

Abstract
DNA glycosylases help maintain the genome by excising chemically modified bases from DNA. Escherichia coli 3-methyladenine DNA glycosylase I (TAG) specifically catalyzes the removal of the cytotoxic lesion 3-methyladenine (3mA). The molecular basis for the enzymatic recognition and removal of 3mA from DNA is currently a matter of speculation, in part owing to the lack of a structure of a 3mA-specific glycosylase bound to damaged DNA. Here, high-resolution crystal structures of Salmonella typhi TAG in the unliganded form and in a ternary product complex with abasic DNA and 3mA nucleobase are presented. Despite its structural similarity to the helix–hairpin–helix superfamily of DNA glycosylases, TAG has evolved a modified strategy for engaging damaged DNA. In contrast to other glycosylase-DNA structures, the abasic ribose is not flipped into the TAG active site. This is the first structural demonstration that conformational relaxation must occur in the DNA upon base hydrolysis. Together with mutational studies of TAG enzymatic activity, these data provide a model for the specific recognition and hydrolysis of 3mA from DNA.
Keywords: base excision, DNA repair, glycosylase, helix–hairpin–helix, 3-methyladenine
Introduction
The information encoded within the sequence and structure of DNA is vital to the survival of any organism. The integrity of the genome is constantly threatened by the chemical reactivity of the nucleobases, which are modified by a variety of alkylation, oxidation or radiative processes (reviewed in Friedberg et al, 2006). DNA alkylation by cellular metabolites, environmental toxins, or chemotherapeutic agents produces a wide spectrum of aberrant nucleotides that are cytotoxic or mutagenic, and hence can lead to cell death and heritable disease. A large number of alkylated purines, including cytotoxic 3-methyladenine (3mA), 7-methylguanine (7mG), and the highly mutagenic lesion 1,N6-ethenoadenine (ɛA), have been detected in humans after exposure to various carcinogens (Shuker et al, 1987). As a safeguard against alkylation damage, cells have devised a number of DNA repair strategies to remove these modifications and restore the DNA to an undamaged state. The base excision repair pathway is the principal mechanism by which alkylpurines are eliminated from the genome. DNA glycosylases initiate this pathway by locating and removing a specific type of modified base from DNA through cleavage of the C1′–N glycosylic bond.
Alkylpurine DNA glycosylases have been shown to be essential for the survival of both eukaryotic and prokaryotic organisms (Clarke et al, 1984; Chen et al, 1989), and have been identified in humans, yeast, and bacteria. Among these are Escherichia coli 3mA DNA glycosylase I (TAG) and II (AlkA), Thermotoga maritima methylpurine DNA glycosylase II (MpgII), Helicobacter pylori 3mA DNA glycosylase (MagIII), yeast methyladenine DNA glycosylase (MAG), and human alkyladenine DNA glycosylase (AAG) (Riazuddin and Lindahl, 1978; Thomas et al, 1982; Chen et al, 1990; O'Connor and Laval, 1991; Begley et al, 1999; O'Rourke et al, 2000). Although structurally unrelated, the human and bacterial alkylpurine glycosylases have evolved a common base-flipping mechanism for gaining access to damaged nucleobases in DNA (reviewed in Roberts and Cheng, 1998; Hollis et al, 2000b). The bacterial enzymes TAG, AlkA, and MagIII belong to the helix–hairpin–helix (HhH) superfamily of DNA glycosylases (Labahn et al, 1996; Nash et al, 1996; Drohat et al, 2002; Eichman et al, 2003). The HhH motif is used by hundreds of repair proteins for binding DNA in a sequence-independent manner (Doherty et al, 1996). Crystal structures of HhH glycosylases AlkA, hOgg1, EndoIII, and MutY in complex with DNA illustrate how the HhH motif is used as a platform for base flipping to expose damaged bases in DNA (Bruner et al, 2000; Hollis et al, 2000a; Fromme and Verdine, 2003b; Fromme et al, 2004).
Alkylpurine DNA glycosylases from bacteria have widely varying substrate specificities despite their structural similarity. TAG and MagIII are highly specific for 3mA (Bjelland et al, 1993; O'Rourke et al, 2000), whereas AlkA is able to excise 3mA, 7mG, and other alkylated or oxidized bases from DNA (McCarthy et al, 1984; Bjelland et al, 1994; Saparbaev et al, 1995). The importance of specificity during base excision is underscored by the fact that glycosylases must identify subtle alterations in base structure amidst a vast excess of normal DNA. Recognition of the substrate base must occur at two steps—interrogation of the DNA duplex during a processive search and direct read-out of the target base that has been flipped into the active site of the enzyme (Stivers and Jiang, 2003; Banerjee et al, 2006). Our structural understanding of 3mA processing by bacterial alkylpurine DNA glycosylases is currently limited to structures of TAG and MagIII bound to alkylated bases in the absence of DNA. Crystal structures of MagIII bound to 3mA and ɛA revealed that direct contacts to nucleobase substituent atoms are not necessary for binding alkylpurines in the binding pocket (Eichman et al, 2003). NMR studies of E. coli TAG bound to 3mA demonstrated that TAG makes specific contacts to the base, and that the enzyme lacks the hallmark catalytic aspartic acid present in all other HhH glycosylases (Nash et al, 1996; Drohat et al, 2002; Cao et al, 2003). Given the lack of DNA in these structures, the mechanism by which specific 3mA glycosylases locate and excise their target bases from DNA is currently a matter of speculation.
Presented here are the crystal structures of Salmonella typhi TAG alone and in complex with abasic DNA and 3mA, together with mutational studies of TAG enzymatic activity. TAG binds damaged DNA in a manner similar to other HhH glycosylases, but uses a different strategy to intercalate the DNA in order to gain access to the damage site. Surprisingly, the abasic ribose adopts two specific conformations, neither of which is fully flipped into the active site pocket as has been observed in all other glycosylase product complexes. Extensive interactions with the bases on both DNA strands provide a structural rationale for how TAG detects 3mA lesions within DNA. Inside the base binding pocket, a conserved glutamic acid has been identified to play a significant role in catalysis of base excision. A comparison of structures of HhH alkylpurine DNA glycosylases provides a basis for understanding the unique mechanisms by which 3mA is selected and removed from DNA.
Results and discussion
TAG from the bacterium S. typhi is 82% identical and 91% conserved overall to the E. coli enzyme. S. typhi TAG was crystallized alone and in complex with 3mA base and DNA containing a tetrahydrofuran (THF) abasic site analog. The THF moiety is isosteric with enzymatically generated apurinic sites, but is not prone to ring opening owing to the lack of a C1′ hydroxyl group (Takeshita et al, 1987). The crystal structures of TAG and the TAG/THF-DNA/3mA complex were determined using experimental phases from multi- and single-wavelength anomalous dispersion (MAD, SAD) experiments, respectively (Table I). A crystallographic model of the free protein, which consists of two TAG molecules in the asymmetric unit, was built into 1.5-Å MAD electron density (Supplementary Figure S1) and refined to a crystallographic residual of 0.161 (Rfree=0.196). Likewise, the model of the TAG/THF-DNA/3mA product complex (Figure 1) was built into 1.85-Å SAD experimental electron density (Supplementary Figure S1) and refined to a crystallographic residual of 0.175 (Rfree=0.198).
Table 1.
