salt-bridge network in the native state that is disrupted in the transition states. Asp-23 does not interact with Lys-2 and Glu-7 in three of four wild-type transition state models. Asp-23 contributes adversely to the stability of the isolated single α-helix in the transition state due to close interactions with unpaired carbonyl groups at the end of the helix and the lack of compensatory favorable interactions after the N terminus pulls away. The models were drawn using the programs molscript and raster3d (54–56).
Figure 2.

Electrostatic interactions in the protease-binding loop region of CI2. The figure shows residues Arg-46, Arg-48, Arg-62, and the C-terminal carboxyl group (labeled with a C in italics) in the native protein, the four wild-type transition state models (14) and the RF48 transition state structure (475 ps). Replacement of Arg-48 by Phe leads to the diminution of charge repulsion in the transition state and heightened hydrophobic packing interactions. The models were drawn by using the programs molscript and raster3d (54, 55).
MATERIALS AND METHODS
Helix Propensity Calculations.
The percentage helicity of the α-helix of CI2 as an isolated peptide sequence was calculated by using the semi-empirical helix propensity algorithm AGADIR (32, 33). The wild-type helical peptide comprises the helical region (residues 12–24 of CI2) plus five- and four-flanking residues at the N- and C-termini, respectively. Predictions were made for the helical region excluding the flanking residues, at pH 6.3, 298 K, a free N terminus, and an amidated C terminus. We determined the peptide helicity experimentally by titrating the peptides with trifluoroethanol and measuring the change in the ellipticity at 222 nm (34).
Mutagenesis and Protein Purification.
Mutant CI2 proteins were engineered by inverse-PCR mutagenesis (35), using the pJB2 CI2 expression vector (36) as the DNA template. The sequenced DNA vectors were transformed in Escherichia coli strain NM554, which was grown at 37°C to an absorbance of 0.5 before overnight induction with 1 mM isopropyl β-d-thiogalactoside. The wild-type and mutant proteins were purified to homogeneity as published (6) and stored in the liquid phase of nitrogen. Molecular masses were checked by both matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) and electrospray ionization (ESI) mass spectrometry (A.G.L. and P. Skelton, Cambridge University Chemical Laboratory Mass Spectrometry Service).
Equilibrium Denaturation and Folding Kinetics.
All CI2 proteins were analyzed kinetically and thermodynamically as described (6, 26).
CI2 Sequence Homology Searches.
The wild-type, truncated CI2 gene was aligned to its sequence homologues by using the program phd (37). Two sequences with a low sequence similarity scoring were removed (the two genes removed were a E. coli chaperonin GroEL fragment and one from DNA polymerase Klenow fragment). Our sequence alignments therefore differ from earlier reported sequence alignments (30).
MD Simulations.
The four wild-type transition states (TS1–TS4) and a control simulation have been described (14). New simulations of the DA23, KR18/DR23, and RF48 mutants have been performed by using the same procedures. The potential energy function and protocols for MD (38, 39), as implemented within encad (40), have been described. The system under simulation consists of the protein and surrounding water molecules. The crystal structure (1YPC.pdb, 1.7-Å resolution, ref. 41) was used as the starting point for the mutant denaturation simulations. The mutants were constructed by swapping in the new residue for the wild-type residue while maintaining the side chain conformation. All simulations were at neutral pH (Lys and Arg residues were positively charged and Asp and Glu residues were negatively charged). The starting structures were minimized for 500 cycles to reduce any bad contacts before performing MD. After minimization, water molecules were added around the protein to fill a rectangular box, with walls at least 8 Å away from any protein atom (≈2,400 waters). The density of the solvent was set to the experimental value for 498 K, 0.829 g/Å3 (42, 43) by adjusting the volume of the box, resulting in at least a 10-Å shell of waters. Periodic boundary conditions were used to minimize boundary effects. Because solvent molecules were explicitly present, no macroscopic dielectric constant was needed.