Data collection, phasing, and refinement statistics
| Unliganded TAG | TAG/THF-DNA/3mA | ||||
|---|---|---|---|---|---|
| Native | Se peak | Se remote | Se inflection | Se peak | |
| Cell parameters | |||||
| a | 57.5 Å | 57.5 Å | 102.0 Å | ||
| b | 63.7 Å | 63.9 Å | 102.0 Å | ||
| c | 62.1 Å | 62.1 Å | 55.5 Å | ||
| α | 90° | 90° | 90° | ||
| β | 106.9° | 107.0° | 90° | ||
| γ | 90° | 90° | 120° | ||
| Space group | P21 | P21 | P63 | ||
| Molecules/asu | 2 | 2 | 1 | ||
| Data collectiona | |||||
| Wavelength (Å) | 1.000 | 0.9793 | 0.9718 | 0.9795 | 0.9793 |
| Resolution (Å) | 50–1.5 | 50–1.5 | 50–1.5 | 50–1.5 | 50–1.85 |
| Unique reflections | 64 266 (4195) | 121 469 (7253) | 125 767 (8960) | 122 660 (7290) | 54 963 (5415) |
| Completeness (%) | 93.6 (61.1) | 89.7 (53.4) | 92.8 (65.8) | 90.4 (53.5) | 99.6 (97.9) |
| Redundancy | 7.0 (4.6) | 3.7 (2.6) | 3.8 (2.9) | 3.7 (2.6) | 6.4 (5.3) |
| Rsymb | 0.064 (0.247) | 0.089 (0.185) | 0.097 (0.176) | 0.095 (0.192) | 0.053 (0.433) |
| 〈I〉/〈σ(I)〉 | 23.8 (4.8) | 17.6 (4.1) | 17.2 (4.6) | 17.5 (3.8) | 27.2 (3.4) |
| Phasing | |||||
| Resolution (Å) | 50–1.5 | 50–1.5 | 50–1.5 | 50–1.85 | |
| Phasing powerc | 1.41 | 1.55 | — | — | |
| Rcullis (c/a)d | 0.63/0.69 | 0.59/0.65 | — | — | |
| Rcullis (anom) | 0.60 | 0.73 | 0.65 | 0.77 | |
| Refinement | |||||
| Resolution | 50–1.5 | 50–1.85 | |||
| No. of reflections | 60 990 | 26 778 | |||
| Rcryste | 0.161 | 0.175 | |||
| Rfreee | 0.196 | 0.198 | |||
| Number of atoms | |||||
| Protein | 2903 | 1450 | |||
| DNA | 0 | 518 | |||
| Solvent | 399 | 160 | |||
| Average B- factor (Å2) | 31.6 | 34.3 | |||
| R.m.s.d. bonds (Å) | 0.014 | 0.018 | |||
| R.m.s.d. angles (deg) | 1.427 | 1.868 | |||
| Values in parentheses refer to data in the highest resolution shell. | |||||
| Rsym=∑hkl∑j∣Ij−〈I〉∣/∑hkl∑j Ij where 〈I〉 is the mean intensity of j observations of reflection hkl and its symmetry equivalents. | |||||
| Phasing power=∑hkl FH/∑hkl∣FPH−FPH,calc∣. Value shown is the weighted average of centric and acentric data. | |||||
| Rcullis=(∑hkl∣∣FPH±FP∣−FH,calc∣)/∑hkl∣FPH−FP∣. c/a, centric/acentric data. | |||||
| Rcryst=∑hkl∣Fobs−kFcalc∣/∑hkl∣Fobs∣. Rfree=Rcryst for 5% of reflections that were not used in refinement (Brünger, 1992). | |||||
Figure 1.
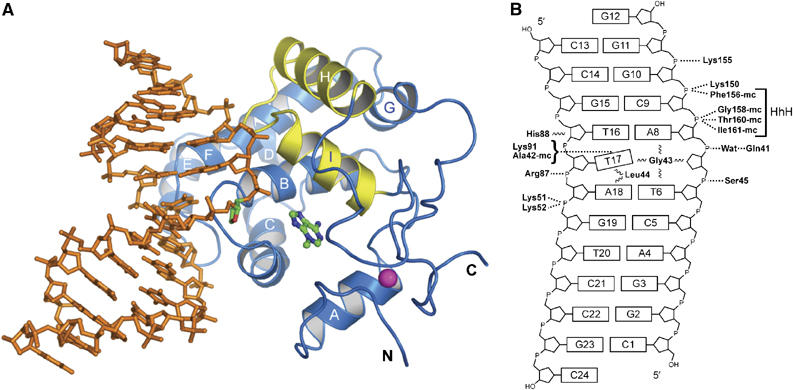
The structure of the TAG–DNA complex. (A) The crystallographic model of TAG (blue ribbons; yellow HhH motif) bound to DNA (orange sticks) and 3mA (ball-and-stick). The abasic site in the DNA is highlighted in green and the coordinated Zn2+ ion is shown as a magenta sphere. (B) Schematic representation showing the electrostatic (dashed lines) and van der Waals (wavy lines) interactions between protein side-chain and main-chain (mc) atoms with the DNA.
The crystal structures of S. typhi TAG are consistent with NMR structures of the E. coli enzyme that identified TAG as a member of the HhH superfamily of DNA glycosylases (Drohat et al, 2002). TAG adopts a globular fold consisting of an α-helical domain that contains the HhH motif (helices H and I) and a second, unique Zn2+-binding domain that tethers the N- and C-termini (Figure 1A) (Kwon et al, 2003). The 3mA binding pocket is located at the interface between the two domains (Figure 1A) (Cao et al, 2003). Superposition of the S. typhi (crystal) and E. coli (NMR) structures shows that the protein backbones and positions of bound 3mA are virtually identical (with an r.m.s. deviation of 1.8 Å for all main-chain atoms; Supplementary Figure S2). Surprisingly, the largest differences between the two structures occur in the positions of two conserved tryptophan side chains in the 3mA binding pocket. Each of the indole rings of Trp 6 and Trp 21 are rotated ∼120° between the two models (Supplementary Figure S2). Based on the high degree of sequence and structural conservation between S. typhi and E. coli TAG, these differences are likely an artifact of structure determination and not inherent differences between the two orthologs.
DNA binding by TAG
The HhH glycosylases use a common mechanism for binding DNA. These proteins anchor both strands of the DNA duplex from the minor groove side through van der Waals and polar interactions with the bases and the phosphate backbone. Main-chain atoms from the HhH hairpin form hydrogen bonds with two phosphate groups immediately 3′ to the lesion, whereas positively charged side chains from a conserved protein loop engage the non-lesioned strand. An intercalating side chain occupies (or ‘plugs') the gap in the DNA left by the flipped-out nucleotide, and a second side chain wedges into the non-lesioned DNA opposite the flipped-out nucleotide. Collectively, these interactions stabilize a 60–70° bend in the duplex and help the protein gain access to the modified base.
TAG binds DNA similarly to other HhH glycosylases (Bruner et al, 2000; Hollis et al, 2000a; Fromme and Verdine, 2003b; Fromme et al, 2004), with subtle unique differences that categorize TAG as a divergent member of the superfamily and that likely result in its high specificity for positively charged 3mA bases. The DNA is anchored to the protein by three hairpin loops formed from helices B/C, E/F, and the HhH motif (Figure 1A). Basic side-chain and main-chain atoms from the HhH motif bind the phosphate groups 3′ to the abasic site, whereas basic residues from the E/F loop contact the DNA backbone on the non-lesioned strand (Figure 1B). The loop between helices B and C inserts into the abasic gap in the DNA duplex, and the details will be discussed below. The DNA is kinked at the THF site by ∼62°, with the two duplex arms on either side of the bend primarily B-form DNA (Figure 2B). Interestingly, there are no protein–DNA contacts with the 5 base pairs upstream of the lesion (C1·G23–C5·G19), and the B-factors for the DNA are significantly higher at that end. The structures of TAG in the free state and when bound to product DNA are essentially identical, with r.m.s. deviations of 0.6 Å (backbone atoms only) and 1.0 Å (all atoms) (Supplementary Figure S3). Thus, no significant protein movement is necessary to engage the DNA.
Figure 2.
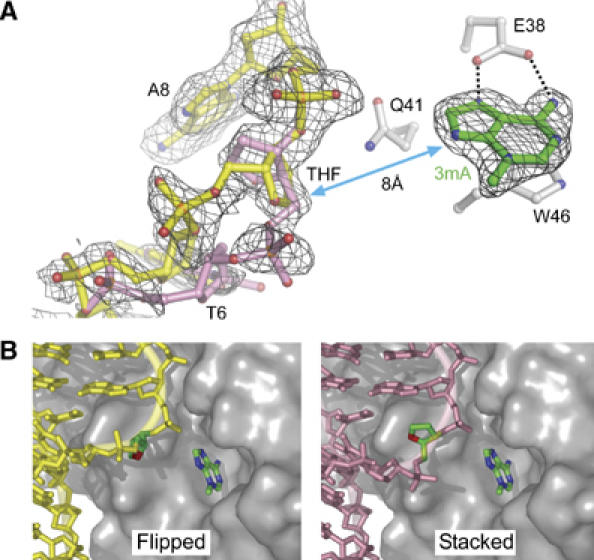
Two conformations of the abasic DNA backbone. (A) Refined models showing the flipped (yellow) and stacked (pink) conformations of the THF abasic site are superimposed on an unbiased composite omit electron density map (contoured at 2σ). Only density corresponding to nucleic acid is shown for clarity. The proximity of the DNA to the 3mA (green sticks) is highlighted by a double-sided arrow. (B) The flipped and stacked DNA conformations are shown separately against a van der Waals surface representation of the protein. THF and 3mA carbon atoms are highlighted in green.
TAG contains a unique HhH motif that accounts for about half of the polar interactions with the DNA backbone. Amide nitrogens from Phe156, Gly158, Thr160, and Ile161 form hydrogen bonds to the phosphate groups 3′ to the THF site (cytosine C9 and guanine G10) (Figure 1B). In contrast to DNA complexes of AlkA, hOgg1, and EndoIII, TAG does not coordinate a cation (Na+, Mg2+, or Ca2+) at the hairpin (Bruner et al, 2000; Hollis et al, 2000a; Fromme and Verdine, 2003b). Instead, a water molecule links the hairpin with the DNA backbone by coordinating in a tetrahedral arrangement only four ligands: the main-chain nitrogen of Val157, the amino Nζ nitrogen of Lys150, the O1P phosphate oxygen of guanine G10, and a water molecule (Supplementary Figure S4). Despite its structural divergence from other HhH glycosylases, TAG's HhH motif serves the same functional role of anchoring the protein to the DNA.
The abasic site in two conformations
One surprising aspect of the TAG/DNA complex structure is the conformational flexibility of the THF abasic site. This residue exists in two discrete orientations in the crystal (Figure 2). Both experimental MAD and unbiased composite omit electron density maps clearly show two equally occupied trajectories for the DNA backbone at residues T6 and THF7 (Figure 2A). In one orientation, the THF ribose ring is partially rotated ∼90° out of the helical base stack and into the minor groove toward the protein (‘flipped' conformation; Figure 2B). In the second orientation, the THF ribose remains stacked into the helix in its normal position in B-DNA. The shift in the position of the THF moiety is accompanied by a concomitant rotation of the DNA backbone that forces the THF 5′-phosphate to point either away from (flipped conformation) or toward (stacked conformation) the protein (Figure 2B). The largest deviations in the DNA backbone occur predominantly as rotations around the C3′–O3′ bonds (ɛ torsion angle) of nucleotides T6 and THF7 and around the O3′–P (ζ) bond, although the entire backbone of nucleotides C5, T6, and THF7 significantly deviates from that of B-DNA (Figure 2A). In addition to torsional rotation, the two DNA conformations differ by a 2 Å translation around thymine T6, a movement that affects the positions of both the backbone and thymine base. The slight positional disorder in thymine T6 is reflected in the discontinuous electron density and high B-factors of this residue.
The multiple conformations of the phosphate backbone are likely a consequence of the sharp kink in the DNA and the lack of specific protein–DNA contacts at the abasic site and in the duplex 5′ to the lesion. Surprisingly, both flipped and stacked orientations of the ribose ring make only nonspecific van der Waals contacts with TAG. Even in the flipped conformation, the abasic ribose is only partially rotated out of the DNA duplex and is located ∼8 Å away from the 3mA base bound in the active site pocket (Figure 2A). This unflipped ribose is in stark contrast to the structures of all other HhH glycosylases bound to abasic DNA. In these structures, the ribose is rotated a full 180° around the backbone and forms specific polar interactions inside the active site (Hollis et al, 2000a; Norman et al, 2001; Fromme and Verdine, 2003b). The structure of hOgg1 bound to THF-DNA shows the THF moiety in the same position as the ribose ring in the hOgg1/8-oxoG-DNA substrate complex (Norman et al, 2001), indicating that the protein–DNA interactions necessary to stabilize the flipped nucleotide in the hOgg1 active site need not involve the 8-oxoG base itself. In contrast, the TAG/THF-DNA/3mA structure suggests that the intact glycosylic bond is necessary for TAG to hold 3mA DNA substrate in a specific extrahelical orientation, and that the bound abasic DNA product relaxes its conformation after 3mA excision.
Interrogation of a DNA lesion
The HhH glycosylases use a common strategy for probing the DNA bases within the double helix. A bulky, intercalating side chain (typically, Leu, Asn, Gln, or Arg) plugs the gap in the DNA left by the flipped-out nucleotide, and a second side chain (Phe, Tyr, Leu, or Pro) wedges between the bases opposite the flipped-out nucleotide. Both plug and wedge residues are important for stabilizing the conformation of the DNA necessary to accommodate an extrahelical nucleotide. It has recently been suggested that the wedge residue is important for locating damaged DNA during the search process (Banerjee et al, 2006).
TAG interacts with the DNA bases in a manner different from the other HhH glycosylases. Most notable is the intercalation of Gly43 at the tip of the B/C loop into the abasic gap (Figure 3). To our knowledge, this is the first reported case of a base-flipping enzyme that intercalates backbone atoms, as opposed to a bulky side chain, into the DNA base stack. Second, the side chain of Leu44 serves as the wedge residue and intercalates between thymine T17 and adenine A18 bases on the non-lesioned strand. Interestingly, both plug and wedge residues are located on the same secondary structure element (B/C loop), and not on both the B/C-(plug) and E/F-(wedge) loops, as is observed in all other HhH glycosylase structures (Supplementary Figure S5). Thus, TAG uses a modified strategy to form the plug and wedge interactions present in all DNA glycosylases (Barrett et al, 1998; Parikh et al, 1998; Bruner et al, 2000; Hollis et al, 2000a; Fromme et al, 2004; Banerjee et al, 2006). The conservation of this base intercalation mechanism in divergent protein architectures highlights the importance of this interaction in DNA glycosylase function.