The resulting systems were then prepared for MD. A total of 1,000 cycles of conjugate-gradient minimization followed by 1,000 steps of MD were performed on the water molecules. The water molecules were then minimized again for 1,000 cycles. Following that, the protein was minimized for 1,000 cycles, which was followed by minimization of the entire system for 1,000 cycles. After the above preparations, atoms in the system were assigned velocities according to a Maxwellian distribution. Atoms were allowed to move according to Newton’s equations of motion, and the velocities of the atoms were adjusted intermittently until the system reached the desired temperature, which usually takes about 2–3 ps. No further velocity scaling was needed after that point. A 2-fs (1 fs = 10–15 second) time step was used for both the preparation steps and the full simulation. An 8-Å nonbonded cutoff was used, and the nonbonded list was updated every five cycles. The mutant simulations were carried out for 1,000 ps each, which was sufficient to unfold the protein and yield Cα root-mean-square deviations of 7–10 Å, which is in line with what is observed for simulations of the wild-type protein (13). Transition states for the mutants were identified by using a conformational clustering procedure, as described previously (11–13).
RESULTS AND DISCUSSION
Improving Local Interactions Along the Helix in the Transition State.
Lys-2 forms salt bridges with Glu-7 in strand β1 and Asp-23 at the C terminus of the α-helix in the native state (Fig. 1). This highly conserved salt bridge contributes to the stability of the native state; the mutation DA23 decreases stability by 0.96 kcal mol−1 (Table 1). The MD simulations on the unfolding of CI2 (12–14, 19) predict that the α-helix is well formed in the transition state, but one of the earliest events in the simulated unfolding is the displacement of the N-terminal residues 1–11 from the C terminus from the helix (Fig. 1). As a result, the Lys-2–Asp-23 interaction is not formed in three of our four rate-determining transition states for unfolding (and hence for folding). The C terminus of the helix is stabilized primarily by local interactions in the transition state. In this structural context, residue Asp-23 makes unfavorable interactions with the helix dipole (44–46) or more precisely the unpaired, main chain carbonyl groups at the C terminus of the helix.
Table 1.
Equilibrium denaturation of wild-type and mutant CI2
| CI2 protein* | Predicted helicity¶, % | Determined, helicity†, % | mD-N‡, kcal mol−1 M−1 | [GdmCl]50%‡, M | ΔΔGD-NH2O§, kcal mol−1 |
|---|---|---|---|---|---|
| Wild type | 2.3 | 3.2 ± 1.1 | 1.90 ± 0.03 | 4.00 ± 0.01 | 0 |
| Mutations at the C terminus of the helix | |||||
| KR18 | 3.3 | n.d. | 2.34 ± 0.07 | 3.89 ± 0.01 | 0.21 ± 0.03 |
| KR18/DR23 | 10.0 | 8.1 ± 1.9 | 1.94 ± 0.03 | 3.03 ± 0.01 | 1.87 ± 0.04 |
| DA23 | 4.5 | 5.2 ± 1.0 | 1.87 ± 0.05 | 3.51 ± 0.01 | 0.96 ± 0.03 |
| Mutations in the protease-binding loop region | |||||
| RF48‖ | 1.70 ± 0.05 | 4.85 ± 0.08 | −1.65 ± 0.16 | ||
| RF48/FL50 | 1.88 ± 0.07 | 3.71 ± 0.03 | 0.56 ± 0.05 | ||
Equilibrium denaturation experiments with guanidinium chloride (GdmCl) were performed as described (6). The free energy of unfolding, ΔGD-N, varies according to denaturant concentration as follows: ΔGD-N = ΔGD-NH2O − mD-N [GdmCl].
Determined by titration of the CI2 peptides with trifluoroethanol in 10 mM sodium phosphate, pH 6.3, 25°C (34). This procedure gives the free energy of formation of the helix, ΔG, which is related to the equilibrium constant, K, by ΔG = −RT ln K. The fraction of formed helix is then defined as K/(K + 1).
mD-N is the denaturant dependence of the equilibrium unfolding transition and [GdmCl]50% is the denaturant concentration at which 50% of the protein is unfolded.
The change in free energy of equilibrium unfolding of the mutant and wild-type proteins, Δ(ΔGD-NH2O), was calculated using an average mD-N value of 1.94 kcal mol−1 M−1 (6).
The peptide helicity is calculated by the prediction program agadir (32, 33). The wild-type peptide sequence ELVGK-(SVEEAKKVILQDK)-PEAQ is the input sequence for the program and the conditions are: pH 6.3, 298 K, free N terminus and amidated C terminus. The helicity shown above is the average helicity in the helical region (indicated by the sequence in brackets), which corresponds to residues (12–24) in the intact CI2 protein. The residues shown in bold are those mutated in this study.