Figure 3.
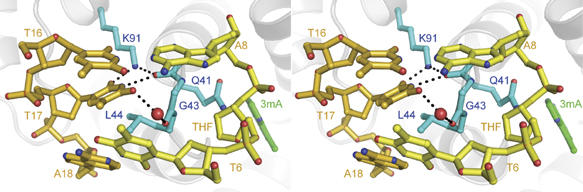
TAG interrogation of the DNA base stack. TAG binds the DNA damage site by intercalating side chains (blue) into both the lesioned (yellow) and non-lesioned (gold) DNA strands. Hydrogen bonds to the estranged thymine are shown as dashed lines and the 3mA base is colored green.
The functional significance of the Gly43 plug and Leu44 wedge identified in the TAG/DNA crystal structure was tested by measuring the glycosylase activity of TAG site-directed mutants. The rate of 3mA excision was measured using genomic DNA treated with the alkylating agent N-methyl-N-nitrosourea (Bjelland et al, 1994). This agent primarily produces 7mG and 3mA lesions in DNA, and TAG selectively excises 3mA but not 7mG (Bjelland et al, 1993). Substituting Gly43 with a leucine residue decreased the glycosylase activity by two orders of magnitude (Table II). This decrease may partially be a result of reduced stability of the Gly43Leu protein, which is ∼50% denatured under the conditions of our assay (Table II and Supplementary Figure S6). It is likely that the remaining 50-fold decrease in 3mA excision activity, which is measured by necessity under subsaturating conditions (see Materials and methods), is a result of compromised DNA binding activity of Gly43Leu. The reciprocal experiment using the closely related enzyme MagIII showed that removal of the bulky asparagine plug (Asn42Ala) enhanced DNA binding (Eichman et al, 2003). It is interesting to note that TAG and MagIII, both highly specific for 3mA, show greater base excision or DNA binding activity in the absence of a bulky side-chain plug.
Table 2.
Glycosylase activities for wild-type and active site mutants of TAG
| Mutant | Conservationa | 3mA excision rateb (10−3 min−1 μM−1) | Relative activity (mutant/WT) |
|---|---|---|---|
| WT | — | 233.6±16.4 | (1.0) |
| L44A | Invariant | 6.5±4.4 | 2.8 × 10−2 |
| G43L | Invariant | 2.5±0.8 | 1.1 × 10−2 |
| K91A | Invariant | 26.9±1.5 | 1.1 × 10−1 |
| Q41A | Invariant | 36.2±3.1 | 1.5 × 10−1 |
| E38A | High | 0.7±0.1 | 3.0 × 10−3 |
| Y16F | Invariant | 19.0±4.6 | 8.1 × 10−2 |
| W46A | Invariant | 20.7±7.4 | 8.9 × 10−2 |
| W6A | Moderate | 29.2±16.9 | 1.2 × 10−1 |
| Y13F | Low | 54.8±39.3 | 2.3 × 10−1 |
| T160V | Moderate | 273.8±68.3 | 1.2 |
| S45A | Moderate | 230.0±7.8 | 1.0 |
| Q167A | Moderate | 107.2±55.7 | 4.6 × 10−1 |
| High, moderate, and low conservation refers to 1, 2–20, and >20 amino-acid substitutions, respectively, out of 250 total aligned TAG sequences (Altschul et al, 1997). | |||
| Second-order rate constants for [3H]3mA release from N-[3H]methyl-N-nitrosourea-treated genomic DNA. | |||
Substitution of Leu44 with alanine decreased the glycosylase activity 36-fold in comparison to wild-type TAG (Table II). A comparable effect of the wedge residue on DNA binding and glycosylase activity has been observed for MagIII (Eichman et al, 2003) and MutY (Chmiel et al, 2003). The predominance of phenylalanine or tyrosine wedge residues in DNA glycosylases MutY, hOgg1, and MutM suggests that aromatic stacking is important for intercalation of the bases opposite the lesion. However, the presence of leucine wedges in TAG and EndoIII (Fromme and Verdine, 2003b) and the observation that an E. coli MutY Tyr82Leu wedge mutant has similar activity compared to wild-type MutY (Livingston et al, 2005) demonstrate that van der Waals contacts are sufficient in this capacity.
As a result of the Leu44 wedge interaction, the estranged thymine T17 is highly distorted opposite the abasic site (Figure 3). This distortion is manifest as a large tilt (τ=−22.5°) and twist (ω=−8.4°) for the T16/T17 base step as compared to B-DNA (τB-DNA=−0.1°; ωB-DNA=36.0°). Such a large distortion in the estranged base has been observed in the structures of MutY and MutM bound to DNA (Fromme and Verdine, 2003a; Fromme et al, 2004; Banerjee et al, 2006). The estranged thymine is held in this distorted conformation in the TAG/DNA complex through an extensive hydrogen bond network involving lysine 91 at the N-terminal end of helix F and the B/C loop backbone (Figure 3). The Nζ amino group of Lys91 donates hydrogen bonds to the O2 keto oxygen of thymine T17 and to the backbone carbonyl oxygen of Ala42. The Ala42 backbone oxygen also accepts a hydrogen bond from the N3 nitrogen of thymine T17 to form a closed T17-Lys91-Ala42 network. Substituting Lys91 with alanine reduced the rate of 3mA excision eight-fold (Table II). In addition to the T17-Lys91-Ala42 network, a water-mediated hydrogen bonding interaction links the Gly43 carbonyl oxygen from the B/C loop to the estranged thymine T17 at its O4 keto oxygen. Thus, TAG makes intimate and specific contacts with the estranged thymine base in addition to the van der Waals interactions from the intercalating residues.
The extensive interactions between TAG and the estranged base help explain the specificity of this enzyme for 3mA and 3mG residues. The same hydrogen bonds between TAG and thymine observed in the crystal structure can be formed with a cytosine but not a purine base. A model constructed with a cytosine in place of the thymine shows that a cytosine would be slightly rotated toward the minor groove of the DNA to make favorable van der Waals contacts with the surface of the protein. Alternatively, purine bases are clearly sterically excluded from this position. Specific interactions between the protein and the estranged nucleobase commonly account for HhH glycosylase substrate specificity. For example, the specificity of hOgg1 for 8oxoG·C base pairs can be rationalized by the extensive contacts between the estranged cytosine and Asn149, Arg154, and Arg204 (Bruner et al, 2000). AlkA, on the other hand, does not form hydrogen bonds with the estranged base, which partially accounts for its broad specificity (Hollis et al, 2000a).
The effect of Leu44 on the estranged base and on TAG glycosylase activity contributes to the growing body of evidence suggesting that this wedge interaction helps the enzyme find damaged base pairs among a vast excess of unmodified DNA. It has been shown that DNA glycosylases search for damage by a processive mechanism of sliding along DNA (Francis and David, 2003; Blainey et al, 2006). Recently, a series of crystal structures of MutM in complex with undamaged DNA demonstrate that a phenylalanine wedge (Phe114) intercalates into the base stack and severely buckles the surrounding base pairs (Banerjee et al, 2006). These structures suggest that such a probe in the nucleobase stack might serve as an early test of base-pair stability and thus allow the enzyme to flip into the active site only those bases whose Watson–Crick pairing has been destabilized by the presence of a modification. The distortion to the estranged thymine imposed by the TAG Leu44 wedge is consistent with the idea that TAG uses this residue to probe for DNA damage. The network of hydrogen bonds to the estranged base would help lock the protein in place to facilitate base flipping into the active site.
3mA selection and hydrolysis in the TAG active site
The active site clefts of the HhH glycosylases have distinct chemical and physical characteristics that are suited for a particular nucleobase substrate and are located adjacent to the DNA-binding elements described above. The location of the active site with respect to the DNA lesion is important when considering how glycosylases couple damage recognition, nucleotide flipping, substrate specificity in the binding pocket, and base excision. The proximity of the TAG base binding cleft to the DNA lesion was identified by co-crystallization of all three components in the TAG/THF-DNA/3mA ternary product complex. The 3mA base was clearly observed in the experimental electron density to reside deep in the active site pocket (Figures 2A and 4). The addition of free 3mA to the crystallization experiment increased the size and quality of the crystals, suggesting that the ternary complex with bound 3mA is more stable than a binary TAG/THF-DNA complex.
Figure 4.
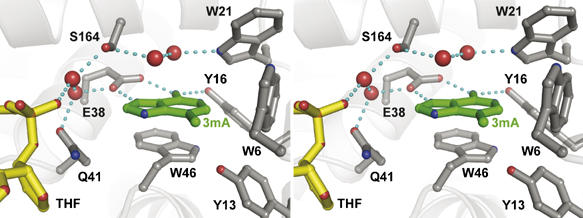
The nucleobase binding pocket of TAG. Protein residues are shown in gray, 3mA in green, DNA in yellow, and waters as red spheres. Hydrogen bonds are depicted as blue dashed lines.
The TAG active site is perfectly shaped to accommodate 3mA. An unbiased composite omit electron density map clearly distinguishes the exocyclic 3-methyl and 6-amino substituents, indicating that the base binds in one orientation (Figure 2A). The nucleobase ring nitrogen N9 that is linked to the ribose before catalysis points toward the bound DNA, suggesting that the crystal structure reflects a catalytically competent orientation of 3mA. The 3mA is constrained by hydrogen bonding and aromatic stacking interactions with active site residues (Figure 4). As observed in the NMR structure of E. coli TAG bound to 3mA (Cao et al, 2003), the side chains of Glu38 and Tyr16 line the back of the active site pocket and form hydrogen bonds to the Hoogsteen and Watson–Crick faces of 3mA, respectively. The side chains of Trp46 and Trp6 pack against one face and edge of the nucleobase ring, whereas the opposite face is contacted by water molecules held in place by hydrogen bonds from peripheral active site residues.
Despite the 8 Å distance and lack of direct contacts between the THF moiety and 3mA, the DNA damage/abasic site is linked to the base binding pocket through a series of interactions that provide insight into the base-flipping step. A water-mediated hydrogen bond network extends from Glu38 in the active site to the phosphate 3′ to the THF moiety (Figure 4). Importantly, an invariant glutamine (Gln41) residue is positioned directly between 3mA and THF (Figures 2A and 4), and is located on the B/C loop that plugs the abasic gap (Figure 3). Substitution of this residue with alanine reduces the rate of base excision ∼6-fold with respect to wild-type TAG (Table II). On the basis of its location at the active site/THF interface and its effect on TAG activity, it is intriguing to speculate that Gln41 is involved in guiding 3mA into the base binding pocket during base flipping. Independent of whether 3mA rotates around the phosphate backbone through major or minor grooves, the modified nucleobase will likely make its first contact with Gln41. Interestingly, this is the only side chain in the base binding pocket that shifts position upon DNA binding.
The aromatic character and shape of TAG's nucleobase binding pocket is particularly well suited for interactions with alkylated purines. Electron-rich aromatic active sites that stack against electron-deficient, ring-substituted purines are common among the bacterial and human 3mA DNA glycosylases, and this feature has been shown to be important for 3mA specificity (Labahn et al, 1996; Hollis et al, 2000a; Lau et al, 2000; Eichman et al, 2003). In TAG, substitution of Trp46 with alanine had a 10-fold effect on base excision activity (Table II). A Trp6Ala mutant, on the other hand, was severely destabilized with respect to wild-type TAG (Supplementary Figure S6), suggesting that Trp6 is important for the structural integrity of the active site.
Despite the similarities in aromaticity among 3mA base binding pockets, TAG's active site differs significantly from other glycosylases in two aspects. First, TAG lacks the conserved aspartic acid that is located 8–9 residues C-terminal to the HhH motif (Figure 5) and that is essential to the base excision activity in other HhH glycosylases (Thayer et al, 1995; Labahn et al, 1996; Guan et al, 1998; Mol et al, 2002; Norman et al, 2003). The lack of this catalytic residue has led to the suggestion that excision of a destabilized 3mA lesion does not require the same catalytic assistance as other more stable alkylpurines (Drohat et al, 2002; Stivers and Jiang, 2003), and that TAG must therefore use a unique mechanism of 3mA excision (Cao et al, 2003). Second, specific hydrogen bonds between 3mA and active site residues analogous to Glu38 and Tyr16 in TAG were not observed in a MagIII/3mA complex (Eichman et al, 2003), nor were they predicted from structures of AlkA or AAG (Hollis et al, 2000a; Lau et al, 2000). It seems likely, therefore, that the 3mA-specific contacts from Glu38 and Tyr16 contribute to TAG's narrow substrate specificity (Drohat et al, 2002). Indeed, the Glu38 side chain has been shown to sterically exclude N7-substituted methylpurine bases from E. coli TAG (Figure 4 and Cao et al, 2003).
Figure 5.
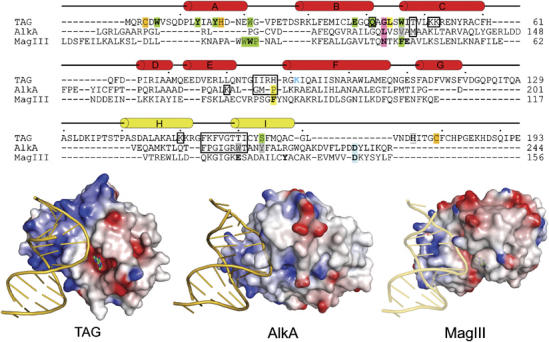
Comparison of 3-methyladenine DNA glycosylases. Top: structure-based sequence alignment of TAG, AlkA, and MagIII shows the relative positions of residues important for DNA binding and base excision. TAG secondary structure elements are shown schematically, with the HhH motif colored yellow. Residues contacting the DNA backbone are boxed, intercalating plug (pink) and wedge (yellow) residues are highlighted, and side chains contacting the estranged base are labeled blue. Side chains confirmed (green) or postulated (gray) to contact 3mA in the base binding pocket are highlighted. Residues verified biochemically to affect substrate binding or catalysis are shown in boldface and the catalytic aspartates in AlkA and MagIII are shaded blue. TAG residues that coordinate Zn2+ are shaded orange. Bottom: crystal structures of TAG/DNA/3mA, AlkA/DNA, and MagIII/3mA (with modeled DNA) are shown. Protein solvent-accessible surfaces are colored according to the electrostatic potential (blue, positive; red, negative). An alternate version of this figure showing all HhH glycosylase/DNA complexes is available as Supplementary data.