The determination of the stability of CI2 RF48 is not very reliable because the protein is very stable. Using the stronger denaturant guanidinium isothiocyanate (Gdm SCN) we derive a ΔΔGD-NH2O of −1.18 ± 0.20 kcal mol−1.
The single α-helix of CI2 is only weakly populated in the denatured state of the full-length protein, as probed by NMR (J. Y. Tan, K. B. Wong, and A.R.F., unpublished results) and MD (S. Kazmirski and V.D., unpublished results), and in peptide models (47–49). Using the helix propensity algorithm AGADIR (32, 33), we have improved the propensity of CI2 peptides corresponding to the α-helix to adopt helical conformations by mutating the C terminus of the helix, Asp-23 to Ala or Arg and also Lys-18 to Arg, which are all stabilizing mutations. The wild-type peptide is predicted to show 2% helicity, close to the experimental value of 3 ± 1% for the model peptide (Table 1). The mutations DA23 and KR28/DR23 stabilize the isolated helix. The mutant KR18/DR23, for example, is calculated to be 10% helical, and we find 8 ± 2%.
We predict that the DA23 and KR18/DR23 mutations should stabilize the transition state for folding, thereby leading to faster folding than the wild-type protein. This is found: there is an increase in the refolding rate constant, which is well beyond experimental error, from 56.5 ± 2.5 for wild type to 83.9 ± 1.7 for DA23 and 89.8 ± 3.6 s−1 for KR18/DR23 (Table 2). These increases are especially significant considering that overall destabilization of CI2 generally leads to a decrease in the rate constant for folding (6).
Table 2.
Kinetics of refolding and unfolding of wild-type and mutant CI2
| CI2 protein | kfH2O*, s−1 | mkf†, M−1 | ΔΔG‡-DH2O‡ kcal mol−1 | kuH2O*, ×104 s−1 | mku†, M−1 | ΔΔG‡-NH2O‡ kcal mol−1 |
|---|---|---|---|---|---|---|
| Wild type | 56.5 ± 2.5 | −1.82 ± 0.12 | — | 1.2 ± 0.1 | 1.31 ± 0.01 | — |
| Mutations at the C terminus of the helix | ||||||
| KR18 | 66.0 ± 2.4 | −1.91 ± 0.05 | −0.09 ± 0.03 | 1.2 ± 0.2 | 1.26 ± 0.03 | −0.01 ± 0.11 |
| KR18/DR23 | 89.8 ± 3.6 | −2.10 ± 0.06 | −0.27 ± 0.04 | 28 ± 3 | 1.27 ± 0.02 | −1.87 ± 0.01 |
| DA23 | 83.9 ± 1.7 | −2.16 ± 0.06 | −0.23 ± 0.03 | 3.8 ± 0.9 | 1.38 ± 0.04 | −0.69 ± 0.05 |
| Mutations in the protease-binding loop region | ||||||
| RF48 | 2300 ± 200 | −1.89 ± 0.04 | −2.18 ± 0.06 | n.d. | n.d. | −0.75 ± 0.02§ |
| RF48/FL50 | 375 ± 10 | −1.92 ± 0.03 | −1.12 ± 0.03 | 18 ± 3 | 1.14 ± 0.03 | −1.59 ± 0.10 |
The rates of folding, kfH2O, and unfolding, kuH2O, of the protein were measured as described (6). The values of kfH2O measured directly in water (data not shown) and those derived by extrapolation from GdmCl (shown here) are identical within experimental error.
mkf and mku are calculated from the denaturant dependency of the folding and unfolding rate constants according to: ln kf[GdmCl] = ln kfH2O + mkf [GdmCl] and ln ku[GdmCl] = ln kuH2O + mku [GdmCl].
ΔΔG‡-DH2O and ΔΔG‡-NH2O are the changes in free energies of activation of folding and unfolding, respectively, relative to the wild-type protein. They are calculated according to ref. 6.
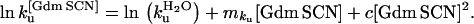 |
The kinetic parameters were calculated at 4 M Gdm SCN. Wild-type CI2 unfolds with a rate constant of 31.5 ± 1 s−1 in 4 M Gdm SCN relative to 113 ± 3 s−1 for RF48 CI2. We can thus calculate ΔΔG‡-D4M Gdm SCN for the RF48 CI2 mutant to be −0.75 ± 0.02 kcal mol−1. The unfolding mku[Gdm SCN]-values in Gdm SCN are 1.69 ± 0.03 M−1 and 1.58 ± 0.04 M−1, for wild-type CI2 and RF48 CI2 respectively.