To gain insight into nucleobase specificity and excision by TAG, the enzymatic contributions of Glu38 and Tyr16 were examined by mutational studies. Steric exclusion of 7mG by Glu38 was tested by measuring the ability of a Glu38Ala mutant to excise 7mG from a defined oligonucleotide substrate. Wild-type TAG is unable to cleave 7mG from this substrate (Supplementary Figure S7). Removal of the glutamate side chain in the Glu38Ala mutant did not result in any measurable 7mG release even after 24 h (Supplementary Figure S7). Thus, TAG's inability to excise 7mG is not merely a consequence of steric exclusion by Glu38. Using the genomic DNA substrate, we found that substitution of Glu38 with alanine reduced the rate of 3mA excision 333-fold with respect to wild-type TAG, whereas a Tyr16Phe mutant had 12-fold reduced activity (Table II). Although the observed rate of 3mA excision in our assay reflects both binding and catalysis (see Materials and methods), it is unlikely that substrate binding fully accounts for the reduced activity of the Glu38Ala mutant. We do not expect the large difference in observed rate constants between Glu38 and Tyr16 mutants to be a consequence of the one additional hydrogen bond that Glu38 contributes to 3mA, as Glu38Ala and Tyr16Ala mutations each reduced the 3mA binding affinity for E. coli TAG by ∼15-fold (Cao et al, 2003). Likewise, the observation that wild-type E. coli TAG binds weaker to positively charged 3,9-dimethyladenine base than to neutral 3mA (Cao et al, 2003) suggests that the reduction in base excision by the Glu38Ala mutant is not due to a loss of the electrostatic interaction between Glu38(−) and DNA-3mA(+). Although the precise mechanism for 3mA excision remains to be determined, these data clearly demonstrate that Glu38 has a significant effect on base excision, and are consistent with the idea that TAG is specific for destabilized 3mA lesions simply because it lacks the catalytic power to remove the more stable alkylpurine adducts from DNA (Stivers and Jiang, 2003).
Comparison of alkylpurine DNA glycosylases
The structures of TAG and AlkA bound to DNA (Figure 5) highlight important features that provide a physical basis for substrate selectivity by alkylpurine glycosylases. First, the TAG–DNA contact surface is more extensive than that of AlkA. TAG forms additional van der Waals and electrostatic interactions with the non-lesioned strand that are not present in AlkA. Additionally, DNA bound to TAG shows less backbone distortion and a closer resemblance to canonical B-DNA than in any of the other DNA complexes of HhH superfamily members (Supplementary Figure S5). This difference is not likely an artifact of the abasic THF moiety as DNA containing this analog was observed in structures of EndoIII and hOgg1 to be highly distorted as a consequence of being pulled into the active site (Norman et al, 2001; Fromme and Verdine, 2003b). The base binding pockets of TAG and MagIII are highly electronegative and provide a snug fit for 3mA, in contrast to AlkA's electropositive, shallow active site surface (Figure 5). This difference helps to explain the exquisite specificity of TAG and MagIII for positively charged 3mA, and suggests that the most important requirement for 3mA excision is a high-affinity binding pocket.
Based on the structures of TAG and AlkA bound to DNA, we constructed a model for TAG in complex with a 3mA-DNA substrate that illustrates a likely mechanism for 3mA excision (Figure 6). The model was constructed by superposition of the DNA from the AlkA-DNA complex (Hollis et al, 2000a) onto the TAG/DNA/3mA structure, while retaining the position of the estranged thymine, flanking base pairs, and 3mA base from the TAG structure. This model confirms that the positions of 3mA and abasic DNA in the TAG crystal structure are aligned in biologically relevant orientations with respect to one another. The redirection of the phosphate backbone necessary to link the damage site to the 3mA base illustrates that the structure of the DNA in the TAG/THF-DNA/3mA product complex is relaxed relative to the substrate complex before hydrolysis of the glycosylic bond. This supports a previously described ground-state destabilization mechanism for catalysis of base excision (Cao et al, 2003; Stivers and Jiang, 2003). Collectively, TAG's enhanced interactions with both the non-lesioned strand and the 3mA base, together with the large distance between the abasic moiety and TAG's active site in the product complex argue that the 3mA glycosylic bond is strained in the substrate complex. This strain would be relieved upon cleavage of the glycosylic bond, allowing the DNA to relax to the position observed in the crystal structure.
Figure 6.
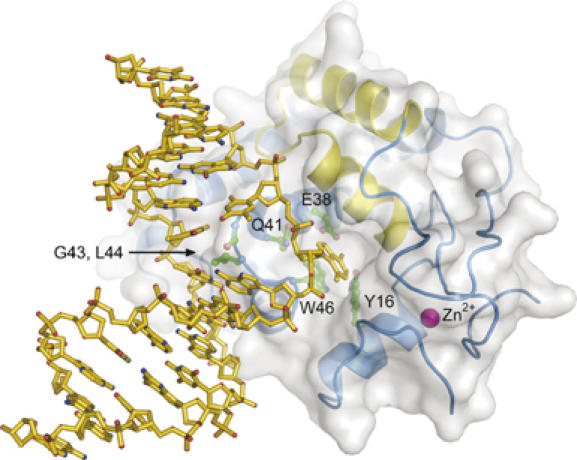
Model of a TAG/3mA-DNA substrate complex. TAG side chains important for DNA intercalation (Gly43, Leu44) and 3mA binding (Glu38, Trp46, Tyr16, Gln41) are shown as green ball-and-sticks. See text for details.
Conclusions
The crystal structures of S. typhi TAG alone and bound to abasic-DNA and 3mA base provide the first structural information for how a highly specific alkylpurine DNA glycosylase engages damaged DNA. In contrast to other glycosylase-DNA structures, the abasic ribose in the TAG complex is not fully rotated into the active site, suggesting that a conformational relaxation in the DNA takes place after base hydrolysis. TAG stabilizes damaged DNA differently than other HhH glycosylases by inserting a single hairpin loop into both strands of the DNA duplex. Specific interactions with the DNA bases help to explain the enzyme's preference for 3mA bases. Results from mutagenesis studies illustrate the functional significance of key residues identified in the crystal structure, and reveal an important catalytic dependence on a highly conserved glutamate residue (Glu38) in the base binding pocket. The crystal structures and mutational data are consistent with a model in which conformational strain in the 3mA DNA substrate drives base excision by destabilizing the ground state of the reaction.
Materials and methods
TAG purification and crystallization
S. typhi was expressed as an N-terminal His10-fusion protein from a pET-19b plasmid (Novagen). E. coli C41 cells transformed with the TAG/pET-19b plasmid were propagated in LB media supplemented with 5 μM ZnSO4, and protein was overexpressed for 4 h at 25°C upon addition of 0.5 mM IPTG. Cells were harvested in 50 mM Tris buffer (pH 7.5), 500 mM NaCl, and 10% glycerol and lysed with an Avestin Emulsifier C3 homogenizer operating at ∼20 000 psi. TAG protein was purified using Ni-NTA (Qiagen) affinity chromatography. After cleavage of the His10 tag, TAG was further purified by heparin affinity and gel filtration chromatography to >99% homogeneity as estimated by Coomassie staining. Protein was concentrated to 8 mg/ml and stored in 20 mM Tris (pH 8.5), 5% glycerol, 100 mM NaCl, 2 mM DTT, and 0.1 mM EDTA.
Selenomethionyl-substituted (SeMet) TAG was prepared similar to wild-type protein, except that the protein was overexpressed under conditions that suppress normal methionine biosynthesis (Van Duyne et al, 1993). Briefly, SeMet TAG was overexpressed for 16 h at 25°C in C41 cells grown in minimal media supplemented with 70 mg/ml selenomethionine (Acros Organics). After the Ni-NTA step, 5 mM methionine and 20 mM DTT were added to all buffers for the remainder of the purification.
Crystals of unliganded TAG were grown at 21°C by vapor diffusion, in which drops containing equal volumes of protein (8 mg/ml) and reservoir (30% PEG 200, 5% PEG 3000, 100 mM MES pH 6.0) were equilibrated against the reservoir. Crystals grew as single blocks (25 × 25 × 25 μm3) and were used as microseeds for a second crystallization experiment using a reservoir solution containing 16% PEG 200, 5% PEG 3000, and 100 mM MES pH 6.0. Crystals grown from seeds appeared as larger single blocks (100 × 100 × 100 μm3) after 1–2 days, and were flash-frozen in liquid nitrogen for X-ray data collection. To crystallize the TAG/THF-DNA/3mA complex, 0.23 mM TAG was preincubated for 15 min at 4°C with 0.27 mM DNA (d(CGGACTXACGGG)/d(CCGTTAGTCCGC), where X is a THF abasic analog) and 2 mM 3mA. Crystals were grown at 21°C by vapor diffusion using equal volumes of protein/DNA/3mA and reservoir (2 M (NH4)2SO4, 2% PEG 400, 100 mM HEPES pH 7.5) solutions. The crystals grew as hexagonal rods (75 × 75 × 500 μm3) in 1–2 days, and were soaked in 2 M sodium malonate (pH 7.5) before flash-freezing.
X-ray data collection, phasing, and structure refinement
X-ray diffraction data (Table I) on flash-frozen TAG and TAG/THF-DNA/3mA crystals were collected at beamline 22-ID at the Advanced Photon Source (Argonne, IL) and processed using the HKL 2000 package (Otwinowski and Minor, 1997). Data collection statistics are summarized in Table I.
Experimental X-ray phases for unliganded and DNA-bound TAG structures were obtained from MAD and SAD experiments, respectively, using crystals grown with SeMet-substituted protein. Diffraction data (Table I) were collected at energies corresponding to the selenium peak, inflection point, and high-energy remote settings (TAG) and at the peak energy only (TAG/DNA). Selenium positions (nine in TAG and five in TAG/DNA) in the asymmetric unit were located and refined using the program SOLVE (Terwilliger and Berendzen, 1999). Density modification and phase calculation were carried out using RESOLVE (Terwilliger, 2000). The protein chain was built de novo into 1.5-Å electron density from the TAG-only crystals. This model was docked into experimental SAD density for the TAG/DNA complex, followed by manual building of the DNA and 3mA portions of the model.
The models were refined using experimental phases and amplitudes from native (TAG, 50–1.5 Å) and SeMet (TAG/DNA, 50–1.85 Å) diffraction data against a maximum likelihood target as implemented in REFMAC 5.1 (Murshudov et al, 1997). Improvements to the models were guided by manual inspection of σA-weighted 2mFo−DFc and mFo−DFc electron density maps, and were judged successful by a decrease in Rfree during refinement. Anisotropic B-factors were refined explicitly for each atom in the TAG structure, and translation/libration/screw-rotation (TLS) refinement in REFMAC was used to model anisotropic motion of four protein/DNA domains in the complex. Individual anisotropic B-factors were derived from the refined TLS parameters and held fixed during subsequent rounds of refinement, which resulted in a decrease in both R and Rfree and a noticeable improvement in the electron density maps. Protein and DNA models were validated using PROCHECK (Laskowski et al, 1993), and DNA parameters were analyzed using CURVES 5.2 (Lavery and Sklenar, 1988).
The model coordinates, experimental phases, and structure factor amplitudes for TAG and TAG/THF-DNA/3mA structures have been deposited in the Protein Data Bank under accession numbers 2OFK and 2OFI, respectively.
Mutagenesis and enzyme activity assays
Mutations were made in the TAG expression plasmid using the Quik Change Site-Directed Mutagenesis kit (Stratagene), and they were verified by DNA sequencing. Mutant proteins were expressed and purified in the same manner as the wild-type enzyme, but without the final gel filtration step.
DNA glycosylase activity assays for 3mA excision were performed similar to the method described previously (O'Rourke et al, 2000; Eichman et al, 2003). The reaction mixture (50 μl) contained 6 μM enzyme and 3000 c.p.m. of N-[3H]methyl-N-nitrosourea-treated calf thymus DNA in activity buffer (50 mM HEPES pH 7.5, 100 mM KCl, 10 mM DTT, 1 mM EDTA). Reactions were incubated at 37°C and terminated at various time points by ethanol precipitation of the DNA. The release of radioactive bases into the soluble fraction was quantitated by liquid scintillation counting. Rate constants were determined from single-exponential fits to data from three different experiments for each mutant and then corrected for the concentration of each enzyme. For this assay, the enzyme concentration was subsaturating with respect to substrate at the highest concentration of enzyme tested (41 μM). The failure to saturate might be caused by nonspecific binding of TAG to the vast excess of unmodified bases in the genomic DNA substrate. The observed second-order rate constants were shown to be linearly dependent on enzyme concentration up to at least 40 μM, and therefore reflect both binding and catalysis under these conditions.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Eric Warren and Jami O'Quin for their assistance during X-ray data collection, and also Kyle Nordquist, Joel Harp, and the staff at the Southeast Regional Collaborative Access Team (SER-CAT) at the Advanced Photon Source (Argonne, IL). We thank Patrick O'Brien for reading the manuscript, and Tom Ellenberger, James Stivers, and Barry Gold for stimulating discussions. This work was funded by the American Cancer Society—Research Scholar Grant (RSG-07-063-01-GMC to BFE) and Institutional Research Grant (IRG-58-009-47) to the Vanderbilt-Ingram Cancer Center, and by the Sartain-Lanier Family Foundation.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Santos WL, Verdine GL (2006) Structure of a DNA glycosylase searching for lesions. Science 311: 1153–1157 [DOI] [PubMed] [Google Scholar]
- Barrett TE, Savva R, Panayotou G, Barlow T, Brown T, Jiricny J, Pearl LH (1998) Crystal structure of a G:T/U mismatch-specific DNA glycosylase: mismatch recognition by complementary-strand interactions. Cell 92: 117–129 [DOI] [PubMed] [Google Scholar]
- Begley TJ, Haas BJ, Noel J, Shekhtman A, Williams WA, Cunningham RP (1999) A new member of the endonuclease III family of DNA repair enzymes that removes methylated purines from DNA. Curr Biol 9: 653–656 [DOI] [PubMed] [Google Scholar]
- Bjelland S, Birkeland NK, Benneche T, Volden G, Seeberg E (1994) DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the AlkA enzyme in Escherichia coli. J Biol Chem 269: 30489–30495 [PubMed] [Google Scholar]
- Bjelland S, Bjoras M, Seeberg E (1993) Excision of 3-methylguanine from alkylated DNA by 3-methyladenine DNA glycosylase I of Escherichia coli. Nucleic Acids Res 21: 2045–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS (2006) A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc Natl Acad Sci USA 103: 5752–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner SD, Norman DP, Verdine GL (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 403: 859–866 [DOI] [PubMed] [Google Scholar]
- Brünger AT (1992) Free R-value—a novel statistical quantity for assessing the accuracy of crystal-structures. Nature 355: 472–475 [DOI] [PubMed] [Google Scholar]
- Cao C, Kwon K, Jiang YL, Drohat AC, Stivers JT (2003) Solution structure and base perturbation studies reveal a novel mode of alkylated base recognition by 3-methyladenine DNA glycosylase I. J Biol Chem 278: 48012–48020 [DOI] [PubMed] [Google Scholar]