GdmCl, guanidine hydrochloride; Gdm SCN, guanidinium isothiocyanate.
The destabilization of the protein upon mutation (Table 1) is reflected in increased rate constants for unfolding of all mutants (Table 2). The denaturant dependencies of the free energy of unfolding and of the refolding and unfolding rate constants (“m-values”) are not significantly affected by the mutations (Tables 1 and 2), suggesting that the degree of solvent exposure of the denatured, native, and transition states has not been altered substantially (50–52). In short, the DA23 mutation increases the stability of the helix and thereby increases the rate of folding, but the mutation prevents stabilizing tertiary interactions in the native state, resulting in destabilization of the protein.
Although the experimental studies support the predictions based on the MD transition state structures, we further tested the predictions by performing explicit simulations of the mutant proteins. For both DA23 and KR18/DR23, the transition state structures are similar to the wild-type transition state models (Fig. 1). The Cα root-mean-square deviations between the wild-type structures in Fig. 1 are between 3.6 and 4.3 Å. Comparison of the mutant transition states with the wild-type structures yields slightly higher values: 4.1–5.5 Å for DA23 and 4.3–4.9 Å for KR18/DR23. Consistent with the m values for unfolding and refolding, the solvent accessible surface areas are also similar: 5,740 Å2 for DA23, 5,278 Å2 for KR18/DR23, and 5,260–5,763 Å2 for the wild-type structures. No new interactions upon mutation are evident in the DA23 mutant transition state structures. This mutation acts by elimination of close interactions between the carboxyl side chain and the main chain carbonyl oxygens of residues 20, 23, and 24. In the KR18/DR23 mutant, however, in addition to elimination of unfavorable charge interactions, favorable interactions are now possible at the C terminus of the helix and furthermore Arg-23 forms a salt bridge with Glu-7 (Fig. 1).
Removing Unfavorable Electrostatic Interactions in the Loop Region.
Arg-48 extends from the main body of the protein toward the reactive site loop in the native protein, making electrostatic interactions with Thr-39 and the carboxylate of Gly-64 (Fig. 2). The side chain of Arg-48 is largely buried with only NH1 and NH2 partly solvent exposed (the solvent accessible surface areas are 10 and 14 Å2, respectively, as calculated by using naccess (53). Arg-48 is found in 16 of 19 CI2 homologues. Arg-46 is inaccessible to solvent except for NH1 (12 Å2), and this residue is conserved in all 19 CI2 homologues.
The protease-binding loop region in CI2 is expanded and more loosely packed in the wild-type transition state (6, 14, 19). Although many of the electrostatic interactions in the loop region are retained in the simulated transition state models, some are weakened or nonnative (Fig. 2). All four transition state models contain two or three of the Arg residues in close proximity. The orientation of the side chains differs in the four transition state models, however, resulting in an heterogeneous ensemble with many of the native electrostatic interactions in the loop weakened. In addition, there are nonnative interactions between Arg-62 and Arg-48 in the transition state. In the native protein Arg-62 forms a salt bridge with Glu-4, and its distance to Arg 48 is >10 Å. Yet, in 3 of 4 transition state models Arg-62, together with Arg-48, forms nonnative interactions with the C terminus of Gly 64 (Fig. 2). The MD simulations thus predict that the native salt bridges and electrostatic interactions are not well optimized in the transition state. It is also noteworthy that the aliphatic portions of the Arg residues remain relatively buried in the transition state models and interact with Ile-30, Leu-32, and Phe-50 (Fig. 2). The removal of some electrostatically repulsive interactions and the improvement of nonpolar packing would therefore be expected to lower the activation energy for folding and increase the rate of folding.