- Chen J, Derfler B, Maskati A, Samson L (1989) Cloning a eukaryotic DNA glycosylase repair gene by the suppression of a DNA repair defect in Escherichia coli. Proc Natl Acad Sci USA 86: 7961–7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Derfler B, Samson L (1990) Saccharomyces cerevisiae 3-methyladenine DNA glycosylase has homology to the AlkA glycosylase of E. coli and is induced in response to DNA alkylation damage. EMBO J 9: 4569–4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmiel NH, Livingston AL, David SS (2003) Insight into the functional consequences of inherited variants of the hMYH adenine glycosylase associated with colorectal cancer: complementation assays with hMYH variants and pre-steady-state kinetics of the corresponding mutated E. coli enzymes. J Mol Biol 327: 431–443 [DOI] [PubMed] [Google Scholar]
- Clarke ND, Kvaal M, Seeberg E (1984) Cloning of Escherichia coli genes encoding 3-methyladenine DNA glycosylases I and II. Mol Gen Genet 197: 368–372 [DOI] [PubMed] [Google Scholar]
- Doherty AJ, Serpell LC, Ponting CP (1996) The helix–hairpin–helix DNA-binding motif: a structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res 24: 2488–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drohat AC, Kwon K, Krosky DJ, Stivers JT (2002) 3-Methyladenine DNA glycosylase I is an unexpected helix–hairpin–helix superfamily member. Nat Struct Biol 9: 659–664 [DOI] [PubMed] [Google Scholar]
- Eichman BF, O'Rourke EJ, Radicella JP, Ellenberger T (2003) Crystal structures of 3-methyladenine DNA glycosylase MagIII and the recognition of alkylated bases. EMBO J 22: 4898–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis AW, David SS (2003) Escherichia coli MutY and Fpg utilize a processive mechanism for target location. Biochemistry 42: 801–810 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T (2006) DNA Repair and Mutagenesis. Washington, DC: ASM Press [Google Scholar]
- Fromme JC, Banerjee A, Huang SJ, Verdine GL (2004) Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature 427: 652–656 [DOI] [PubMed] [Google Scholar]
- Fromme JC, Verdine GL (2003a) DNA lesion recognition by the bacterial repair enzyme MutM. J Biol Chem 278: 51543–51548 [DOI] [PubMed] [Google Scholar]
- Fromme JC, Verdine GL (2003b) Structure of a trapped endonuclease III–DNA covalent intermediate. EMBO J 22: 3461–3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA (1998) MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol 5: 1058–1064 [DOI] [PubMed] [Google Scholar]
- Hollis T, Ichikawa Y, Ellenberger T (2000a) DNA bending and a flip-out mechanism for base excision by the helix–hairpin–helix DNA glycosylase, Escherichia coli AlkA. EMBO J 19: 758–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollis T, Lau A, Ellenberger T (2000b) Structural studies of human alkyladenine glycosylase and E. coli 3-methyladenine glycosylase. Mutat Res 460: 201–210 [DOI] [PubMed] [Google Scholar]
- Kwon K, Cao C, Stivers JT (2003) A novel zinc snap motif conveys structural stability to 3-methyladenine DNA glycosylase I. J Biol Chem 278: 19442–19446 [DOI] [PubMed] [Google Scholar]
- Labahn J, Schärer OD, Long A, Ezaz-Nikpay K, Verdine GL, Ellenberger TE (1996) Structural basis for the excision repair of alkylation-damaged DNA. Cell 86: 321–329 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Macarthur MW, Moss DS, Thornton JM (1993) Procheck—a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T (2000) Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci USA 97: 13573–13578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery R, Sklenar H (1988) The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J Biomol Struct Dyn 6: 63–91 [DOI] [PubMed] [Google Scholar]
- Livingston AL, Kundu S, Henderson Pozzi M, Anderson DW, David SS (2005) Insight into the roles of tyrosine 82 and glycine 253 in the Escherichia coli adenine glycosylase MutY. Biochemistry 44: 14179–14190 [DOI] [PubMed] [Google Scholar]
- McCarthy TV, Karran P, Lindahl T (1984) Inducible repair of O-alkylated DNA pyrimidines in Escherichia coli. EMBO J 3: 545–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol CD, Arvai AS, Begley TJ, Cunningham RP, Tainer JA (2002) Structure and activity of a thermostable thymine-DNA glycosylase: evidence for base twisting to remove mismatched normal DNA bases. J Mol Biol 315: 373–384 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica D53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nash HM, Bruner SD, Scharer OD, Kawate T, Addona TA, Spooner E, Lane WS, Verdine GL (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr Biol 6: 968–980 [DOI] [PubMed] [Google Scholar]
- Norman DP, Bruner SD, Verdine GL (2001) Coupling of substrate recognition and catalysis by a human base-excision DNA repair protein. J Am Chem Soc 123: 359–360 [DOI] [PubMed] [Google Scholar]
- Norman DP, Chung SJ, Verdine GL (2003) Structural and biochemical exploration of a critical amino acid in human 8-oxoguanine glycosylase. Biochemistry 42: 1564–1572 [DOI] [PubMed] [Google Scholar]
- O'Connor TR, Laval J (1991) Human cDNA expressing a functional DNA glycosylase excising 3-methyladenine and 7-methylguanine. Biochem Biophys Res Commun 176: 1170–1177 [DOI] [PubMed] [Google Scholar]
- O'Rourke EJ, Chevalier C, Boiteux S, Labigne A, Ielpi L, Radicella JP (2000) A novel 3-methyladenine DNA glycosylase from Helicobacter pylori defines a new class within the endonuclease III family of base excision repair glycosylases. J Biol Chem 275: 20077–20083 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA (1998) Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J 17: 5214–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin S, Lindahl T (1978) Properties of 3-methyladenine-DNA glycosylase from Escherichia coli. Biochemistry 17: 2110–2118 [DOI] [PubMed] [Google Scholar]
- Roberts RJ, Cheng X (1998) Base flipping. Annu Rev Biochem 67: 181–198 [DOI] [PubMed] [Google Scholar]
- Saparbaev M, Kleibl K, Laval J (1995) Escherichia coli, Saccharomyces cerevisiae, rat and human 3-methyladenine DNA glycosylases repair 1, N6-ethenoadenine when present in DNA. Nucleic Acids Res 23: 3750–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuker DE, Bailey E, Parry A, Lamb J, Farmer PB (1987) The determination of urinary 3-methyladenine in humans as a potential monitor of exposure to methylating agents. Carcinogenesis 8: 959–962 [DOI] [PubMed] [Google Scholar]
- Stivers JT, Jiang YL (2003) A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem Rev 103: 2729–2759 [DOI] [PubMed] [Google Scholar]
- Takeshita M, Chang CN, Johnson F, Will S, Grollman AP (1987) Oligodeoxynucleotides containing synthetic abasic sites. Model substrates for DNA polymerases and apurinic/apyrimidinic endonucleases. J Biol Chem 262: 10171–10179 [PubMed] [Google Scholar]
- Terwilliger TC (2000) Maximum likelihood density modification. Acta Crystallogr D D56: 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D D55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA (1995) Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J 14: 4108–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas L, Yang CH, Goldthwait DA (1982) Two DNA glycosylases in Escherichia coli which release primarily 3-methyladenine. Biochemistry 21: 1162–1169 [DOI] [PubMed] [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol 229: 105–124 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information