Because both Arg 46 and 48 are highly conserved, we used sequence homologies between CI2 and its homologues to identify suitable mutations at Arg-48. Arg-48 is conserved in 16 CI2 homologues, two homologues have Phe-48 and the other has Trp-48. Mutation of Arg-48 to Phe retains the possibility of hydrophobic interactions between side chains but removes unfavorable electrostatic interactions between the guanidinium groups. Mutation of Arg-48 to Phe increases the rate of folding of CI2 from 56.5 s−1 to 2,300 s−1 (Table 2). The solvent exposure of the transition state is not altered because the m-values for refolding are unchanged relative to the wild-type protein (Table 2). The activation energy for the folding of the RF48 mutant is reduced by 2.2 (± 0.1) kcal mol−1, which is even greater than the effects of the mutation on overall stability (1.6 ± 0.2 kcal mol−1, Table 1). The use of sequence homology data for CI2 has thus allowed for the selection of the fastest folding and most stable CI2 protein yet described.
The activation energy for unfolding also is reduced in the RF48 mutant, by 0.7 (±0.1) kcal mol−1, consistent with two-state folding. As with refolding, the equilibrium and unfolding m-values are not changed by the mutation (Tables 1 and 2). Mutations of residue Arg-46 and/or Arg-48 to Ala or Cys also accelerate the rate of folding of CI2 considerably, up to a folding rate constant of 400 s−1 (D. E. Otzen, A.G.L., and A.R.F., data not shown). Although these mutations diminish charge repulsion in the transition state, they also remove favorable nonpolar-packing interactions, resulting in less acceleration than RF48. Also, unlike the mutation of Arg-48 to Phe, the mutation of Arg-46 or Arg-48 to Ala or Cys destabilizes CI2 because of the disruption of hydrophobic packing in the native state (D. E. Otzen, A.G.L., and A.R.F., unpublished results). The Arg mutants are not as effective protease inhibitors as the wild-type protein (A.G.L., D. E. Otzen, and A.R.F., data not shown). The conservation of arginine residues at positions 46 and 48 is thus likely to be important for function but detrimental to the rate of folding.
Given the experimental results, the predictions based on the wild-type transition state models appear to be correct. Mutation of Arg-48 to Phe should accelerate folding by improving nonpolar packing interactions with residues nearby in the binding loop and by removing electrostatically repulsive interactions between Arg residues. Consistent with these expectations, Phe-48 forms more extensive packing interactions, particularly with Ile-30 and Phe-50 than Arg-48 (Fig. 2) in the transition state structures. Removal of the positively charged Arg-48 obviously will eliminate some charge repulsion. Also, however, in the absence of Arg-48, Arg-46 interacts with the C-terminal carboxylate, pulling it down and away from the other Arg residues, adding another mechanism for diminishing charge repulsion (Fig. 2). Consistent with the m-values for unfolding and refolding, the solvent accessible surface area of the RF48 transition state is within the range of values observed for the wild-type transition state structures (5,555 Å2 vs. 5,260–5,763 Å2).
CONCLUSIONS
We have used the general structural features of the transition states derived from MD simulations of the unfolding of wild-type CI2 to design faster folding mutants. In doing so, we have, in effect, tested predictions from the simulations and found them satisfactory. Furthermore, we have performed simulations of the mutants and identified mutant transition state structures that allow for further tests of the predictions from the wild-type transition states. This completes the cycle and provides detailed explanations of the effects of the mutations on the rate-limiting transition state for folding/unfolding. The consistency of MD simulations of CI2 and of barnase (20), benchmarked with suitable experimental results, clearly have considerable predictive power for describing the energy landscape of protein folding.
Acknowledgments
This work was supported by the Medical Research Council of Great Britain and in part by the National Institutes of Health (Grant GM 50789 to V.D.). A.G.L. is supported by the Boehringer Ingelheim Fonds (Stuttgart, Germany) and L.S.I. by a fellowship from the Beit Memorial Fund (London, United Kingdom).
ABBREVIATIONS
- CI2
chymotrypsin inhibitor 2
- MD
molecular dynamics
References
- 1.Matouschek A, Kellis J T, Jr, Serrano L, Fersht A R. Nature (London) 1989;342:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 2.Viguera A R, Serrano L, Wilmanns M. Nat Struct Biol. 1996;3:874–880. doi: 10.1038/nsb1096-874. [DOI] [PubMed] [Google Scholar]
- 3.López-Hernández E, Serrano L. Fold Des. 1996;2:43–55. [PubMed] [Google Scholar]
- 4.Parker M J, Sessions R B, Badcoe I G, Clarke A R. Fold Des. 1996;1:145–156. doi: 10.1016/S1359-0278(96)00023-5. [DOI] [PubMed] [Google Scholar]
- 5.Burton R E, Huang G S, Daugherty M A, Calderone T L, Oas T G. Nat Struct Biol. 1997;4:305–310. doi: 10.1038/nsb0497-305. [DOI] [PubMed] [Google Scholar]
- 6.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 7.Udgaonkar J B, Baldwin R L. Nature (London) 1988;335:700–704. [Google Scholar]
- 8.Roder H, Elöve G A, Englander S W. Nature (London) 1988;335:694–699. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoeltzli S D, Frieden C. Proc Natl Acad Sci USA. 1995;92:9318–9322. doi: 10.1073/pnas.92.20.9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balbach J, Forge V, Vannuland N, Winder S L, Hore P J, Dobson C M. Nat Struct Biol. 1995;2:865–870. doi: 10.1038/nsb1095-865. [DOI] [PubMed] [Google Scholar]
- 11.Li, A. & Daggett, V. (1998) J. Mol. Biol. 487–506. [DOI] [PubMed]
- 12.Li A J, Daggett V. Proc Natl Acad Sci USA. 1994;91:10430–10434. doi: 10.1073/pnas.91.22.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li A J, Daggett V. J Mol Biol. 1996;257:412–429. doi: 10.1006/jmbi.1996.0172. [DOI] [PubMed] [Google Scholar]
- 14.Daggett V, Li A J, Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1996;257:430–440. doi: 10.1006/jmbi.1996.0173. [DOI] [PubMed] [Google Scholar]
- 15.Caflisch A, Karplus M. Proc Natl Acad Sci USA. 1994;91:1746–1750. doi: 10.1073/pnas.91.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caflisch A, Karplus M. J Mol Biol. 1995;252:672–708. doi: 10.1006/jmbi.1995.0528. [DOI] [PubMed] [Google Scholar]
- 17.Tirado-Rives J, Orozco M, Jorgensen W L. Biochemistry. 1997;36:7313–7329. doi: 10.1021/bi970096i. [DOI] [PubMed] [Google Scholar]
- 18.Karplus M, Sali A. Curr Opin Struct Biol. 1995;5:58–73. doi: 10.1016/0959-440x(95)80010-x. [DOI] [PubMed] [Google Scholar]
- 19.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 20.Bond C J, Wong K B, Clarke J, Fersht A R, Daggett V. Proc Natl Acad Sci USA. 1997;94:13409–13413. doi: 10.1073/pnas.94.25.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matouschek A, Fersht A R. Methods Enzymol. 1991;202:82. doi: 10.1016/0076-6879(91)02008-w. [DOI] [PubMed] [Google Scholar]
- 22.Wüthrich K. Curr Opin Struct Biol. 1994;4:93–99. [Google Scholar]
- 23.Shortle D R. Curr Opin Struct Biol. 1996;6:24–30. doi: 10.1016/s0959-440x(96)80091-1. [DOI] [PubMed] [Google Scholar]
- 24.Duggleby R G, Dennis D T. J Biol Chem. 1974;249:167–174. [PubMed] [Google Scholar]
- 25.Fersht A R. Enzyme Structure and Mechanism. New York: Freeman; 1995. [Google Scholar]
- 26.Jackson S E, Fersht A R. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 27.Otzen D E, Itzhaki L S, elMasry N F, Jackson S E, Fersht A R. Proc Natl Acad Sci USA. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fersht A R, Itzhaki L S, elMasry N F, Matthews J M, Otzen D E. Proc Natl Acad Sci USA. 1994;91:10426–10429. doi: 10.1073/pnas.91.22.10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abkevich V I, Gutin A M, Shaknovich E I. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 30.Shakhnovich E, Abkevich V, Ptitsyn O. Nature (London) 1996;379:96–98. doi: 10.1038/379096a0. [DOI] [PubMed] [Google Scholar]
- 31.Shoemaker B A, Wang J, Wolynes P G. Proc Natl Acad Sci USA. 1997;94:777–782. doi: 10.1073/pnas.94.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muñoz V, Serrano L. Nat Struct Biol. 1994;1:399–409. doi: 10.1038/nsb0694-399. [DOI] [PubMed] [Google Scholar]
- 33.Muñoz V, Serrano L. J Mol Biol. 1995;245:275–296. doi: 10.1006/jmbi.1994.0023. [DOI] [PubMed] [Google Scholar]
- 34.Jasanoff A, Fersht A R. Biochemistry. 1994;33:2129–2135. doi: 10.1021/bi00174a020. [DOI] [PubMed] [Google Scholar]
- 35.Innis M A, Gelfand D H. In: PCR protocols: A Guide to Methods and Applications. Innis M A, Gelfand D H, Sninsky J, White T J, editors. San Diego: Academic; 1990. pp. 3–12. [Google Scholar]
- 36.Stott K, Blackburn J M, Butler P J G, Perutz M F. Proc Natl Acad Sci USA. 1995;92:6509–6513. doi: 10.1073/pnas.92.14.6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rost B, Sander C, Schneider S. Comput Appl Biosci. 1994;10:53–60. doi: 10.1093/bioinformatics/10.1.53. [DOI] [PubMed] [Google Scholar]
- 38.Levitt M, Hirshberg M, Sharon R, Daggett V. Comput Phys Commun. 1995;91:215–231. [Google Scholar]
- 39.Levitt M, Hirshberg M, Sharon R, Laidig K E, Daggett V. J Phys Chem. 1997;101:5051–5061. [Google Scholar]
- 40.Levitt M. encad, Computer Program. Molecular Applications Group, Palo Alto, CA: Energy Calculations and Dynamics; 1990. [Google Scholar]
- 41.Harpaz Y, Elmasry N, Fersht A R, Henrick K. Proc Natl Acad Sci USA. 1994;91:3–15. doi: 10.1073/pnas.91.1.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kell G S. J Chem Eng Data. 1967;12:66–68. [Google Scholar]
- 43.Haar L, Gallagher J S, Kell G S. NBS/NRC Steam Tables: Thermodynamic and Transport Properties and Computer Programs for Vapor and Liquid States of Water in SI Units. Washington, D.C.: Hemisphere Publication; 1984. [Google Scholar]
- 44.Serrano L, Fersht A R. Nature (London) 1989;342:296–299. doi: 10.1038/342296a0. [DOI] [PubMed] [Google Scholar]
- 45.Serrano L, Sancho J, Hirshberg M, Fersht A R. J Mol Biol. 1992;227:544–559. doi: 10.1016/0022-2836(92)90906-z. [DOI] [PubMed] [Google Scholar]
- 46.Serrano L, Neira J L, Sancho J, Fersht A R. Nature (London) 1992;356:453–455. doi: 10.1038/356453a0. [DOI] [PubMed] [Google Scholar]
- 47.Itzhaki L S, Neira J L, Ruiz-Sanz J, Prat Gay G d, Fersht A R. J Mol Biol. 1995;254:289–304. doi: 10.1006/jmbi.1995.0617. [DOI] [PubMed] [Google Scholar]
- 48.Neira J L, Davis B, Ladurner A G, Buckle A, Prat Gay G d, Fersht A R. Fold Des. 1996;3:189–208. doi: 10.1016/s1359-0278(96)00031-4. [DOI] [PubMed] [Google Scholar]
- 49.Neira J L, Itzhaki L S, Ladurner A G, Davis B, Prat Gay G d, Fersht A R. J Mol Biol. 1997;268:185–197. doi: 10.1006/jmbi.1997.0932. [DOI] [PubMed] [Google Scholar]
- 50.Matouschek A, Otzen D E, Itzhaki L S, Jackson S E, Fersht A R. Biochemistry. 1995;34:13656–13662. doi: 10.1021/bi00041a047. [DOI] [PubMed] [Google Scholar]
- 51.Smith C K, Bu Z M, Anderson K S, Sturtevant J M, Engelman D M, Regan L. Protein Sci. 1996;5:2009–2019. doi: 10.1002/pro.5560051007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muñoz V, Cronet P, López-Hernández E, Serrano L. Fold Des. 1996;1:167–178. doi: 10.1016/s1359-0278(96)00029-6. [DOI] [PubMed] [Google Scholar]
- 53.Hubbard S J, Thornton J M. naccess, Computer Program. University College, London: Department of Biochemistry and Molecular Biology; 1993. [Google Scholar]
- 54.Kraulis P. J Appl Crystallogr. 1991;24:946. [Google Scholar]
- 55.Merritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 56.Bacon D J, Anderson W F. J Mol Graph. 1988;6:219–220. [Google Scholar]